


Abstract
The previously reported 11C-labeled GluN2B PET radioligand 11C-Me-NB1 served as a starting point for derivatization and led to the successful development of a radiofluorinated analog designated (R)-18F-OF-Me-NB1. Given the short physical half-life of 20.3 min for 11C, (R)-18F-OF-Me-NB1 with a physical half-life of 109.8 min would allow satellite distribution to nuclear medicine facilities without an on-site cyclotron. Methods: Two fluorinated Me-NB1 derivatives, OF-Me-NB1 and PF-Me-NB1, were synthesized. On chiral resolution, the respective enantiomers were radiolabeled with 11C and assessed in a proof-of-concept study by applying in vitro autoradiography on rodent brain sections. On the basis of the autoradiograms, (R)-OF-Me-NB1 was selected for radiofluorination and preclinical evaluation by ex vivo autoradiography, PET imaging, biodistribution, and metabolite studies on Wistar rats. To rule out off-target binding to the σ1 receptor (σ1R), brain uptake of (R)-18F-OF-Me-NB1 in wild-type mice was compared with that in σ1R knock-out mice. Results: Autoradiographic assessment revealed that both enantiomers of 11C-PF-Me-NB1 distributed homogeneously across all brain regions on rodent brain sections. In contrast, the 2 enantiomers of 11C-OF-Me-NB1 exhibited an entirely different behavior. Although (S)-11C-OF-Me-NB1 bound to virtually all brain regions, with considerable σ1R binding, (R)-11C-OF-Me-NB1 exhibited high selectivity and specificity for the GluN2B-rich rat forebrain. These findings were confirmed for the radiofluorinated analog (R)-18F-OF-Me-NB1, which was obtained via copper-mediated radiofluorination in radiochemical yields of 13%–25% and molar activities ranging from 61 to 168 GBq/μmol. PET imaging and biodistribution studies on Wistar rats indicated an appropriate pharmacokinetic profile and high in vivo specific binding of (R)-18F-OF-Me-NB1 as revealed by blocking studies with the GluN2B antagonist CP101,606. Off-target binding to the σ1R was excluded by PET imaging of σ1R knock-out mice. Half-maximal receptor occupancy by CP101,606 occurred at 8.3 μmol/kg (intravenous). Conclusion: (R)-18F-OF-Me-NB1 is a promising radiofluorinated probe that exhibits specificity and selectivity for the GluN2B-containing N-methyl-d-aspartate complex and enables in vivo target occupancy studies on rodents.
Excessive glutamatergic stimulation leads to the activation of proapoptotic neuronal signaling pathways contributing to neurodegeneration (1). This excitotoxicity state is associated with numerous pathologic conditions of the central nervous system, including Parkinson disease, Alzheimer disease, neuropathic pain, and depression, as well as ischemic brain injury (2,3). Because of their high permeability for calcium ions, N-methyl-d-aspartate (NMDA) receptors are particularly implicated in glutamate-mediated neurotoxicity. Among the NMDA receptor subunits, GluN2B has emerged as a promising target for subtype-selective modulation. Accordingly, therapeutic interventions by GluN2B antagonists have proven to be effective for Parkinson disease, cerebral ischemia, neuropathic pain, and depression in preclinical trials (4–7). In contrast to broad-spectrum NMDA channel blockers, selective GluN2B antagonists have an improved safety profile with lower cognitive side effects, as GluN2B is hardly expressed in the cerebellum, a brain region that regulates cognitive functions through mapping of cerebral association networks (8,9). Since the discovery of the N-terminal domain binding pocket of GluN2B-carrying NMDA receptors, several antagonists have been developed, and promising candidates such as CP101,606 and CERC-301 (MK-0657) have been evaluated in clinical trials (9–11). Despite high expectations that arose from animal studies, initial euphoria has dissipated as increasing numbers of clinical trials with GluN2B-selective drug candidates have shown limited efficacy (11–13). It is therefore crucial to guide these clinical trials with diagnostic tools such as PET, which allows one to predict and assess target engagement at a given drug dose in vivo (14).
We recently reported on the development of 11C-Me-NB1, a 3-benzazepine–based GluN2B PET tracer with promising attributes that make it possible to address the several issues that limited previously reported GluN2B imaging probes (15). A major limitation of 11C-Me-NB1 remains the short physical half-life of 11C (20.3 min). Particularly, this limitation confines the use of 11C-Me-NB1 to facilities with an on-site cyclotron. To overcome this limitation, we envisioned the development of a suitable 18F (physical half-life, 109.8 min)-labeled analog of 11C-Me-NB1. Therefore, we synthesized 2 fluorinated analogs, PF-Me-NB1 and OF-Me-NB1 (Fig. 1), and evaluated their utility as imaging agents for the GluN2B subunit of the ionotropic NMDA receptor. Because of the distinct enantiomeric behavior observed for 11C-Me-NB1, both fluorinated derivatives were separated into their enantiomerically pure (R) and (S) forms. We initially labeled the 4 enantiomers (R)-OF-Me-NB1, (S)-OF-Me-NB1, (R)-PF-Me-NB1, and (S)-PF-Me-NB1 with 11C and assessed in a proof-of-concept study their performance characteristics by in vitro autoradiography using rodent brain sections. On the basis of the autoradiograms, (R)-OF-Me-NB1 was selected for radiofluorination and preclinical evaluation. The results of the preclinical study showed that (R)-18F-OF-Me-NB1 is a promising radioligand for imaging GluN2B receptors and, furthermore, that it can be used to study the in vivo receptor occupancy of CP101,606, the only existing GluN2B antagonist with reported clinical efficacy (4,16,17). Derivatives of 3-benzazepin-1-ol have been shown to generally exhibit high selectivity over a series of central nervous system receptors, including the phencyclidine-binding site and the σ2 receptor; however, off-target binding to σ1 receptor (σ1R) has been reported, wherein a slight structural modification can shift the binding profile from a GluN2B binder to a σ1R-selective ligand (18). Therefore, to rule out off-target binding to the σ1R, the in vivo brain uptake of (R)-18F-OF-Me-NB1 in wild-type mice was compared with σ1R-knock-out (σ1R-KO) mice.
Chemical structures of 11C-Me-NB1, PF-Me-NB1, and OF-Me-NB1.
MATERIALS AND METHODS
The syntheses of precursors and reference compounds—including 1H- and 13C-nuclear MR; high-resolution mass spectrometry characterizations; and high-performance liquid chromatography conditions, chiral separations, and circular dichroism spectra—are presented as supplemental material (available at http://jnm.snmjournals.org). Animal studies were performed in compliance with the Swiss Animal Protection Law and the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines and after approval from the local veterinary office of the Canton Zurich, Switzerland. Wistar rats and CD1 mice were purchased from Charles River, and σ1R-KO mice were provided by Envigo. All animals were kept under standard conditions as previously reported (15).
Radiochemistry
18F-fluoride ions were produced by bombardment of 98% enriched (18O) water via the 18O(p,n)18F nuclear reaction in a Cyclone 18/9 cyclotron (18-MeV; IBA) with a starting activity of approximately 50 GBq in 0.94 mL of 18O-enriched water. The activity was trapped on an anion-exchange cartridge (Waters SepPak Accell QMA cartridge, carbonate) and subsequently eluted with a solution of Kryptofix 222 (6.3 mg/mL, 16.7 μmol/mL; Merck), K2C2O4 (1.0 mg/mL, 5.4 μmol/mL), and K2CO3 (0.1 mg/mL, 0.7 μmol/mL) in a mixture of acetonitrile and H2O (4:1, 0.9 mL), followed by azeotropic drying with acetonitrile (3 × 1 mL) (19). A borosilicate glass Reacti-Vial (Wheaton Industries, 6 mL) sealed with a plastic cap containing a Teflon (DuPont)-faced rubber septum was purged with air (20 mL), and a solution of 6–8 mg (12–16 μmol) of boronic ester (R)-7 and 14 mg (21 μmol) of Cu(OTf)2(py)4 in 0.3 mL of dry dimethylacetamide was added. The resulting solution was stirred at 120°C for 20 min and diluted with 1.5 mL of acetonitrile/H2O (1:1). On addition of 0.4 mL of aqueous NaOH (10 M, 400 mg/mL), the mixture was stirred at 90°C for 15 min. The final product was purified by semipreparative high-performance liquid chromatography, and the final product was formulated with 5% ethanol in saline. This vehicle was used for all biologic experiments. The identities of the radioligands were confirmed by coinjection with corresponding nonradioactive reference compounds. 11C-labeled probes were synthesized using the same procedure as previously reported for 11C-Me-NB1 (15).
In Vitro Autoradiography
Rodent brain tissue was embedded in Tissue-Tek optimal-cutting-temperature compound (Sakura Finetek ). Coronal rat and mouse brain sections of 10-μm thickness were prepared on a cryostat (Cryo-Star HM 560 MV; Microm/Thermo Scientific). The tissue sections were mounted to SuperFrost Plus slides (Menzel) and stored at −20°C until further use. Before the autoradiography experiments, brain slices were initially thawed for 15 min on ice and subsequently preconditioned for 10 min at 0°C in assay buffer 1 (pH 7.4) containing 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (30 mM, 7.1 mg/mL), MgCl2 (0.56 mM, 0.05 mg/mL), NaCl (110 mM, 6.4 mg/mL), CaCl2 (3.3 mM, 0.4 mg/mL), KCl (5 mM, 0.4 mg/mL), and 0.1% fatty acid–free bovine serum albumin. On drying, the tissue sections were incubated with 1 mL of the respective radioligand (3 nM) for 15 min at 21°C in a humidified chamber. For σ1R blockade, 1 μM SA4503 (half-maximal inhibitory concentration [IC50] for σ1R, 17.4 nM (20)), fluspidine (inhibition constant for σ1R, 0.59 nM (21)), or (+)-pentazocine (IC50 for σ1R, 13.7 nM (20)) was added to the radiotracer solution. For GluN2B blockade, 1 μM CERC-301 (IC50 for GluN2B, 3.6 nM (22)), EVT 101 (IC50 for GluN2B, 12 nM (23)), or CP101,606 (IC50 for GluN2B, 11 nM (24)) was added to the radiotracer solution. The brain slices were washed for 5 min in assay buffer 1 and further washed twice 3 min in assay buffer 2 (assay buffer 1 without bovine serum albumin). Tissue sections were dipped twice in distilled water, were subsequently dried, and were exposed to a phosphor imager plate (Fuji) for 30 min. The films were scanned in a BAS5000 reader (Fuji), and images were generated using AIDA software (version 4.50.010; Raytest Isotopenmessgeräte GmbH).
Metabolite Study
Wistar rats were injected with 242–704 MBq (14.8–27.5 nmol/kg) of (R)-18F-OF-Me-NB1. Samples of the brain extracts at predefined times (15, 30, and 60 min) and plasma (5, 15, 30, 45, and 60 min) were obtained and analyzed by radio-ultra-performance liquid chromatography (UPLC) and radio-thin-layer chromatography.
Ex Vivo Biodistribution
Eight male Wistar rats (4 baseline and 4 blockade animals) were administered (R)-18F-OF-Me-NB1 (5–16 MBq, 2.0–4.9 nmol/kg) via tail-vein injection and sacrificed by decapitation under isoflurane anesthesia at 30 min after injection. For blockade experiments, a 2 mg/kg dose of eliprodil in a vehicle of aqueous glucose (5%), NaCl (0.45%), and citric acid (1 mM) was injected shortly before tracer administration. Organs were dissected and weighed, and radioactivity was measured in a γ-counter (Perkin Elmer). Biodistribution results are reported as percentage normalized injected dose per gram of tissue.
In Vivo PET and Dose–Response Studies
CD1 mice, σ1R-KO mice, and Wistar rats were anesthetized with isoflurane and scanned in a PET/CT scanner (Super Argus; Sedecal) on tail-vein injection of (R)-18F-OF-Me-NB1 (15–38 MBq, 0.6–1.7 nmol/kg [rats, scan time of 0–90 min], or 3–16 MBq, 4.3–19.7 nmol/kg [mice, scan time of 1–90 min]). For anatomic orientation, PET scans were followed by CT scans. For blockade studies, a 2 mg/kg dose of eliprodil was injected into CD1 and σ1R-KO mice a few seconds before the radioligand. Dose–response and receptor occupancy studies in Wistar rats were conducted by tail-vein injection of different doses (0.1, 1, 2, 3, and 10 mg/kg) of CP101,606 shortly before tracer administration. The obtained data were reconstructed in user-defined time frames with a voxel size of 0.3875 × 0.3875 × 0.775 mm as previously described by our group (15). Time–activity curves were generated by PMOD (version 3.7; PMOD Technologies) with predefined regions of interest. The results are given as SUVs. Receptor occupancy evaluations were performed using the area under the curve of the SUVs calculated from 4 to 90 min as previously reported by our group (15).
Statistical Analysis
An independent 2-tailed Student test assuming normal distribution of the dataset was used to calculate statistical probability values.
RESULTS
Chemistry and Binding Affinity
Reference compounds, OF-Me-NB1 and PF-Me-NB1, were obtained in chemical yields of 41% and 49%, respectively (Supplemental Fig. 1). By competitive binding assays with 3H-ifenprodil and rat brain homogenates, inhibition constants for the GluN2B binding site of 37 nM ((rac)-OF-Me-NB1), 56 nM ((rac)-PF-Me-NB1), and 4 nM ((R)-OF-Me-NB1) were determined. Applying the same competitive binding assay with (+)-3H-pentazocine and rat brain homogenates, inhibition constants obtained for σ1R were 12 nM, 32 nM, and 100 nM for (rac)-PF-Me-NB1, (rac)-OF-Me-NB1, and (R)-OF-Me-NB1, respectively. The respective phenolic precursors, OF-NB1 and PF-NB1, were obtained by treatment of OF-Me-NB1 and PF-Me-NB1 with BBr3 in 41% and 53% chemical yields, respectively. The synthetic strategy, chiral separations, and circular dichroism results are described in Supplemental Figures 2 and 3. The multistep synthesis of boronic pinacol ester precursor 7 for radiofluorination is provided in Supplemental Figure 4.
11C Labeling of PF- and OF-Me-NB1 and Preliminary In Vitro Autoradiography
(R)- and (S)-11C-PF-Me-NB1, as well as (R)- and (S)-11C-OF-Me-NB1, were obtained by chiral resolution of the respective racemic phenolic precursors followed by 11C labeling using 11C-MeI. The radiochemical yields ranged from 7% to 36% (decay-corrected, calculated from the 11C-CO2 starting activity), with molar activities between 54 and 220 GBq/μmol. The radiochemical purity was greater than 99% for all 4 radioligands. Introduction of 18F at the para position of the phenyl moiety resulted in a homogeneous binding across all brain regions on coronal rat brain sections for both enantiomeric forms (Fig. 2A). In contrast, the introduction of fluorine at the ortho position resulted in a high and specific accumulation of the (R)-enantiomer in GluN2B-rich forebrain regions (Fig. 2B). To further investigate the underlying causes for the differences in the regional accumulation of the 4 different radioligands, we performed autoradiography studies on mouse brain sections using GluN2B (specificity assessment) and σ1R ligands (selectivity assessment) as blockers. We found that both para-fluorinated enantiomers (R)- and (S)-11C-PF-Me-NB1 showed considerable σ1R binding (Supplemental Figs. 5 and 6). Remarkably, the ortho-fluorinated enantiomers (R)- and (S)-11C-OF-Me-NB1 revealed an entirely different receptor binding behavior (Fig. 2C). Although (R)-11C-OF-Me-NB1 exhibited GluN2B specificity and selectivity over the σ1R, (S)-11C-OF-Me-NB1 was found to bind predominantly to the σ1R. Consequently, (R)-OF-Me-NB1 was selected for radiofluorination and further preclinical evaluation.
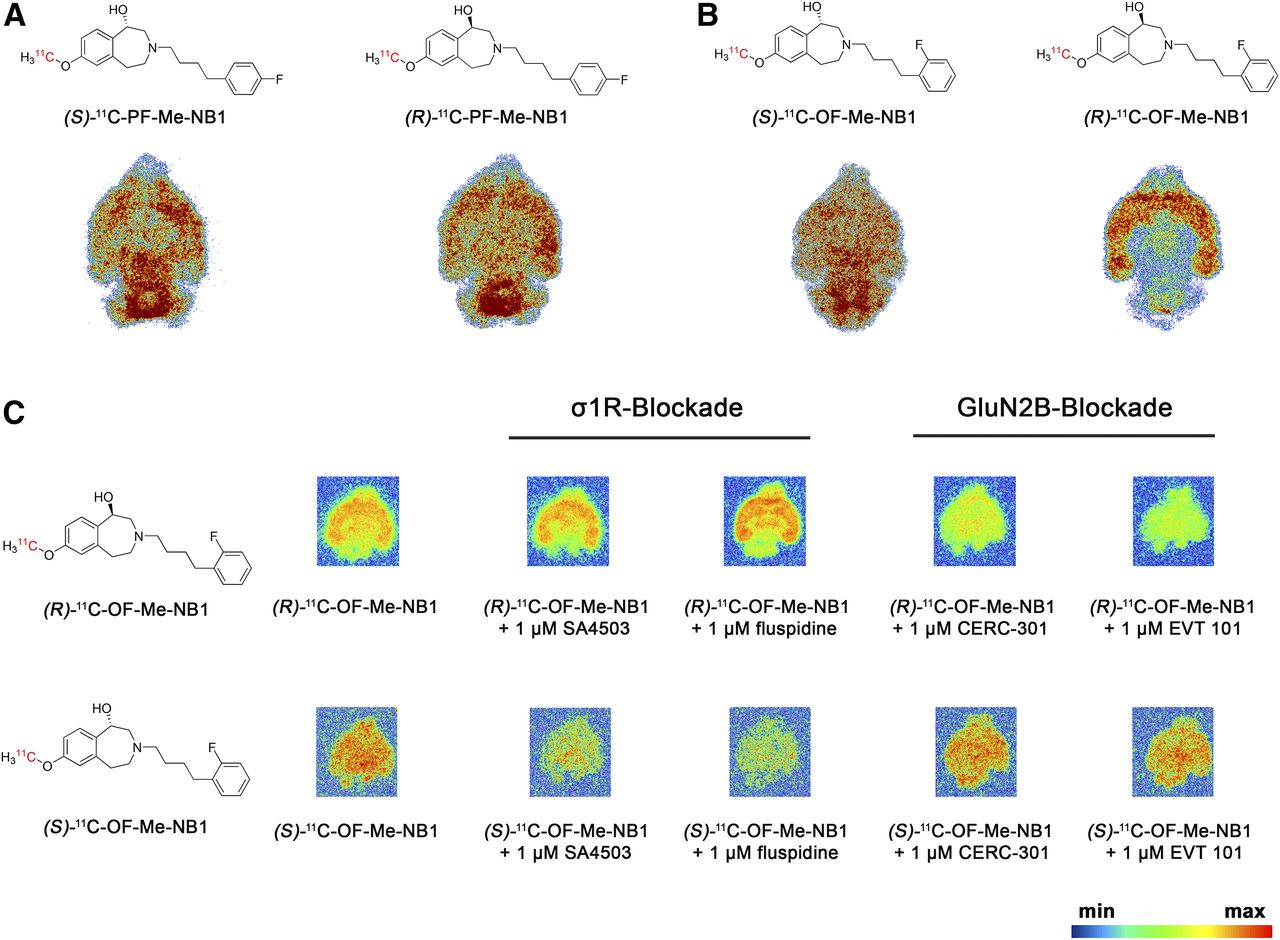
Representative in vitro autoradiograms with 11C-labeled probes on coronal rat and mouse brain sections. (A) Para-fluorinated derivatives (R)- and (S)-11C-PF-Me-NB1 revealed binding across all rat brain regions, with cerebellum showing highest accumulation. (B) Ortho-fluorinated derivatives (R)- and (S)-11C-OF-Me-NB1 exhibited distinct binding patterns. Although (S)-enantiomer distributed uniformly across all rat brain regions, (R)-enantiomer revealed high selectivity for GluN2B-expressing forebrain regions. (C) Distinct binding patterns of (R)- and (S)-11C-OF-Me-NB1 were further confirmed on mouse brain sections. Blocker screening revealed GluN2B specificity for (R)-enantiomer and considerable σ1R binding of (S)-enantiomer.
Radiofluorination and In Vitro Autoradiography with (R)-11C-OF-Me-NB1
To accomplish the radiofluorination, aryl boronic ester precursor 7 was synthesized as shown in Supplemental Figure 4. The radiosynthesis presented in Figure 3A was performed according to the procedure recently reported by Preshlock et al., but with slight modifications (19). (R)-18F-OF-Me-NB1 was obtained in 13%–25% radiochemical yields (n = 8, based on high-performance liquid chromatography analysis of the crude product), and radiochemical purity was more than 99%. Molar activities ranged from 61 to 168 GBq/μmol at the end of the synthesis. Enantiopurity of the radioligand was assessed by chiral high-performance liquid chromatography. A logD7.4 value of 2.2 ± 0.1 (n = 4) was obtained using the shake-flask method. (R)-18F-OF-Me-NB1 revealed a high selectivity for the GluN2B-expressing cortex, striatum, thalamus, and hippocampus over the GluN2B-deficient cerebellum on rat brain autoradiograms (Fig. 3B). Blockade studies were conducted with the σ1R ligands (+)-pentazocine, SA4503, and fluspidine to elucidate their ability to compete with (R)-18F-OF-Me-NB1 binding. To confirm GluN2B specificity, the GluN2B antagonists EVT 101 and CP-101,606 were included in the same experiment. The results of the autoradiography screening with the various ligands confirmed the GluN2B specificity of (R)-18F-OF-Me-NB1, as well as its in vitro selectivity over σ1R.
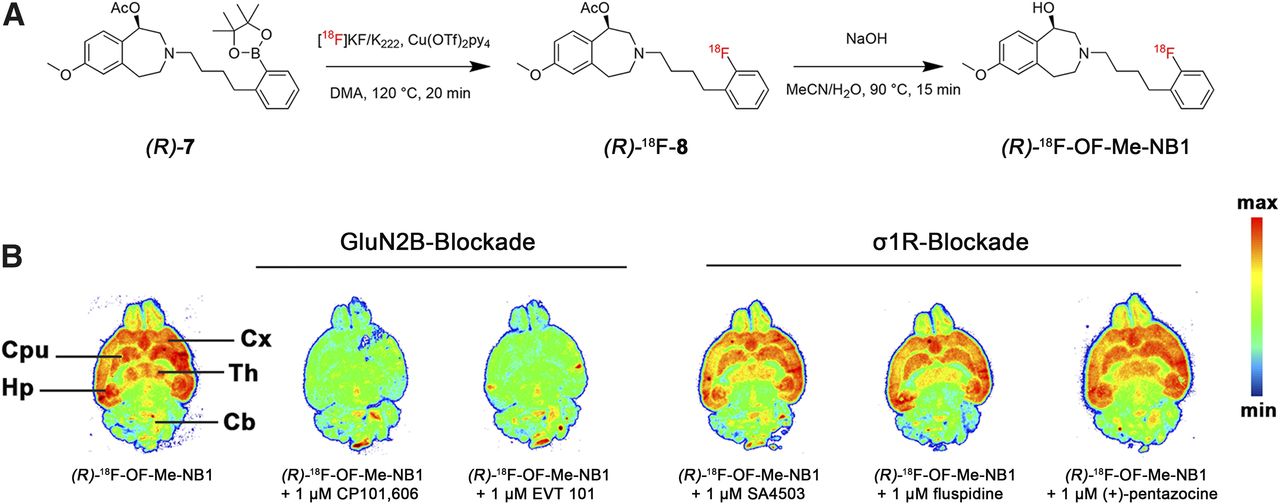
(A) Synthesis of (R)-18F-OF-Me-NB1 using copper-mediated radiofluorination and subsequent deacetylation under basic conditions. (B) Representative in vitro autoradiographic blocker screening in rat brain sections with GluN2B ligands (CP101,606, EVT 101) and σ1R ligands (SA4503, fluspidine, (+)-pentazocine). Cb = cerebellum; CPu = caudate putamen (striatum); Cx = cortex; Hp = hippocampus; Th = thalamus.
Dose-Dependence and Receptor Occupancy Studies
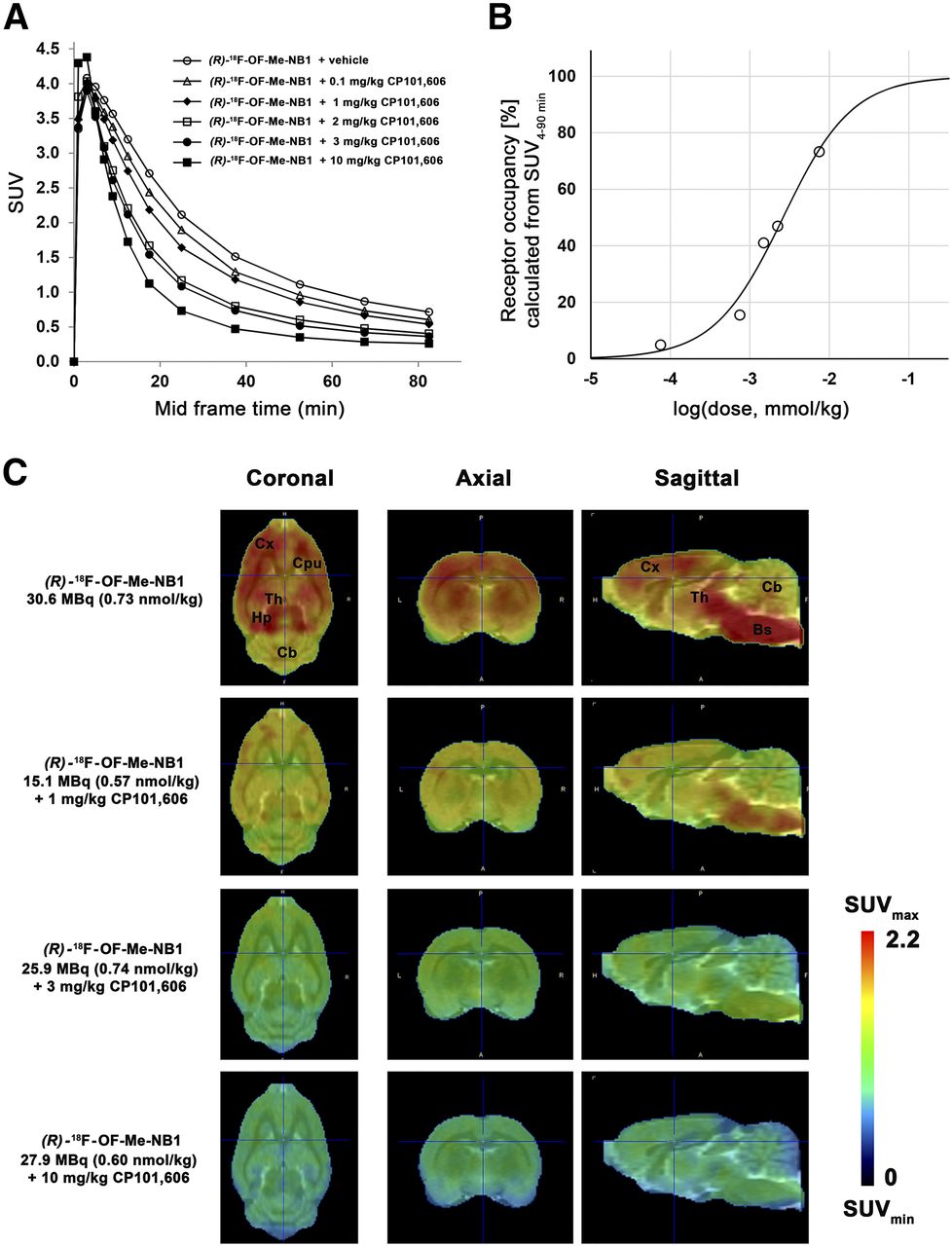
The clinical efficacy of the GluN2B-selective antagonist CP101,606 has been demonstrated in clinical trials (4,16,17). Applying (R)-18F-OF-Me-NB1, we assessed the receptor occupancy of CP101,606 in the rat brain at a dose ranging from 0.1 to 10 mg/kg. A consistent dose-dependent reduction of the SUVs with escalating CP101,606 doses was observed (Fig. 4A). SUVs were further used to determine in vivo receptor occupancy. The CP101,606 dose required for 50% receptor occupancy was 8.3 μmol/kg (Fig. 4B), and the lowest and highest blocker doses showed 4.9% and 73.3% receptor occupancy, respectively. PET images confirming the dose-dependent target engagement are depicted in Figure 4C.
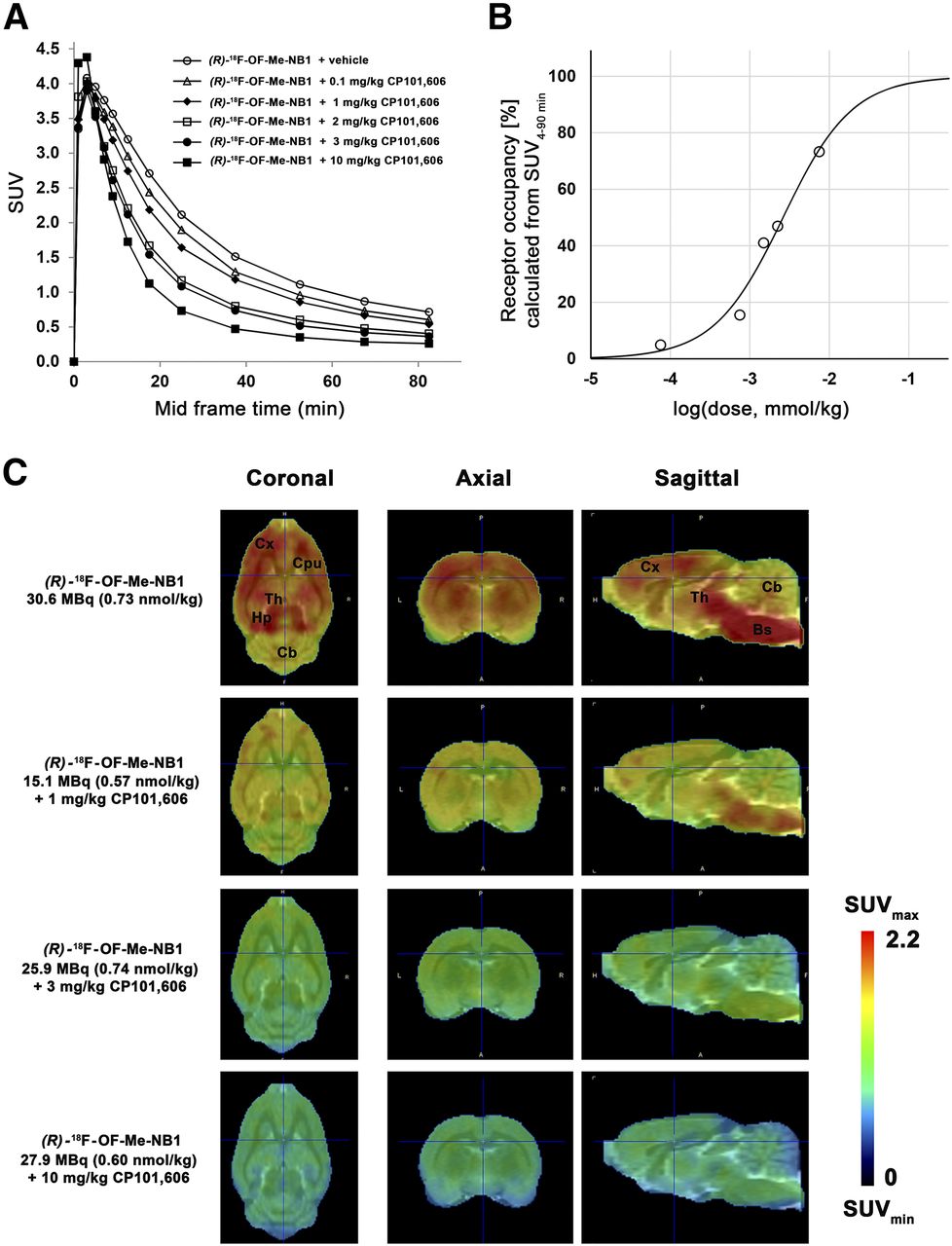
Dose-dependent blocking of (R)-18F-OF-Me-NB1 binding by GluN2B antagonist CP101,606 in Wistar rat brain in vivo and respective calculated receptor occupancy. (A) Time–activity curves for baseline and blockade experiments with escalating injected doses of CP101,606 (0.1–10 mg/kg) are presented as SUVs. (B) Receptor occupancy for each CP101,606 dose calculated from area under curve of SUVs (SUV0–90 min). (C) PET images of rat brain superimposed on PMOD MRI template under baseline and blockade conditions with GluN2B antagonist CP101,606. Images were averaged from 0 to 90 min after injection. Bs = brain stem; Cb = cerebellum; CPu = caudate putamen (striatum); Cx = cortex; Hp = hippocampus; Th = thalamus.
Metabolite Studies
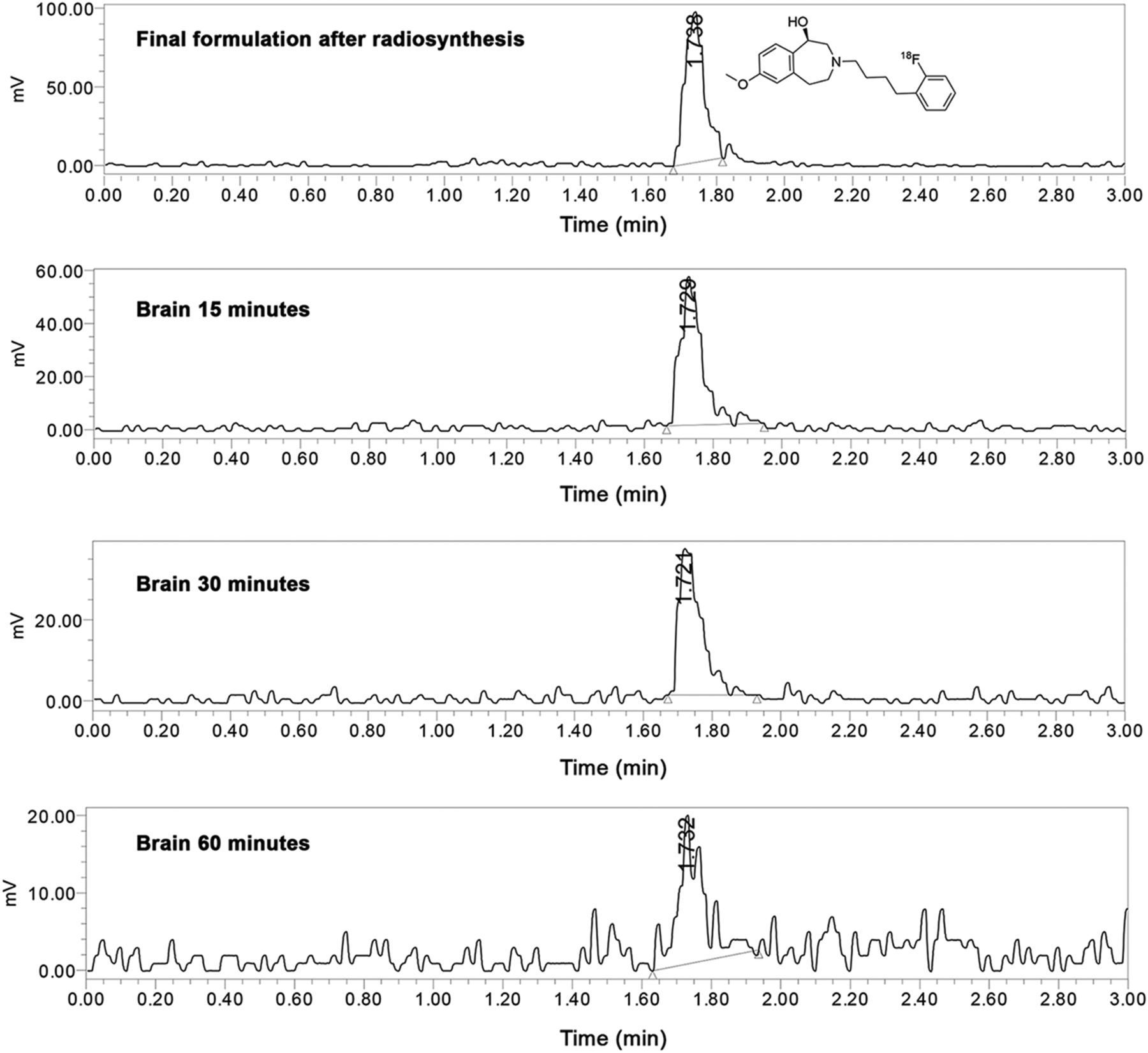
To assess its metabolic stability, (R)-18F-OF-Me-NB1 was injected into male Wistar rats, and plasma radioactivity at 5, 15, 30, 45, and 60 min after injection, as well as brain extracts at 15, 30, and 60 min after injection, were analyzed by radio-UPLC. The analysis revealed the presence of intact tracer in the brain at 15, 30, and 60 min after injection and the absence of radiometabolites up to 30 min after injection (Fig. 5). The chromatogram of the sample obtained at 60 min after injection was rather noisy and did not allow the detection of potential radiometabolites in the brain.

Radio-UPLC analysis of Wistar rat brain extracts. Final tracer formulation after radiosynthesis is depicted, as well as brain samples at 15, 30, and 60 min after injection.
Plasma samples revealed the presence of 2 polar radiometabolites. The fractions of intact parent of total radioactivity found in the plasma at 0, 5, 15, 30, 45, and 60 min after injection are summarized in Table 1. At the last measured time point of 60 min after injection, the fraction of intact tracer was 45%.
Metabolite Study of (R)-18F-OF-Me-NB1 in Male Wistar Rats
Brain Time–Activity Curves and Biodistribution
GluN2B specificity of (R)-18F-OF-Me-NB1 was demonstrated by PET in male Wistar rats on tail-veil injection of the radioligand with the GluN2B antagonists CP101,606 or eliprodil (Fig. 6). Time–activity curves were generally higher in the cortex, hippocampus, striatum, and thalamus than in the cerebellum. These findings were confirmed by ex vivo autoradiography (Supplemental Fig. 7). On injection of CP101,606 or eliprodil (blockers were injected intravenously shortly before the tracer), a blocking effect was observed in all brain regions; however, the reduction of radioactivity accumulation was most pronounced in the GluN2B-rich regions such as the cortex, hippocampus, striatum, and thalamus. Ex vivo biodistribution experiments on male Wistar rats were conducted at 30 min after injection. The time point was based on the in vivo specificity deduced from the time–activity curves of baseline and blockade PET scans with the GluN2B antagonists CP101,606 and eliprodil (Fig. 6). We found a heterogeneous uptake of the radioligand in different brain regions, and radioligand uptake was significantly higher in the GluN2B-expressing regions than in the cerebellum: the cortex-to-cerebellum ratio was 1.37 (P = 0.001), the striatum-to-cerebellum ratio was 1.30 (P = 0.002), the hippocampus-to-cerebellum ratio was 1.36 (P = 0.003), and the thalamus-to-cerebellum ratio was 1.33 (P = 0.007). The ex vivo biodistribution results are summarized in Supplemental Tables 1 and 2. On ex vivo biodistribution and PET imaging studies, the Wistar rat brain stem showed a considerable radioligand uptake, which was efficiently blocked by CP101,606 and eliprodil.
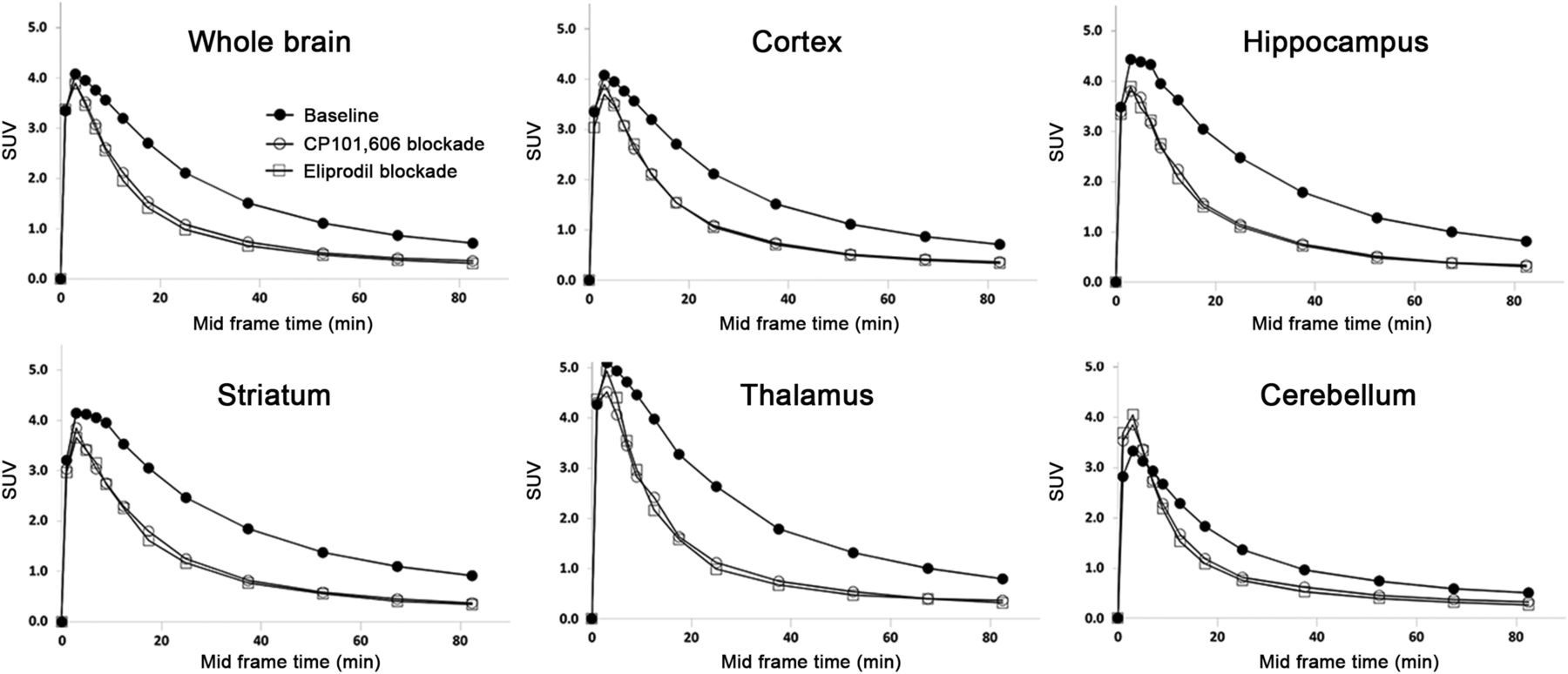
Time–activity curves in different brain regions of Wistar rat are presented as SUVs. Specificity of (R)-18F-OF-Me-NB1 binding was confirmed by injection of GluN2B antagonists CP101,606 (3 mg/kg) and eliprodil (2 mg/kg) shortly before radioligand injection. One rat was scanned for each condition.
PET Experiments with σ1R-KO and Wild-Type Mice
Dynamic PET scans were performed with σ1R-KO and wild-type mice to exclude σ1R binding of (R)-18F-OF-Me-NB1 in vivo. PET images and time–activity curves of radioligand uptake in the brain are presented in Figure 7. There was no difference in the time–activity curves of σ1R-KO mice and the respective wild-type animals (Fig. 7B), indicating that (R)-18F-OF-Me-NB1 brain uptake is independent of the σ1R and, thus, confirming the selectivity of binding of (R)-18F-OF-Me-NB1 to GluN2B receptors over σ1R in vivo. In both σ1R-KO and wild-type mice, injection of a 2 mg/kg dose of the GluN2B antagonist eliprodil resulted in a lower brain uptake and a faster washout of (R)-18F-OF-Me-NB1, indicating specific binding of (R)-18F-OF-Me-NB1 to GluN2B receptors in the mouse brain.
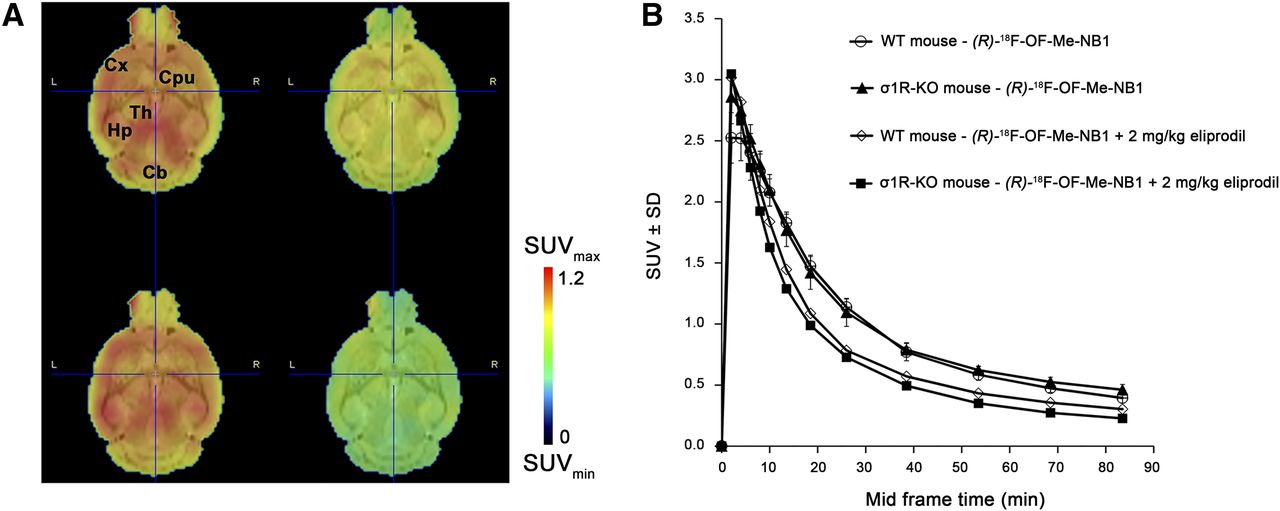
PET study of (R)-18F-OF-Me-NB1 in σ1R-KO and wild-type (WT) mice. For blockade experiments, 2 mg/kg dose of commercially available GluN2B antagonist eliprodil was used. (A) Coronal PET images, averaged from 1 to 90 min after injection, superimposed on MRI template (PMOD). Shown are WT baseline (upper left), WT eliprodil blockade (upper right), σ1R-KO baseline (lower left), and σ1R-KO eliprodil blockade (lower right). (B) Brain time–activity curves are presented as SUV ± SD. Baseline scans were performed on 3 mice and blockade scans on 1 mouse. Cb = cerebellum; CPu = caudate putamen (striatum); Cx = cortex; Hp = hippocampus; Th = thalamus.
DISCUSSION
Given the implication of GluN2B-containing NMDA receptors in various neurologic disorders and the lack of a useful GluN2B-specific PET probe, our research group has dedicated concerted efforts to address this unmet medical need and recently succeeded in developing a suitable 11C-labeled GluN2B PET radioligand, 11C-Me-NB1 (15). The short physical half-life of 11C, however, has prompted us to design analogs that are suitable for 18F labeling. In particular, a radiofluorinated derivative would enable a wider use through satellite distribution to facilities lacking the necessary radiochemistry infrastructure. PF-Me-NB1 and OF-Me-NB1 were synthesized in good chemical yields, and their binding affinities to the rat GluN2B-containing NMDA receptor were shown to be comparable to the 40 nM inhibition constant of lead compound Me-NB1 (15). Because of the differences observed in autoradiography experiments with the enantiomers of 11C-Me-NB1, we initially radiolabeled the 4 candidate compounds, (R)-PF-Me-NB1, (S)-PF-Me-NB1, (R)-OF-Me-NB1, and (S)-OF-Me-NB1, with 11C and assessed their in vitro performance using autoradiography with the aim of finding a suitable GluN2B-specific and -selective candidate for radiofluorination. Autoradiograms of the rodent brain indicated that the introduction of fluorine at the para position of the aryl moiety led to a remarkable reduction in selectivity over the σ1R, possibly because of direct interactions of the fluorine atom with the σ1R. When fluorine was introduced at the ortho position, selectivity over the σ1R was restored, thereby underlining the relevance of the fluorine position within the aromatic ring and supporting the hypothesis of direct fluorine engagement (e.g., via hydrogen bond) in the case of PF-Me-NB1. The promising autoradiography results with (R)-11C-OF-Me-NB1 prompted us to radiolabel (R)-OF-Me-NB1 with 18F. Using a boronic ester precursor, (R)-18F-OF-Me-NB1 was obtained in moderate radiochemical yields, high molar activities, and excellent radiochemical purities. The radiofluorinated probe outperformed 11C-Me-NB1 with regard to forebrain selectivity by in vitro autoradiography (15). In accordance with our previous report on 11C-Me-NB1, we observed a considerable binding of (R)-18F-OF-Me-NB1 to Wistar rat brain stem in vivo, and this binding was abolished by the GluN2B antagonists CP101,606 and eliprodil. Whether this uptake is related to GluN2B or alternative GluN subtypes as suggested for 11C-Me-NB1 remains to be further investigated. Off-target binding to the σ1R was excluded in vivo by PET imaging with σ1R-KO mice. Calculated time–activity curves did not reveal any differences in the brain uptake of σ1R-KO and the respective wild-type mice (n = 3), confirming that the accumulation of (R)-18F-OF-Me-NB1 is independent of the σ1R. Finally, the utility of (R)-18F-OF-Me-NB1 in studying GluN2B target engagement in vivo was demonstrated by receptor occupancy studies with CP101,606, the only existing GluN2B antagonist with documented clinical efficacy (4,17,25). (R)-18F-OF-Me-NB1 is the first radiofluorinated probe that can be successfully used to visualize interactions between drug candidates and the GluN2B-carrying NMDA receptor in vivo. Clinical availability of such a PET radioligand will not only provide the possibility of predicting therapeutic doses but also help to explain why GluN2B antagonists fail to demonstrate clinical efficacy. It remains to be elucidated in future studies whether the 11C version, (R)-11C-OF-Me-NB1, will show in vivo characteristics similar to those of (R)-18F-OF-Me-NB1.
CONCLUSION
We have successfully developed (R)-18F-OF-Me-NB1, which is, to our knowledge, the first radiofluorinated radioligand for imaging GluN2B subunits of the NMDA receptor complex. The data suggest that (R)-18F-OF-Me-NB1 has promising imaging attributes and great potential for receptor occupancy studies with GluN2B drugs. In addition, (R)-18F-OF-Me-NB1 could be a future tool for imaging GluN2B receptors in humans with brain diseases in which the GluN2B receptors have been implicated.
DISCLOSURE
This project was supported by grant 310030E-160403/1 from the Swiss National Science Foundation. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Bruno Mancosu for providing technical assistance with the carbon-11 module and Dr. Jose Miguel Vela for providing the σ1R-KO mice.
Footnotes
Published online Jul. 20, 2018.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication March 28, 2018.
- Accepted for publication July 4, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}