


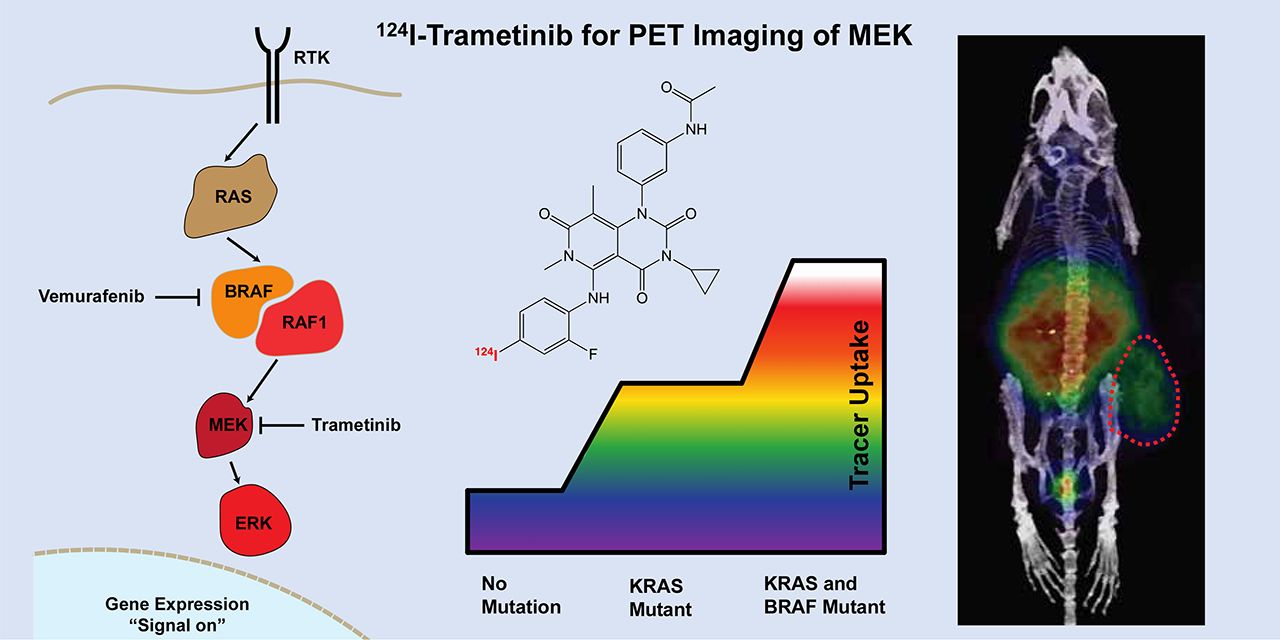
Visual Abstract

Abstract
Trametinib is an extremely potent allosteric inhibitor of mitogen-activated protein kinase (MAPK)/extracellular-signal–regulated kinase (ERK) (MEK) 1/2, which has been approved for treatment of metastatic melanoma and anaplastic thyroid cancer in patients with confirmed BRAFV600E/K mutations. Though trametinib is highly efficacious, adverse side effects, including skin, gastrointestinal, and hepatic toxicity, are dose-limiting and can lead to treatment termination. Development of a noninvasive tool to visualize and quantify the delivery and distribution of trametinib (either as a single agent or in combination with other therapeutics) to tumors and organs would be helpful in assessing therapeutic index, personalizing individual dose, and potentially predicting resistance to therapy. Methods: To address these issues, we have developed a radiolabeled trametinib and evaluated the in vitro and in vivo properties. 123I-, 124I-, and 131I-trametinib, pure tracer analogs to trametinib, were synthesized in more than 95% purity, with an average yield of 69.7% and more than 100 GBq/μmol specific activity. Results: Overall, 124I-trametinib uptake in a panel of cancer cell lines can be blocked with cold trametinib, confirming specificity of the radiotracer in vitro and in vivo. 124I-trametinib was taken up at higher rates in KRAS and BRAF mutant cell lines than in wild-type KRAS cancer cell lines. In vivo, biodistribution revealed high uptake in the liver 2 h after injection, followed by clearance through the gastrointestinal tract over 4 d. Importantly, uptake higher than expected was observed in the lung and heart for up to 24 h. Peak uptake in the skin and gastrointestinal tract was observed between 6 and 24 h, whereas in B16F10 melanoma–bearing mice peak tumor concentrations were achieved between 24 and 48 h. Tumor uptake relative to muscle and skin was relatively low, peaking at 3.4- to 8.1-fold by 72 h, respectively. The biodistribution of 124I-trametinib was significantly reduced in mice on trametinib therapy, providing a quantitative method to observe MEK inhibition in vivo. Conclusion: 124I-trametinib serves as an in vivo tool to personalize the dose instead of using the current single-fixed-dose scheme and, when combined with radiomic data, to monitor the emergence of therapy resistance. In addition, the production of iodinated trametinib affords researchers the ability to measure drug distribution for improved drug delivery studies.
Targeted small-molecule therapy is based on identifying and inhibiting an overactive signaling protein downstream in a pathway such as in the tyrosine receptor kinase. The mitogen-activated protein kinase (MAPK) pathway contains kinase kinases (MAPKKK), which phosphorylate another MAPK kinase (MAPKK), which ultimately activates another MAPK such as an extracellular-signal–regulated kinase (ERK) (1). In the tyrosine receptor kinase pathway, Ras is a G-protein, Raf is a MAPKKK, MAPK/ERK kinase (MEK) is MAPKK, and ERK is MAPK. As this pathway is not unique to cancer cells, side effects can arise in healthy tissue on inhibition. Many cancers possess mutant Braf and Ras kinases, resulting in hyperactive signaling through MEK and ERK, ultimately turning on proliferative genes (Fig. 1) (2). Trametinib (Mekinist; GlaxoSmithKline) is a Food and Drug Administration–approved BRAF-mutation–containing MEK inhibitor for use in metastatic melanoma. As a type III MEK inhibitor, trametinib binds MEK1/2 with a half-maximal inhibitory concentration of 0.9 nM as an allosteric inhibitor outside the adenosine triphosphate–binding pocket, preventing Raf phosphorylation of MEK on S217 and thus ERK, whereas overall MEK levels are unaffected (3). The use of trametinib as an MEK inhibitor has allowed improvement of survival in patients with melanoma and new strategies to treat mutant KRAS lung cancers (4). More recently, activation of natural killer cells when treated in combination with cyclin-dependent kinase 4/6 inhibitors (5) provides even newer uses for MEK inhibitors.
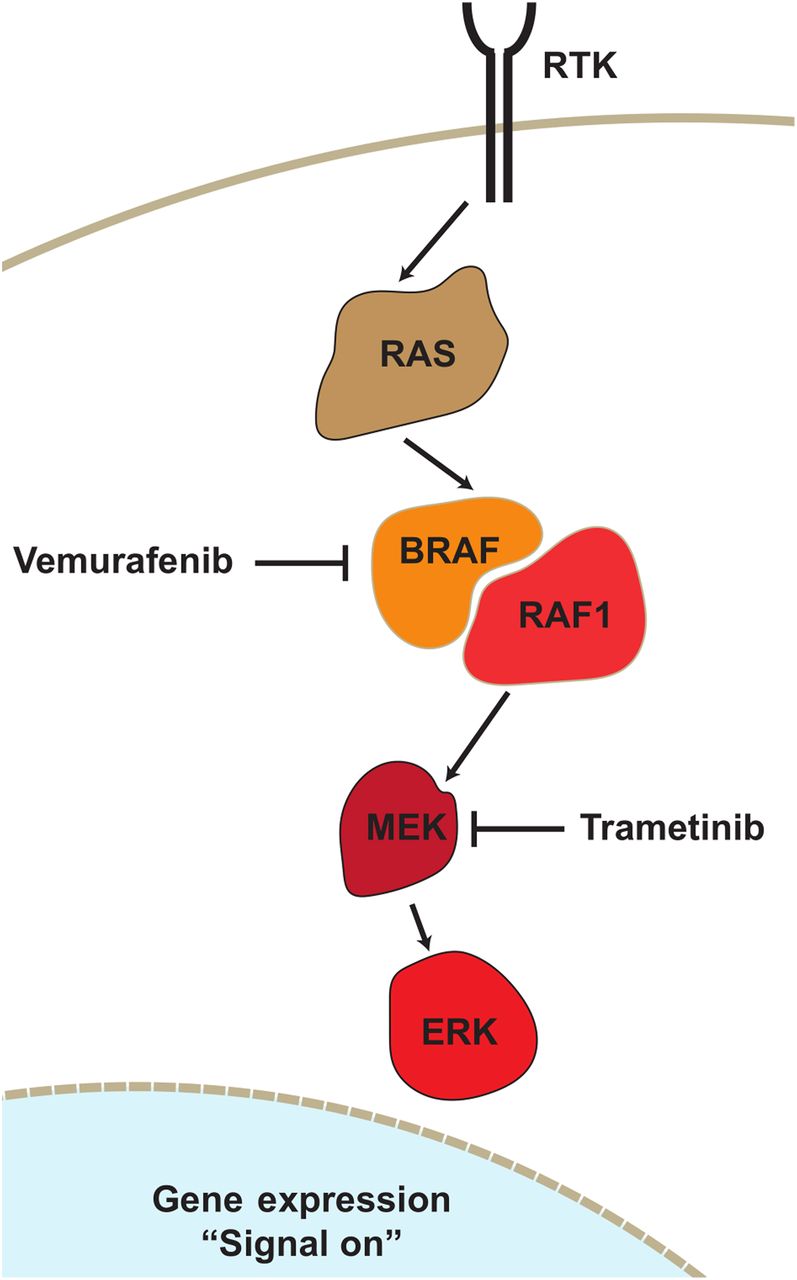
Receptor tyrosine kinase (RTK) pathway containing RAS and BRAF. MEK is found in receptor tyrosine kinase pathway downstream of both KRAS and BRAF. Trametinib is MEK1/2 inhibitor and Food and Drug Administration–approved for BRAFV600E mutant cancers. By inhibiting MEK activity, trametinib can reduce hyperproliferative signaling from KRAS or BRAF mutants, though on-target toxicity is observed even with wild-type KRAS and BRAF cells.
Despite the high potency of trametinib, toxicity in the skin, gastrointestinal tract, and liver reduces compliance with continued use (6). This toxicity is on-target/off-tumor, as healthy tissue also uses the tyrosine receptor kinase pathway; quantification of drug distribution is therefore important for patient dosing, understanding of target tissue and organs, and monitoring of potential resistance to therapy. We aimed to build a radioactive version of trametinib allowing for PET or SPECT imaging. According to the Food and Drug Administration toxicology package, trametinib is not highly metabolized by cytochrome P450 enzymes and is metabolized primarily by deacetylation, demethylation, ketone formation, monooxygenation, and glucuronidation (7). The para-positioned iodine on the 2,4-fluoro-iodoanilino ring of trametinib would be an ideal site for radiotracer placement because the iodine ring directly engages the MEK subpocket (8,9). The para-positioned iodine also remains intact in the major metabolite, known as M5, which is 2-fold less potent for MEK1 and phospho-MEK1 than the parent trametinib, allowing tracking of the metabolite in addition to the parent. Radioiodine is also ideal as a tracer, given the numerous forms readily available for PET or SPECT imaging, and radioiodinated trametinib would be identical to trametinib given therapeutically.
To allow facile radioiodination, the 4-iodo group was replaced with a boronato-pinacol prosthetic group on trametinib. Previous work with (hetero)aryl boron reagents identified radiosynthesis via copper-mediated iodination with high selectivity in a mild 1-step procedure (10–12). Production of 123I-, 124I-, and 131I-trametinib from a boronato-pinacol could be initiated and purified within 1.5 h from the boronato-pinacol-trametinib precursor with approximately 70% relative radiochemical yield. Iodine is not cleaved during metabolism of trametinib, and thus radioiodine will allow in vivo tracking of trametinib in addition to the relevant deacylated and oxidized metabolites, which are still highly potent inhibitors of MEK1 (2- to 10-fold loss in potency from parent) (7). Radiolabeled trametinib is taken up with higher avidity in known BRAF and or KRAS mutant cell lines.
Here, we provide a robust synthesis of radioiodinated trametinib, providing a tool for patient-level pharmacokinetic modeling, patient-specific dosing, and resistance estimation. Radiolabeled trametinib was readily taken up into KRAS and BRAF mutant cell lines and was significantly blocked when coadministered with cold trametinib. The distribution of radiolabeled trametinib in vivo follows target organs known for toxicity such as the skin and gastrointestinal tract but also reveals distribution in the heart and lung at levels not consistent with distribution by the blood. Accumulation in trametinib-naïve tumors peaked at 48 h. Future work could use radiolabeled trametinib as a tool for developing new drug delivery systems, focusing on the distribution of the drug and metabolites. Radioiodinated trametinib can monitor MEK1/2 levels, such as in KRAS and BRAF mutant cancers, thus personalizing dosages needed per patient during therapy and serving as a diagnostic for MEK resistance.
MATERIALS AND METHODS
Trametinib Synthesis
Trametinib was modified using diborylpinacol under palladium catalysis at 130°C with a microwave reactor to contain a bora-pinacol group in lieu of natural iodine, making the trametinib-boronato-precursor. Radioiodination was attempted via 2 methods: chloramine-T and a copper-mediated insertion. Briefly, the copper-mediated insertion uses greater than 37 MBq of 123I-, 124I-, or 131I-NaI in 20–100 μL added to an Eppendorf tube containing the trametinib-boronato precursor (200 μg, 200 μL of ethyl acetate), copper acetate (0.16 mg,73 μL), and 1,10-phenanthroline (0.20 mg, 200 μL) in a methanol:water mixture (4:1). The reaction mixture was heated at 80°C for 35 min and allowed to cool to room temperature. The progress of the reaction was monitored on high-performance liquid chromatography (HPLC) with a Bioscan radiodetector (Phenomenex Luna C-18 column, 250 × 4.6 mm, 5 μm, 100 Å) using a gradient of 5%–95% CH3CN in 0.1% trifluoroacetic acid in water (2–15 min). Iodinated trametinib was isolated by HPLC from reagents in the 95% CH3CN fraction, diluted to 5% with phosphate-buffered saline, and captured onto a Waters Sep-Pak Light C18 cartridge. Iodinated trametinib either was recovered using EtOH and rotary-evaporated to near dryness or was recovered directly with dimethyl sulfoxide.
Radiotracer Production
124I was received with gratitude from the Memorial Sloan Kettering Cancer Center Radiochemistry and Molecular Imaging Probe Facility after production and purification into 0.05 M Na124I. 131I and 123I were purchased from National Diagnostic Products and provided as NaI in 0.05N NaOH or sulfate buffer.
Cell Uptake Studies
Uptake kinetic studies of 124I-trametinib were done in 24-well plates, with cell lines seeded at 1.3 × 105 cells per well with standard culture medium. Cell lines were administered 1.85 kBq of 124I-trametinib per well and incubated at 37°C for up to 72 h. Blocking experiments were done with 1 μg of cold trametinib, and a titration curve of cold trametinib was performed with the B16F10 cell line for 2 h before 0.05% trypsin addition for γ-counting of 124I-trametinib in cells. Activity present in each vial was determined alongside a standard curve by γ-counting on a Perkin Elmer 2480 Wizard 3 with a count time of 60 s per sample. The murine cell lines tested were AKP, B16F10, KP1, and Raw264.7. The human cell lines tested were 22Rv1, ASPC1, Du145, LNCAP, MDA-MB-231, MiaPaca2, PC3, and SW1222.
Animal Studies
All experiments were done in compliance with, and with the approval of, the Memorial Sloan Kettering Cancer Center research animal resource center and the institutional animal care and use committee. Healthy C57BL/6-J mice were purchased from Taconic. The B16F10 melanoma cell line was purchased and cultured from American Type Culture Collection in Dulbecco modified essential medium with 10% fetal bovine serum and 1% penicillin streptomycin; 3 × 105 cells were implanted into the right dorsal foot pad or right flank of the mice. After 2 or 5 wk, the flank-melanoma–bearing or foot-pad-melanoma–bearing mice, respectively, were injected with 124I-trametinib in 10% dimethyl sulfoxide in saline intravenously and imaged or euthanized at the given time point for biodistribution studies. A cold iodine block was administered intraperitoneally up to 1 h before radiotracer injection. For therapy monitoring, a cohort of mice was administered a 6 mg kg−1 dose per mouse per day in 10% dimethyl sulfoxide and 90% saline intraperitoneally for 3 d. Daily dosing intraperitoneally was done to mimic daily oral dosing of trametinib, as well as to preserve tail veins for 124I-trametinib administration. No further therapy was administered after injection of 124I-trametinib. For biodistribution studies, the mice were euthanized in accordance with the research animal resource center and the institutional animal care and use committee. Tissues collected during biodistribution were placed into preweighed tubes for determination of tissue mass. The tissues collected were blood, heart, lungs, liver, spleen, kidneys, complete gastrointestinal tract (stomach through lower intestine, including pancreas), thyroid, thigh muscle, femur bone, ear, lymph nodes (popliteal, inguinal, or mesenteric), and tumor. Activity present in each vial was determined alongside a standard curve by automatic γ-counting on a Perkin Elmer 2480 Wizard 3. Metabolite analysis with 131I-trametinib was performed with a Miltenyi gentle Macs C-dissociation cup, and metabolites were extracted with acetonitrile for analysis by HPLC using the same conditions as for trametinib purification.
Imaging Studies
Mice injected with 124I-trametinib were imaged at 0, 1, 3, 6, 12, 24, and 96 h after injection using a Siemens Inveon PET/CT scanner. A fixed 30-min acquisition was performed for all images. Images represented as percentage injected dose per gram were overlaid onto the CT images for anatomic reference.
RESULTS
Trametinib (1) is not immediately available for direct radioiodination in high yield, requiring a facile precursor for rapid iodination. Through the use of palladium catalysis under elevated temperature and microwave conditions, the stable para-iodine could be removed and exchanged with a boronato-pinacol group. The precursor boronato-trametinib (2) was achieved in a 9:1 ratio with a boronato-hydroxide impurity, which could also be used for radioiodination and attempted via 2 methods. Chloramine-T oxidation was unsuccessful, but the copper (II) acetate–mediated 1,10-phenanthroline iodination (Fig. 2A) was successful after heating at 80°C for 30 min. Iodination was achieved (3) as seen by the HPLC chromatogram, with minimal unreacted radioiodine (Fig. 2B). Synthesis with the additional radiotracers 123I and 124I yielded high reaction selectivity as well, allowing facile iodination across the 3 iodine sources used. The reaction product was purified by HPLC and collected onto a Sep-Pak Light C18 cartridge for elution into either EtOH or dimethyl sulfoxide. Overall, iodination and purification were completed in less than 1.5 h, with an overall radiochemical yield of 69.7% and specific activity of more than 100 GBq/μmol for 124I-trametinib. Because radioiodine replaces cold iodine on the 2,4-fluoro-iodo-analine, radioiodinated trametinib is a pure tracer without perturbation of the structure. With an available method to produce radioiodinated trametinib from a wide variety of radioiodine sources, the focus of the remaining work used 124I-trametinib for facile PET imaging. 124I is a PET tracer with a 23% β+ yield and a 4.18-d half-life, allowing imaging out to 96 h and beyond for long-term biodistribution.

Synthesis and cell line specificity of radioiodinated trametinib. (A) Synthesis scheme started from trametinib into boronato-pinacol precursor readily available for iodination. Radiosynthesis and purification were complete within 1.5 h, with radiochemical yield of 70% for 124I-trametinib and specific activity greater than 100 GBq/μmol. (B) HPLC purification shows that most radioiodinated product is distinct from boronato-precursor and identical to cold trametinib. (C) Trametinib is type-III MEK allosteric inhibitor designed to reversibly inhibit adenosine triphosphate from phosphorylating MEK1/2. 124I-trametinib addition to cell lines expressing mutant Ras and or mutant Braf reveal nearly 10-fold range of uptake after 2 h, all of which is blocked with 1 μg of cold trametinib. Human KRAS mutant (G12V, G12C, or G12D) or BRAFV600E cell lines retained higher percentages of 124I-trametinib, with double KRAS BRAF mutant having highest uptake, differentiating KRAS and BRAF mutant tissue from normal. Murine cancer cell lines were all similarly avid for 124I-trametinib except Raw264.7, representing normal KRAS and BRAF and having lower uptake. DMSO = dimethyl sulfoxide; RCY = radiochemical yield; SA = specific activity; UV/Vis = ultraviolet/visible light.
With 124I-trametinib, radiotracer specificity was determined in human and murine cancer cell lines. The addition of 124I-trametinib at 2 h (Fig. 2C) was readily blocked by the addition of 1 μg of cold, commercially available trametinib. This block persisted through 72 h in the cell lines tested, showing the selectivity of the iodinated trametinib produced and the duration of inhibition in vitro (Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org). Internalization of 124I-trametinib at 2 h varied almost 10-fold, with KRAS mutants having higher internalization percentages. The highest uptake was observed with the MDA-MB-231 human cell line, which has both KRAS- and BRAF-activating mutations. Murine cell lines were similarly avid for 124I-trametinib, as KRAS mutants and the KRAS BRAF wild-type cell line Raw264.7 had the lowest uptake (Fig. 2C). A titration of cold trametinib with 13.4 kBq of 124I-trametinib revealed the dose-dependent inhibition with cold trametinib and a 50% block by 0.03 μM (Supplemental Fig. 2). Selectivity of trametinib blocking was confirmed with other allosteric MEK inhibitors and partial inhibition with BRAF and pan tyrosine receptor kinase inhibitors (Supplemental Fig. 3). KRAS and BRAF mutant lines overall had higher internalization rates than wild-type cancer cell lines, providing a nearly 10-fold range between lines tested. Du145 has a UBE2L3-KRAS fusion gene despite having a normal KRAS variant (13), though uptake studies suggest the fusion gene does not have an effect on 124I-trametinib uptake. The murine melanoma line B16F10, although not a KRAS or BRAF mutant, contains a INK4a/ARF deletion, allowing oncogenes to drive proliferation (14,15). Here, the use of iodinated trametinib can separate KRAS and BRAF mutants from the wild type in naïvely treated cancer cell lines, and blocking studies show that 124I-trametinib uptake can determine the degree of MEK inhibition.
Trametinib distribution and associated organ toxicity have been reported in clinical trials (2,6,16) and in preclinical toxicology findings (7). Despite recorded adverse events, most published studies refer to on-target/off-tumor toxicity of MEK inhibition in tissues but do not correlate the amount of drug delivered to each organ and the toxicity observed. According to the trametinib prescribing information (17), adverse events include hemorrhage, venous thromboembolism, cardiomyopathy, ocular toxicity, interstitial lung disease, serious febrile reactions, serious skin reactions, hyperglycemia, and embryofetal toxicity. The most common reactions include rash, diarrhea, and lymphedema. Metabolism of trametinib is mainly hepatic, with 70%–93% excreted via the feces when given orally (7). Knowing the drug concentration over time in each tissue and the reported adverse interactions establishes a baseline for trametinib biodistribution and a tool to show how new formulations and delivery vehicles could alter and prevent organ toxicity. Healthy C57BL/6-J mice were administered approximately 1.5 MBq of 124I-trametinib for biodistribution studies or 1.85 GBq for imaging studies via the tail vein and euthanized after 1, 2, 6, 12, and 24 h (Fig. 3). The organs collected were blood, heart, lung, liver, spleen, kidneys, entire gastrointestinal tract (stomach, pancreas, small intestine, large intestine, and rectum), thyroid, muscle, bone, bladder, skin, and lymph nodes. Imaging studies showed rapid clearance of the drug from the blood within 2 h, with most activity in the liver (Fig. 3A). Subsequent images showed gastrointestinal clearance and fecal excretion. Image series identified low uptake of 124I-trametinib across the blood–brain barrier and into the brain (7). Biodistribution studies showed the highest uptake in liver and kidneys between 1 and 6 h, at 18% and 11% injected dose per gram (%ID/g), respectively, with the gastrointestinal tract and spleen representing 11 and 8 %ID/g (Fig. 3B). 124I-trametinib was rapidly cleared from the blood, yet 5 %ID/g in the heart and lung did not appreciably clear until 24 h after administration. Skin, the area most notable for toxicity in patients, was found in mice to have an overall low uptake of 1–3 %ID/g, highlighting the sensitivity of the organ to trametinib. Other notable organs included the muscle, at 2–3 %ID/g at each time point observed.

In vivo distribution of 124I-trametinib in healthy mice primarily in liver, with subsequent elimination through gastrointestinal tract. (A) PET imaging at 1, 3, 6, and 24 h shows broad distribution in tissues, with highest uptake in liver, spleen, and kidneys and later gastrointestinal clearance. Scale bar values in injected dose per gram (%ID/g). (B) Terminal biodistributions at up to 12 h show high heart and lung uptake (5 %ID/g) compared with blood (1 %ID/g) or skin (1–2 %ID/g). Peak uptake in skin was observed at 6 h after injection. GI = gastrointestinal.
Next, biodistribution studies were completed in mice bearing B16F10 melanomas on their right dorsal foot pad. This melanoma model is highly metastatic, yielding metastases in the ascending lymph nodes. Primary tumor uptake was visible as early as 1 h (Fig. 4A) after injection, increased through 24 h, and peaked at 4 %ID/g at 48 h, whereas all other organs were appreciably cleared of tracer by 24–72 h. The tumor accumulation over 48 h presents a possibility of imaging melanomas after 24 h, with a nearly 6-fold ratio over skin by 48 h and 8-fold over skin by 72 h, though the greatest benefit is to identify the MEK avidity of naïve tissue and inhibition during trametinib therapy. No significant difference from healthy mice was observed in organ uptake (Supplemental Fig. 4). Uptake in sentinel draining nodes taken from the foot-pad primary tumor was elevated at early time points. Numerous studies have previously shown the success of 18F-FDG in mapping melanoma (18); thus, 124I-trametinib is best suited to other cancers with mutant KRAS and BRAF genotypes that are not 18F-FDG–avid.

Imaging and biodistribution of 124I-trametinib in mice bearing B16F10 melanomas through 72 h. (A) PET imaging of B16F10 melanoma–bearing mice shows slow tumor uptake through 72 h. Scale bar values in injected dose per gram (%ID/g). (B) Terminal biodistribution reveals maximal uptake of 124I-trametinib in B16F10 tumor footpad between 48 and 72 h after injection. Inset: ratio of %ID/g in tumor to skin, blood, muscle, and heart increased after 24 h, showing retention of 124I-trametinib in tumor relative to other tissues. Tumor uptake after 24 h would allow imaging of naïve tumors in addition to monitoring tumor resistance during conventional trametinib therapy.
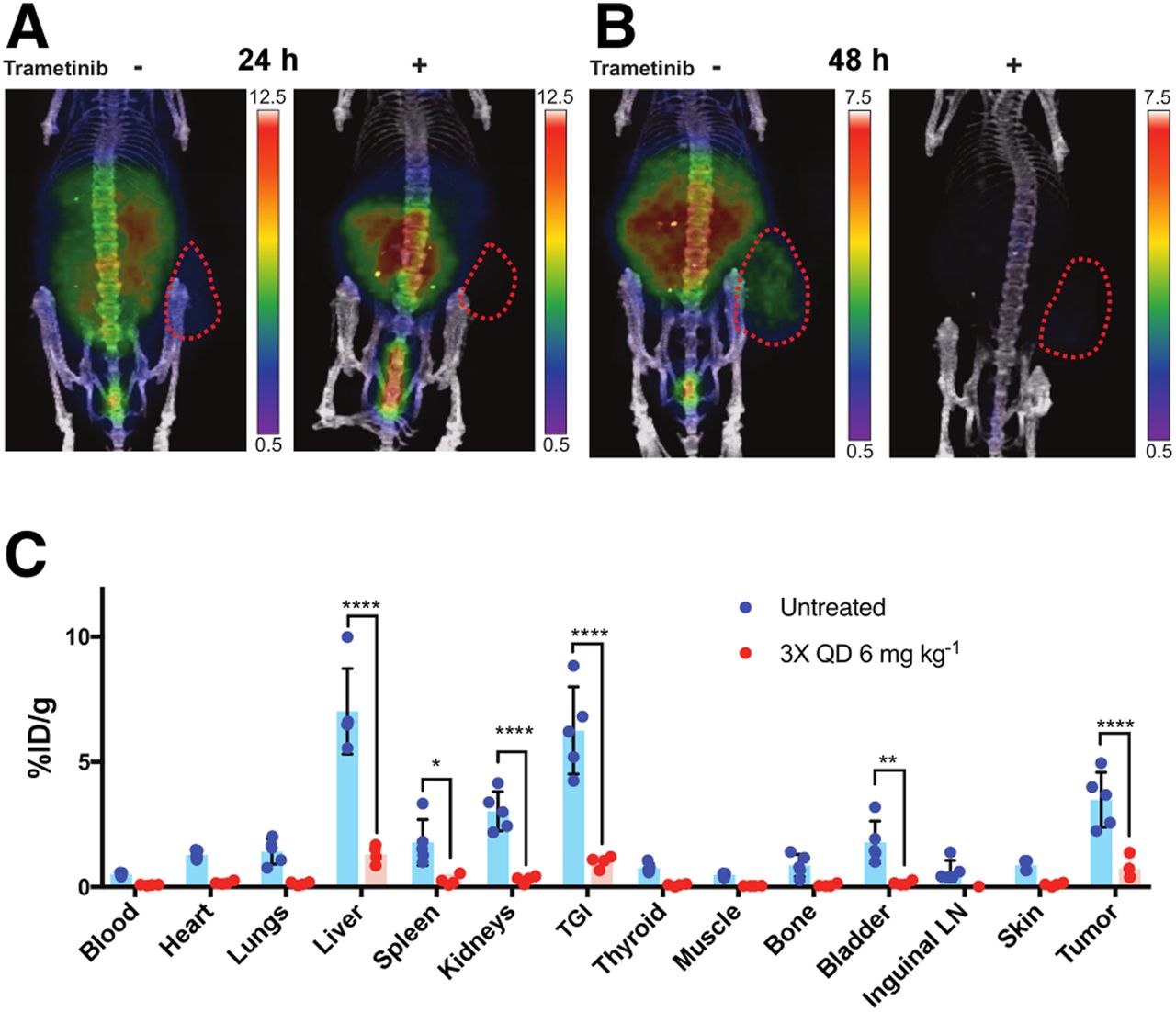
Mice with flank B16F10 melanomas were divided into a naïve group and a trametinib treatment group. Mice under trametinib therapy received trametinib at 6 mg kg−1 once daily for 3 d by intraperitoneal injection. Both cohorts of mice were administered approximately 15 MBq of 124I-trametinib intravenously and imaged through 48 h after injection. PET images showed a significantly lower 124I-trametinib distribution in trametinib-treated mice at 24 h (Fig. 5A), and the tracer was nearly absent from mice by 48 h (Fig. 5B). Terminal biodistribution confirmed that trametinib-treated mice have lower systemic MEK expression, with significant reductions in the liver, gastrointestinal tract, bladder, flank tumor, and inguinal lymph nodes (Fig. 5C). Here, 124I-trametinib can confirm blocking of MEK during trametinib therapy, and when blocked, 124I-trametinib is rapidly cleared.

Trametinib therapy reduces uptake of 124I-trametinib systemically. Prior administration of 6 mg kg−1 trametinib intraperitoneally once per day for 3 d shows, by PET, reduced uptake of 124I-trametinib at 24 h (A) and 48 h (B) after administration. Scale bar values in injected dose per gram (%ID/g). (C) Terminal biodistribution on post–48-h PET scan confirms significant decreases in 124I-trametinib uptake in liver, spleen, kidneys, total gastrointestinal tract, bladder, and right-flank B16F10 tumor. Two-way ANOVA was used for biodistribution analysis of 4 mice receiving ×3 trametinib treatment and 5 naïve mice before 124I-trametinib imaging. ****P < 0.001. **P < 0.01. *P < 0.05. LN = lymph node; TGI = total gastrointestinal tract; QD = once daily.
In another murine model, nude mice with MDA-MB-231 tumors were administered 124I-trametinib. MDA-MB-231 cell lines contain both KRAS- and BRAF-activating mutations, suggesting higher uptake as seen in vitro. Coronal image slices at 24 h after injection revealed higher uptake, at 5 %ID/g, which began clearing by 48 h after injection (Supplemental Fig. 5). To confirm that the uptake observed was due to the parent molecule and not a metabolite, nude mice bearing MDA-MB-231 tumors were administered 131I-trametinib for analysis. Radio-HPLC traces showed trametinib as the parent species extracted from tumor, liver, and intestine (including feces), though minor metabolites were observed in the intestine and at 6 h after injection in the liver (Supplemental Fig. 6).
DISCUSSION
Trametinib is widely used as a therapeutic agent for tumors bearing BRAFV600E/K mutations. The combinatorial success of trametinib with BRAF inhibitors highlights the importance of a targeted blockade in the overactive receptor tyrosine kinase pathway. The importance of trametinib for KRAS mutant cancers has also opened new therapy opportunities for patients with lung cancers (4). Overactive signaling cascades from KRAS and or BRAF mutations use MEK to keep downstream proliferation genes on. Through administration of trametinib and inhibition of MEK, the overactive signaling cascades can be attenuated (Fig. 1). Trametinib alone has been shown to block phosphorylation of MEK and downstream ERK but does not affect total MEK amounts. Dimerization of KRAS with mutant and wild-type copies presents different degrees of MEK phosphorylation, impacting MEK inhibitor sensitivity (19), and MEK monotherapy can lead to allelic mutants resistant to MEK inhibitors (20). In addition, administration of trametinib has presented several on-target toxicities, whereas administration in the clinic remains a fixed dose per patient. By using radiolabeled trametinib, concentration-based information is now possible for each organ, allowing therapy and resistance surveillance.
The design of radioiodinated trametinib allowed for the radiotracer to replace an existing stable iodine isotope. Synthesis of iodinated trametinib using copper-mediated 1,10-phenanthroline provided high selectivity for 123I, 124I, and 131I over chloramine-T oxidation (Figs. 2A and 2B). Purification and solvent exchange for injection provided nearly 70% radiochemical yield and a total synthesis time of about 90 min. This synthesis efficiently uses a variety of iodine isotopes such that trametinib could be imaged on a variety of imaging systems.
The selectivity in vitro of 124I-trametinib across a panel of human and murine cell lines was determined by a reduction in cell uptake on blocking with 1 μg of cold trametinib (Fig. 2C). Human cell lines with the highest 124I-trametinib uptake after 2 h were lines known to have aberrant KRAS or BRAF mutations, whereas lines lowest in uptake had wild-type alleles per milligram of protein with a 10-fold change between extremes. Uptake in several cell lines through 24 h indicates facile accumulation of 124I-trametinib, whereas persistent blocking with cold trametinib dramatically limits internalization (Supplemental Figs. 1–3). Radiolabeled trametinib can be used to determine the avidity of trametinib-naïve tumors as a surrogate measurement for MEK signaling avidity. Furthermore, during trametinib therapy, radiolabeled trametinib can measure inhibition of MEK in the patient’s tumor, allowing dose finding and therapy personalization with new treatment combinations.
Healthy mice showed rapid metabolism of 124I-trametinib in the liver by 2 h, with subsequent gastrointestinal clearance though 24 h (Fig. 3A). Clearance of trametinib is known to be 20% renal and 80% fecal (17), agreeing with observed imaging and biodistribution (Fig. 3B) and metabolite analysis (Supplemental Fig. 6). Uptake in skin was not as extensive as in liver and spleen; however, uptake observed in the lungs and heart could not be explained by activity in the blood. Adverse-event reporting (17) mentions pulmonary and cardiac events, though it is unclear if uptake of 124I-trametinib would correlate with susceptible patients. Mice bearing B16F10 melanomas showed slow uptake in the primary tumor through 48 h (Fig. 4A), with nonsignificant differences in organ uptake compared with healthy mice (Supplemental Fig. 4). Overall, clearance of tissue over tumor by 48 h allowed 124I-trametinib to serve as a tool compound to identify MEK in vivo and thus serve to quantitate the avidity of lesions for trametinib therapy. Lastly, 124I-trametinib administered during traditional trametinib therapy was blocked (Fig. 5A), providing meaningful feedback on systemic therapy for personalized dosing (Fig. 5B) and on therapy resistance. Preclinically, development of radioiodinated trametinib can serve as a tool compound for researchers.
CONCLUSION
Trametinib is widely used for treatment of metastatic melanoma in combination with other BRAF inhibitors, though therapy relies on fixed dosages and suffers from numerous limiting side effects. Here, development of radioiodine trametinib (123I-, 124I-, and 131I-labeled) provides 3 radiotracers with properties identical to those of cold, naïve trametinib, allowing visualization of KRAS mutant cell lines not done previously. 124I-trametinib in vitro was found to be taken up in KRAS and BRAF mutant lines with greater avidity than in wild-type cancer lines, with overall uptake significantly blocked by cold trametinib. 124I-trametinib was widely distributed to skin, liver, and the gastrointestinal tract but also had uptake higher than expected in the heart and lungs. Mice bearing B16F10 melanomas showed the highest uptake in tumors by 48 h after injection. Here, radioiodinated trametinib can be used to determine trametinib avidity in trametinib-naïve tumors and to measure inhibition during trametinib therapy, allowing dose personalization and assessment of potential drug resistance.
DISCLOSURE
This work was supported by National Institutes of Health grants R01CA215700 (to Jan Grimm) and P30 CA08748 (to Memorial Sloan Kettering Cancer Center). Funding for the Organic Synthesis Core is provided through the NCI core grant P30 CA008748 and by NCI R50 CA243895-01 (to Ouathek Ouerfelli). No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can a radioiodinated trametinib serve as a tool radiotracer for identifying levels of MEK in vivo, potentially identifying malignant tissue and MEK-resistant tissue?
PERTINENT FINDINGS: Radioiodinated trametinib was made with a variety of iodine sources in high yield and was taken up most avidly in cell lines expressing KRAS or BRAF mutations. Uptake of radioiodinated trametinib in melanoma tumors increased through 48 h and could be blocked when traditional trametinib was administered before the radiotracer.
IMPLICATIONS FOR PATIENT CARE: 124I-trametinib, a radiotracer specific to MEK and identical to the Food and Drug Administration–approved trametinib, can measure the avidity of malignant tissue by PET and determine the degree of tumor inhibition for patients receiving BRAF and MEK inhibitors, identifying any emergence of resistance.
Footnotes
Published online May 22, 2020.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication January 23, 2020.
- Accepted for publication April 16, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.