


Abstract
Theranostic strategies involve select radionuclides that allow diagnostic imaging and tailored radionuclide therapy in the same patient. An example of a Food and Drug Administration–approved theranostic pair is the 68Ga- and 177Lu-labeled DOTATATE peptides, which are used to image neuroendocrine tumors, predict treatment response, and treat disease. However, when using radionuclides of 2 different elements, differences in the pharmacokinetic and pharmacodynamic profile of the agent can occur. Theranostic agents that incorporate the matched-pair radionuclides of scandium—43Sc/47Sc or 44Sc/47Sc—would guarantee identical chemistries and pharmacologic profiles. The aim of this study was to investigate production of 43,44,47Sc via proton-induced nuclear reactions on titanium nuclei using a 24-MeV cyclotron. Methods: Aluminum, niobium, and tantalum target holders were used with titanium foils and pressed TiO2 to produce scandium radionuclides with proton energies of up to 24 MeV. Irradiated targets were digested using NH4HF2 and HCl in a closed perfluoroalkoxy alkane vessel in 90 min. Scandium radionuclides were purified via ion-exchange chromatography using branched N,N,N′,N′-tetra-2-ethylhexyldiglycolamide. The titanium target material was recovered via alkali precipitation with ammonia solution. Results: Titanium foil and TiO2 were digested with an average efficiency of 98% ± 3% and 95% ± 1%, respectively. The typical digestion time was 45 min for titanium foil and 75 min for TiO2. The average scandium recovery was 94% ± 3%, and the average titanium recoveries from digested titanium foil and TiO2 after precipitation as TiO2 were 108% ± 8% and 104% ± 5% of initial mass, respectively. Conclusion: This work demonstrated a robust method for the cyclotron production of scandium radionuclides that could be used with natural or enriched TiO2 target material.
The element scandium has 3 radionuclides with properties suitable for diagnostic and therapeutic applications in nuclear medicine. 43Sc (half-life [t1/2] = 3.89 h) and 44Sc (t1/2 = 3.97 h) undergo positron decay, with β+mean energies of 476.0 keV (88.1%) and 632.0 keV (94.3%), respectively, making them well suited for PET (1). 47Sc (t1/2 = 3.35 d) undergoes β-decay, with a β−mean energy of 162 keV to 47Ti, followed by emission of a 159-keV γ-ray (68.4%) (1). Thus, 47Sc might be used for targeted radionuclide therapy and SPECT. These radionuclides of scandium might be used to create theranostic agents for diagnostic imaging and targeted radiotherapy in the same patient.
Interest in these radionuclides is partly motivated by the recent clinical success of DOTATATE and results in clinical trials investigating small-molecule inhibitors of prostate-specific membrane antigen (PSMA). DOTATATE, a somatostatin analog, labeled with 68Ga (t1/2 = 67.7 min) or 177Lu (t1/2 = 6.7 d) can be used in theranostics to diagnose and treat SSTR-expressing neuroendocrine tumors (2,3). Similarly, inhibitors of PSMA such as PSMA-11, PSMA-617, DCFPyL, and MIP-1072 labeled with 68Ga, 177Lu, 18F (t1/2 = 109.7 min), 131I (t1/2 = 8.0 d), 123I (t1/2 = 13.2 h), or 99mTc (t1/2 = 6 h) can be used for diagnosis and treatment of PSMA-positive prostate cancer (1,4). Theranostic agents that incorporate radionuclides of different elements can have differences in their in vivo kinetics (5). The selection of a matched pair (e.g., 43/47Sc), guarantees identical pharmacodynamic and pharmacokinetic profiles and provides the truest picture of in vivo distribution before radionuclide therapy. Further, the t1/2 of 43Sc and 44Sc, in contrast to 68Ga, could allow for imaging at extended time points (6). Therefore, development of efficient methods to produce 43,44,47Sc is an important first step to translating these matched-pair radionuclides of scandium to the clinic.
43Sc and 44Sc can be produced via multiple routes. Accelerator production using α-particles, protons, and deuterons on calcium and titanium nuclei has been investigated for the natCa(α,x)43,44,47Sc, 42Ca(d,n)43Sc, 43Ca(p,n)43Sc, 44Ca(p,2n)43Sc, 46Ti(p,α)43Sc, 47Ti(p,α)44Sc, 44Ca(p,n)44Sc, natCa(p,n)44Sc, and 44Ca(d,2n)44Sc nuclear reactions (7–13). The use of calcium and titanium has been shown to provide gigabecquerels and hundreds of megabecquerels of 43Sc and 44Sc, depending on beam current and irradiation time; however, 44Sc produced by these reactions will result in the coproduction of its nuclear isomer, 44mSc (t1/2 = 58.6 h). The presence of this longer-lived metastable state may complicate patient dosimetry. 44Sc may be obtained without its nuclear isomer from a 44Ti (t1/2 = 59.2 y) generator (14).
47Sc can also be produced via multiple routes. Reactor production of 47Sc has been investigated using fast neutrons on enriched titanium via the 47Ti(n,p)47Sc reaction (neutron energy > 1 MeV) or thermal neutrons (neutron energy = 0.025eV) on enriched calcium via the 46Ca(n,γ)47Ca→47Sc reaction for a 47Ca-47Sc (t1/2 = 4.5 d) generator system (15). One challenge facing development of a 47Ca-47Sc generator is the natural abundance of 46Ca (0.004%). 47Sc may also be produced by the 48Ti(γ,p)47Sc reaction using bremsstrahlung radiation, and accelerator production by the 48Ca(p,2n)47Sc reaction was investigated at 18 MeV and resulted in a saturation yield of 12 GBq/μA (16–19).
Unlike production routes using calcium, the use of titanium could allow production on a single proton accelerator using a unified separation methodology that applies identical targetry and target-processing chemistries and results in better radionuclidic purity for 43,44,47Sc. However, the magnitude of the cross section for reactions on titanium are much less than those on calcium at lower energies (16–18 MeV). Favorably, the cross-section values increase at the higher energies (≤24 MeV) used in this study. Multiple challenges must be addressed to produce scandium radionuclides from stable nuclei of titanium using this unified approach. First, the powdery TiO2 targets require a holder that encapsulates the irradiated material to prevent radiologic contamination. Next, the slow TiO2 digestion kinetics and short t1/2 of 43Sc/44Sc necessitate development of a rapid digestion method. Lastly, 43,44,47Sc with high radionuclidic purity can be obtained only using enriched titanium, since 46,47,50Ti has natural abundances of 8.25%, 7.44%, and 5.18%, respectively. Thus, efficient recycling chemistry must be developed to recover and reuse the enriched target material.
To address these challenges, methods were developed to investigate 43,44,47Sc yields and purification chemistry using natural titanium and TiO2. The results from this work suggest that 43,44,47Sc might be produced at energies of up to 24 MeV using enriched TiO2 and purified using a unified methodology to supply quantities needed for the preclinical evaluation of these matched-pair radionuclides.
MATERIALS AND METHODS
Reagents
All chemicals were of analytic or trace metals grade. Deionized 18.2 MΩ-cm water (Milli-Q System; Millipore) was used unless otherwise specified. Ammonium bifluoride (NH4HF2, 99.999%), hydrochloric acid (HCl, 37% by weight, 99.999%), 25% ammonia solution (NH4OH, EMSURE; Merck), TiO2 (anatase form, 99.8%), periodic table analytic standards (mix 1, 10 mg/L, TraceCERT; Sigma-Aldrich), ammonium acetate (CH3CO2NH4, 99.999%), and nitric acid (HNO3, 70% by weight, 99.999%) were purchased from Sigma-Aldrich. Analytic standards containing titanium (1,000 mg/L) and scandium (1,000 mg/L) were purchased from Agilent. Analytic-grade N,N,N′,N′-tetra-2-ethylhexyldiglycolamide (branched DGA resin) was purchased from Eichrom Technologies. DOTA was purchased from Macrocyclic. All glassware was cleaned in a 50% HNO3 bath overnight.
Target Holders
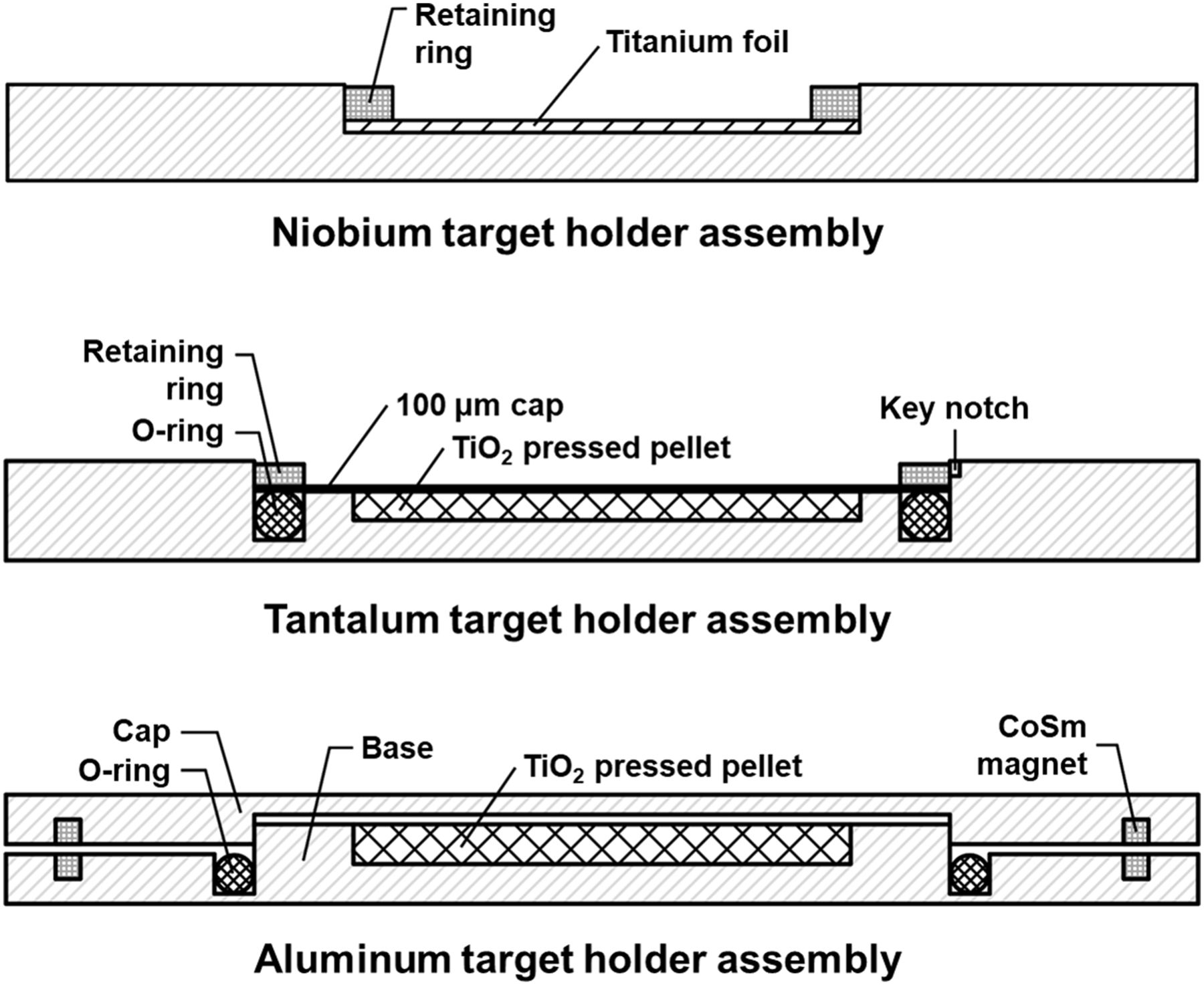
Multiple target holders were developed to mount and irradiate titanium foil and TiO2 target materials. The disk-shaped 2-mm-thick target holders fabricated from aluminum, tantalum, and niobium are shown in Figure 1. The aluminum target holder had a circular base and cap secured by samarium-cobalt magnets. A fluoroelastomer o-ring was integrated between the base and cap to enclose the target material. A bore (∅ = 7 mm) was centered in the base. The niobium target holder contained a centered bore (∅ = 10 mm) holding titanium foil, which was secured against the base using a 10.1-mm single-turn spiral internal retaining ring (Smalley). The tantalum target holder contained a centered bore (∅ = 10 mm) with an integrated o-ring between the base and cap to enclose the target material. The bore held a TiO2 disk that was secured by placing a 100-μm titanium disk atop the bore and o-ring. The titanium disk was held in place by a 14.3-mm spiral retaining ring (Rotor Clip). Stopping and range of ions in matter (SRIM) was used to determine the optimal thickness of each target holder to stop 17- or 24-MeV protons (20).
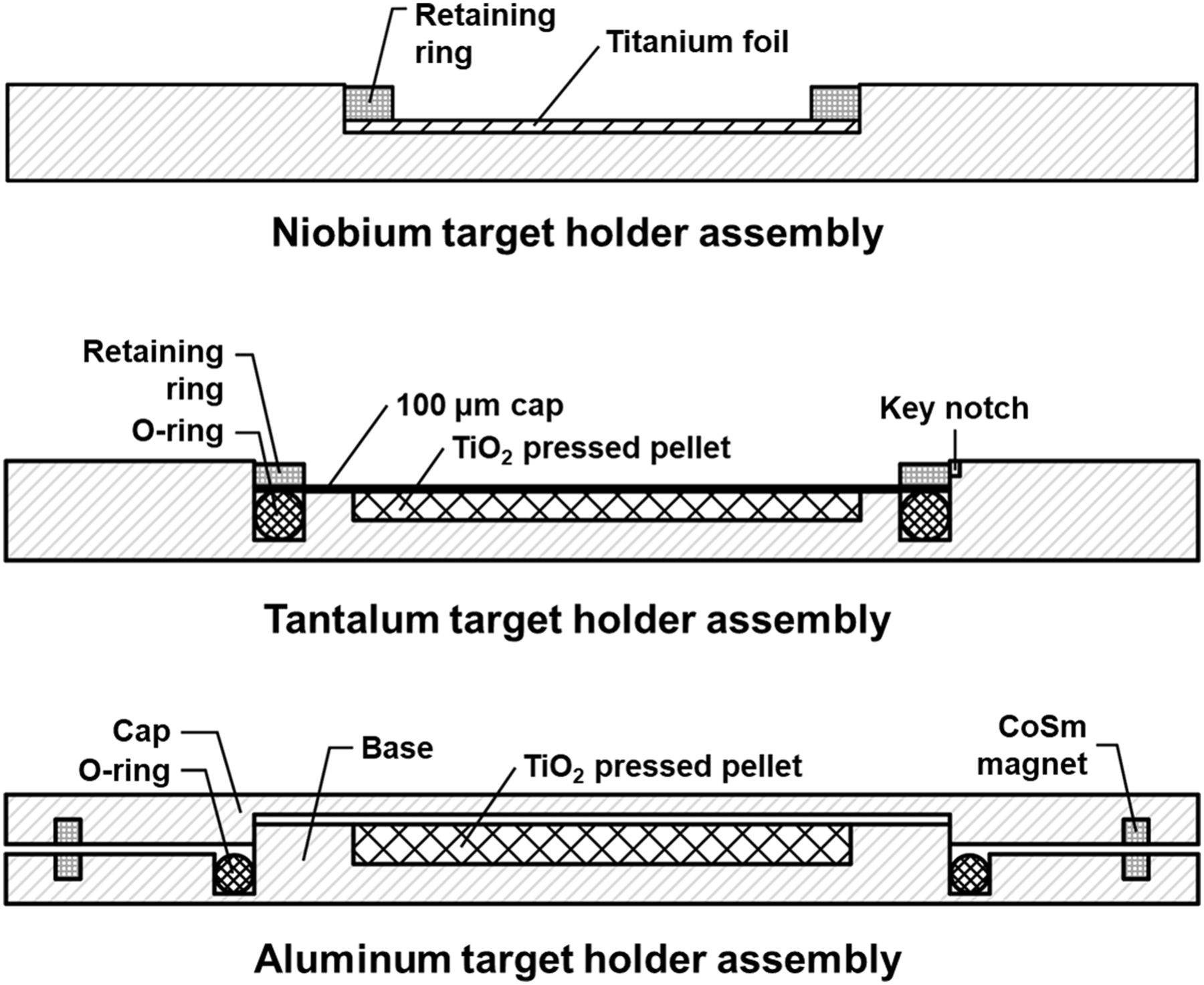
Target holder assemblies tested in this work.
Preparation of Target Material
Titanium foil targets were prepared using a 10-mm punch and die (Precision Brand Products). Before pressing, TiO2 was dried overnight at 230°C in a vacuum oven at 0.3 Pa. Approximately 35 or 70 mg of TiO2 were transferred into a clean 7- or 10-mm die (Specac) and pressed for 5 min at 4 tons of pressure on a manual press (Carver). Additional TiO2 was iteratively added and pressed. This process was repeated twice, with a final target mass of approximately 52 or 110 mg.
Cyclotron Parameters
Irradiation was conducted on a TR-24 cyclotron (Advanced Cyclotron Systems). 43Sc, 44Sc, and 47Sc were produced via the natTi(p,x) nuclear reaction at 17 and 24 MeV. Titanium and TiO2 targets were mounted at a 90° angle and bombarded at beam currents of up to 40 and 30 μA, respectively. The targets were cooled by high-pressure, low-flow helium gas (275–345 kPa, 1 L/min) on the front and pumped water (2–3 L/min) on the back. Bombarded targets were allowed to decay for 1–5 h to allow for the decay of coproduced 47V (t1/2 = 32.6 min).
Production Yields
The radionuclidic yields were determined using a high-purity germanium detector. Predicted yields were calculated using nuclear data retrieved from the Experimental Nuclear Reaction Data database (21,22). The code SRIM was used to model the entrance and exit proton energies. End-of-bombardment yields were determined using Mathcad (version 14.0) and Equation 1, where NA is the Avogadro constant (atoms/mol), I is incident particle flux (particles/s), AT is the atomic weight (g/mol), λ is the decay constant (1/s), t is the irradiation time (s), E0 is the initial proton energy, Ee is the exit proton energy, σ(E) is the energy-dependent reaction cross section (cm2), and S(E) is the total stopping power (MeV/cm2/g) (23).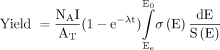 Eq. 1
Eq. 1
Determination of Equilibrium Dissociation Constant
The equilibrium dissociation constant was determined following a previously reported method (24). The dissociation constants were calculated using Equation 2, where Cresin is the concentration of radioactivity adsorbed onto the resin, Caqueous is the concentration of radioactivity in the solvent, Ai is the radioactivity in filtered supernatant (Bq), V is the volume of the filtered supernatant (mL), and m is the mass of the resin (g). Eq. 2
Eq. 2
Digestion of Titanium Foil and TiO2
Irradiated titanium was transferred into a conical 15-mL screw-cap perfluoroalkoxy alkane vial (Savillex) containing approximately 200 mg of NH4HF2 dissolved in 3 mL of 12.1 M HCl. The vial was capped and heated at 230°C for 45 min. Irradiated TiO2 was transferred into a conical 15-mL screw-cap vial and mixed with NH4HF2 (1:3 ratio). The vial was capped and heated to 230°C for 45 min. After heating, the dry residue was dissolved by adding 5 mL of 12.1 M HCl and heating the closed vessel for 45 min in a silicone-oil bath at 160°C. For both digestion methods, samples were taken in triplicate, filtered, and diluted in 2% HNO3 for trace metal analyses. The digestion methods are illustrated in Figure 2.
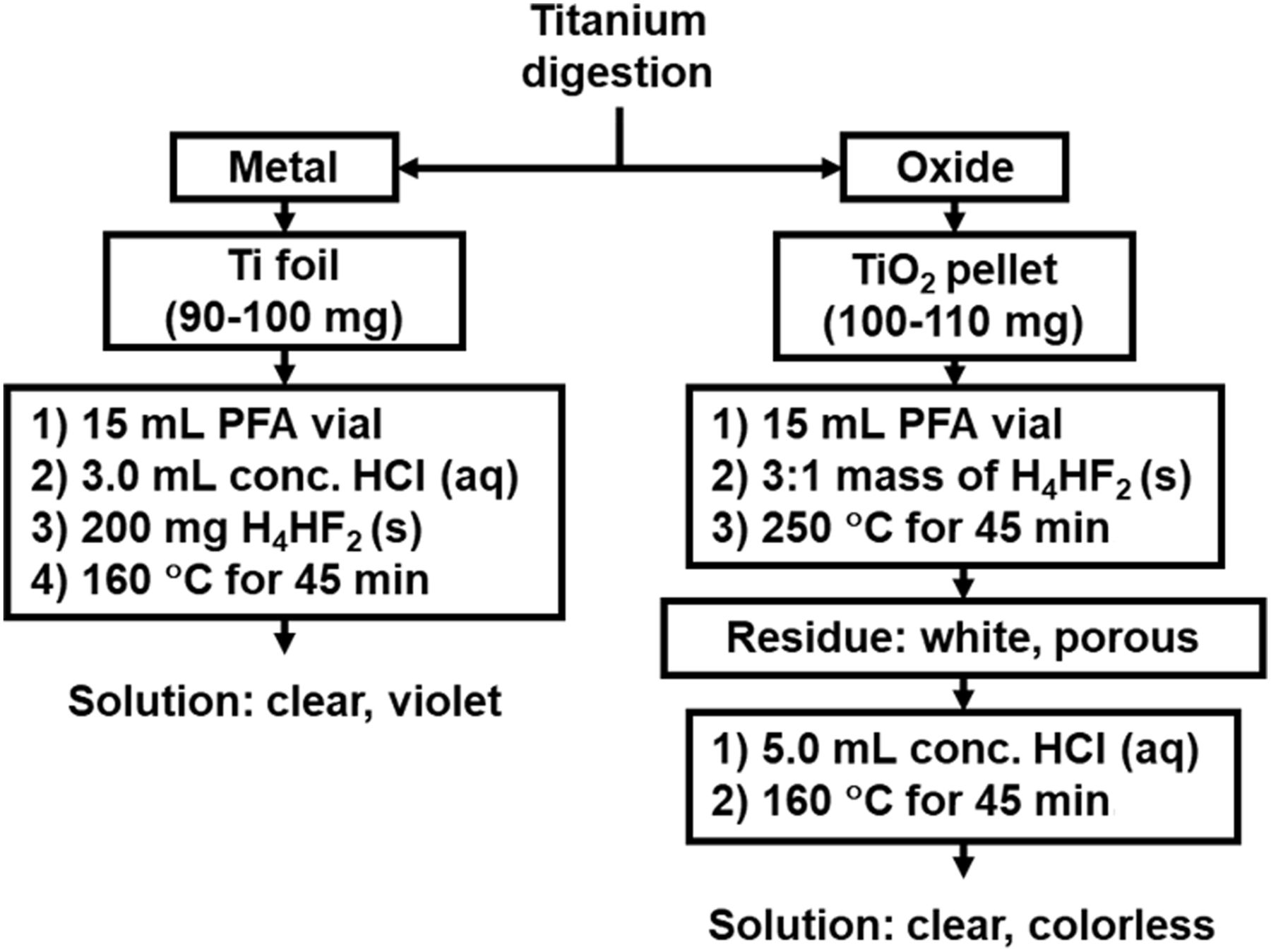
Methods for digestion of titanium foil and TiO2. PFA = perfluoroalkoxy alkane.
Branched DGA Resin Separation
Approximately 110 mg of branched DGA resin were stirred in a vortex mixer with 5 mL of 7 M HNO3. The mixture was transferred into a 0.8 × 9 cm polypropylene column (Bio-Rad), the HNO3 was eluted, and a glass wool plug was placed on the resin bed (7-mm height). A flow rate of 1.0 mL/min was used for each eluent. The resin was washed with 5 mL each of 7 M HNO3, water, 0.1 M HCl, and 7 M HCl. The digestant was loaded onto the column in approximately 7–8 M HCl. Eluent 1 (20 mL, 7 M HCl), eluent 2 (10 mL, 7 M HNO3), and eluent 3 (10 mL, 0.1 M HCl) were passed through the column and collected. The final eluate containing the scandium radionuclides was collected into a 10-mL conical vial and evaporated to dryness under vacuum at 100°C. After evaporation, the purified scandium was reconstituted in 0.5 M CH3CO2NH4 buffer (pH 4.7). The separation method is shown in Figure 3.

Schematic of 43,44,47Sc separation.
Samples were taken from each eluate and analyzed to determine the distribution of titanium and vanadium and the percentage recovery of scandium. The decontamination factor (DA,B), a measure of the scandium purification, was determined using Equation 3, giving the change in ratio of constituent 1 (A) to constituent 2 (B), QA,0 is the initial amount of A, QA is the final amount of A, QB,0 is the initial amount of B, and QB is the final amount of B (25).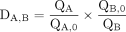 Eq. 3
Eq. 3
43,44,47Sc-PSMA-617 Radiolabeling and Cell Uptake Studies
The bioconjugate PSMA-617 was labeled with 43,44,47Sc (10 MBq/nmol) and characterized by high-performance liquid chromatography using 2 solvents. Solvent A was acetonitrile with 0.1% trifluoroacetic acid, and solvent B was water with 0.1% trifluoroacetic acid. The linear gradient was 5% A:95% B to 80% A:20% B over 15 min at 0.7 mL/min. The specific uptake of the radioligand to PSMA receptors was demonstrated via a blocking study using the PSMA inhibitor 2-(phosphonomethyl)pentanedioic acid. The PSMA-negative PC3 cell line was used as a control. The concentration of the inhibitor was 100 μM.
Alkali Precipitation of Titanium
Eluate containing titanium was diluted to 500 mL with water and heated for 45 min at 160°C. While it was being stirred, the pH was adjusted to 8 by dropwise addition of 19 mL of 25% ammonia solution. The stir bar was removed, and the precipitate was allowed to settle for 1 h. The mixture was vacuum-filtered with a grade 43 ashless filter paper (Cytiva) and rinsed twice with 10 mL of 1% aqueous ammonia solution. The precipitate was washed with 10 mL of acetone. The filter paper was transferred to a zirconia crucible and combusted in a tube furnace at 1,000°C for 24 h.
High-Purity Germanium Spectroscopy
Radioactive samples were characterized by γ-ray spectroscopy using a Canberra GC2018 high-purity germanium detector with an intrinsic efficiency of 24.5% connected to a DSA-100 multichannel analyzer. Canberra Genie 2000 software was used for data acquisition and analysis. An energy and efficiency calibration was performed using a mixed nuclide source in a sealed 1.5-mL microcentrifuge vial prepared by Eckert and Ziegler Analytics. Efficiency-calibration spectra with a minimum photopeak area of 2 × 105 counts were collected and fit to determine the relative detector efficiency. For all measurements, aliquots of sample material were diluted to 1 mL in a 1.5-mL vial and suspended in an acrylic holder at a distance of 5 or 25 mm from the high-purity germanium detector. These geometries were maintained for all sample measurements.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
Samples containing stable elements were preserved and analyzed via ICP-MS. Multielement standards in 2% HNO3 at 0.1, 1, 10, 100, and 1,000 μg/L were used for instrument calibration. Samples were diluted in 2% HNO3 and measured on an Agilent Technologies 7700 ICP-MS device. Agilent Technologies MassHunter software (version 4.3) was used for data acquisition and analyses.
RESULTS
Target Irradiations
The niobium target holder was used to irradiate 250-μm-thick titanium foils (ρ = 4.506 g/cm3) at 24 MeV and tolerated beam currents of up to 40 μA. The average (n = 3 ± 1σ) mass of a titanium foil was 91.5 ± 0.3 mg. A 2-piece aluminum target holder was used to irradiate a 400-μm-thick pressed TiO2 (ρ = 4.23 g/cm3) disk at 17 MeV. The average (n = 3 ± 1σ) mass of the disk target was 52.2 ± 0.4 mg. The magnetization of the samarium-cobalt magnets was confirmed after each irradiation (n = 6). At a beam current of 20 μA, thermal damage was observed on the aluminum cap and base; thus, subsequent irradiations were limited to 15 μA. The tantalum target holder was used to irradiate a 400-μm-thick pressed TiO2 disk at 24 MeV at beam currents of up to 40 μA for 30 min with no damage to the target holder or material. The average (n = 3 ± 1σ) mass of the disk target was 110.1 ± 0.5 mg.
Digestion Methods
Typical digestion times for titanium foils and pressed TiO2 disks were 45 and 90 min, respectively. Titanium foils were digested at 160°C using a mixture of NH4HF2 and 12.1 M HCl. Trace metal analysis of the digestant via ICP-MS showed that the average (n = 3 ± 1σ) percentage of titanium foil dissolved in solution was 98% ± 3%. TiO2 disks were digested in 2 steps, and the average (n = 3 ± 1σ) percentage of TiO2 dissolved in solution was 95% ± 1%. These methods were developed using nonirradiated titanium target material, and data from initial experiments are shown in Supplemental Figure 1 (supplemental materials are available at http://jnm.snmjournals.org).
43Sc, 44g,44mSc, 47Sc, and 48V Yields from natTi Targets
The radionuclides, measured by a high-purity germanium detector, were 48V (t1/2 = 15.97 d, γ1 = 944.1, γ2 = 983.5, γ3 = 1,312.1 keV), 43Sc (372.9 keV), 44gSc (1,157.0 keV), 44mSc (271.2 keV) and 47Sc (159.4 keV). The sample counting time was varied to achieve a minimum photopeak area of 500 counts, and dead time was kept below 5%. The average end-of-bombardment production rate (n = 9 ± 1σ) and predicted rate for each radionuclide are shown in Table 1. These experimental rates were normalized to 1 μA.
Branched DGA Resin Separation
The digestant containing titanium ions, scandium radionuclides, and 48V was loaded onto branched DGA resin. The bulk titanium target material was eluted from the resin and resulted in an average (n = 6 ± 1σ) titanium recovery factor of 96% ± 5% in 12 mL of the load eluate and 20 mL of eluate 1. In contrast, scandium radionuclides were strongly adsorbed onto the resin in 7–8 M HCl. No scandium radionuclides were detected in the eluates from the load, eluent 1, or eluent 2. The scandium radionuclides were desorbed from the resin in 10 mL of eluent 3 (0.1 M HCl). The isolated scandium radionuclides (eluate 3) were free of radionuclidic impurities, including 48V. These data are summarized in Table 2, and they show an average (n = 3 ± 1σ) scandium recovery from titanium foil and TiO2-irradiated targets of 94% ± 1% and 94% ± 3%, respectively.
Average (n = 3 ± 1σ) Initial 47Sc and 48V Radioactivity and Its Recovery in Each Eluate
Samples taken from the final scandium product after reconstitution showed the presence of nonradioactive contaminants. The identity and average (n = 3 ± 1σ) concentration of contaminants in 400 μL of 0.5 M CH3CO2NH4 were iron (1.0 ± 0.3 mg/L), copper (0.8 ± 0.3 mg/L), zinc (1.3 ± 0.3 mg/L), vanadium (0.5 ± 0.3 mg/L), and aluminum (0.8 ± 0.3 mg/L). Scandium was not detected or was below the detection limits of the ICP-MS used in this work. The complete separation and concentration of scandium radionuclides required approximately 180 min.
43,44,47Sc-PSMA-617 Radiolabeling and Specific Uptake in LNCaP Tumor Cells
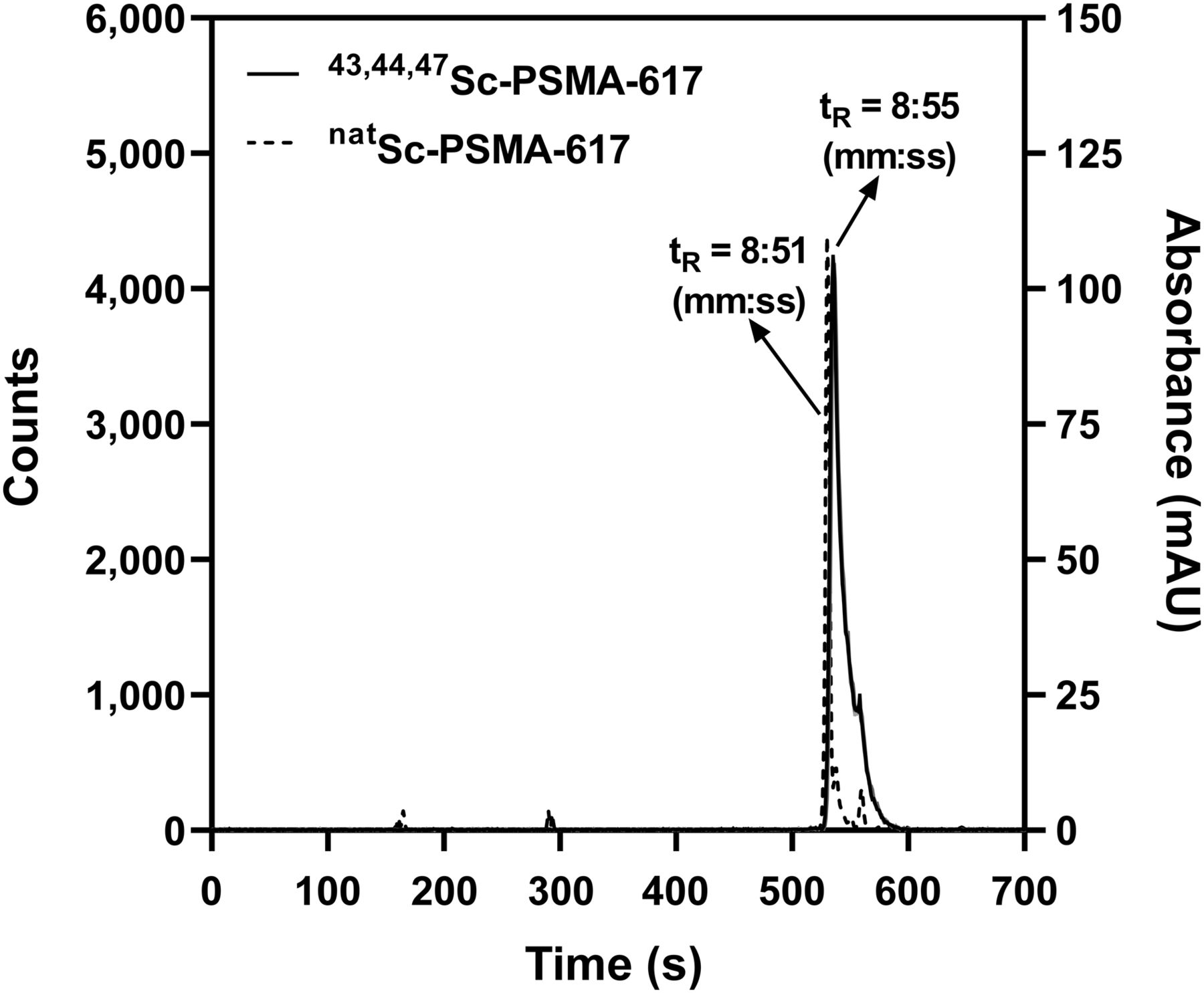
PSMA-617 was labeled with 43,44,47Sc in 0.5 M NH4CH3CO2 at 95°C for 30 min. A more than 99% radiochemical purity (10 MBq/nmol) was measured by high-performance liquid chromatography (Fig. 4). The average (n = 4 ± 1σ) percentage of 43,44,47Sc-PSMA-617 associated with LNCaP cells (Supplemental Fig. 2) was 16.6% ± 1.5% after 2 h and 21.9% ± 1.7% after 4 h of incubation at 37°C. The average (n = 4 ± 1σ) nonspecific uptake in PC3 cells (control) and in the presence of 2-(phosphonomethyl)pentanedioic acid was less than 0.5%.
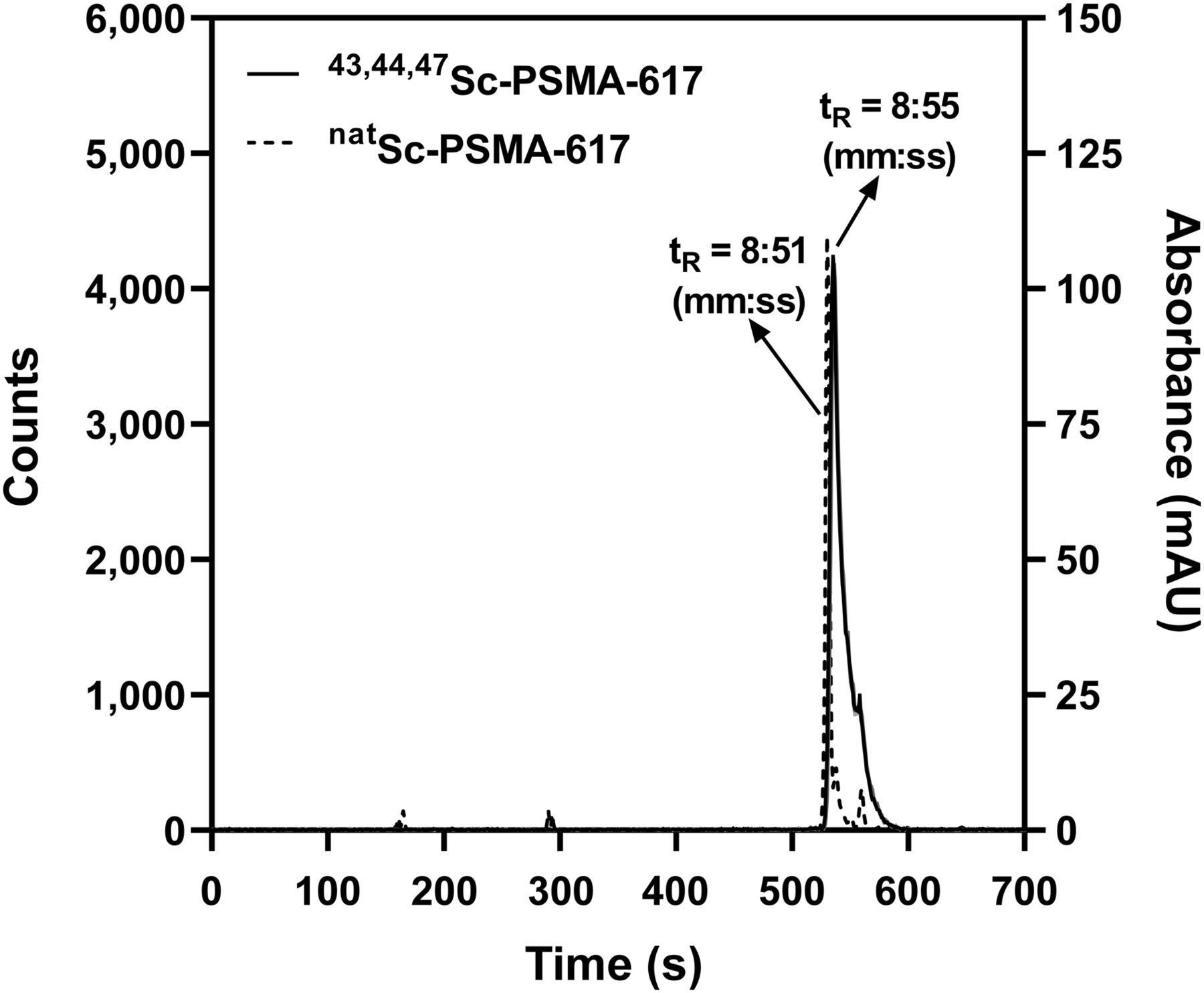
Solid line shows radiochromatogram of 43,44,47Sc-PSMA-617. Dashed line shows absorbance of natSc-PSMA-617 at 280 nm. AU = arbitrary units.
Titanium Recycling
The combined eluates containing titanium were diluted and pH-adjusted. Near pH 8 (verified by pH strip test), a fine white, cloudy precipitate formed. After filtration and combustion, the average (n = 4 ± 1σ) percentage recoveries of titanium were 103% ± 5% and 108% ± 8% from titanium foil and TiO2, respectively.
DISCUSSION
Targets were created for the irradiation of TiO2 at a beam current of up to 40 μA and an energy of up to 24 MeV. The samarium-cobalt magnets (aluminum target) did not lose their magnetism and could play a role in the design of compact cyclotron targets. The tantalum target was robust, and future work will be performed to define the maximum tolerated beam current on TiO2. Further, these data show that 43,44,47Sc can be produced via TiO2 at beam currents greater than those currently used for cyclotron production via CaO or CaCO3 (∼20 μA).
The end-of-bombardment production rate of scandium radionuclides and coproduced 48V at 24 MeV in titanium foils agreed with the predicted rate based on published nuclear datasets for the natTi(p,x) reaction (Table 1). Using the same methodology, the end-of-bombardment yields of 43,44,47Sc from enriched TiO2 target material were predicted for the 46,47,50Ti(p,α) nuclear reactions (Supplemental Fig. 3) at 1, 4, and 24 h (7,26,27). These yields (MBq–GBq) obtained from enriched titanium nuclei would be suitable for preclinical and in some cases clinical investigations.
Previously reported methods for digestion of TiO2 used concentrated hydrogen fluoride, with some methods requiring hours to reach completion (2–6 h). These methods could result in the loss of at least one 43,44Sc t1/2. The digestion methods in this work use pressure, heat, and the in situ generation of approximately 1.0 M hydrogen fluoride via the reaction of NH4HF2 with 12.1 M HCl (28). The results showed that greater than 95% of titanium foil and TiO2 was dissolved in 45 and 90 min, respectively. The closed perfluoroalkoxy alkane vessels used in this work were inexpensive and durable at temperatures of up to 250°C. These data support future efforts to characterize and optimize the digestion parameters to further decrease digestion times for titanium foil and TiO2.
Branched DGA and DGA resins have been used to purify scandium in multiple reports (8,17,29). On the basis of the equilibrium dissociation constants determined in this work for scandium, titanium, and vanadium (Supplemental Fig. 4), the separation method developed in this work was predicted to separate scandium from titanium and vanadium and was confirmed by determining the scandium decontamination factor relative to each contaminant. On the basis of Table 2, the decontamination factor for 47Sc relative to 48V was greater than 6 × 106 and 3 × 106 for scandium isolated from titanium foil and TiO2, respectively. Similarly, titanium present in the recovered scandium fractions was below the ICP-MS detection limits (∼1 μg/L); thus, the data show that the decontamination factor of 47Sc relative to titanium was greater than 2.8 × 105 and 1.6 × 105 for titanium foil and TiO2, respectively. These data show that scandium was isolated with little contamination from titanium and vanadium. The overall scandium recovery factors agreed with previously reported procedures using DGA resins (8,17,30). Trace metal analyses via ICP-MS of the titanium-containing digestants and recovered scandium fractions showed the presence of iron, copper, zinc, vanadium, and aluminum. These transition metal contaminants can reduce radiolabeling yield by competing with scandium radionuclides for ligand binding. The sources of these impurities can be from the target material or environmental contamination. The average concentrations of these impurities were between 0.5 and 1.3 mg/L, which compared well with previous reports (17,30).
To demonstrate the efficacy of these methods, a proof-of-concept study was conducted using PSMA-617 radiolabeled with scandium. The results showed specific targeting of the PSMA receptors on LNCaP cells using 43,44,47Sc-PSMA-617 at a molar activity of 10 MBq/nmol.
Historically and in recent reports, titanium has been precipitated with aqueous ammonia solutions at pH 8 (8,31). In this work, a titanium precipitant was obtained using an aqueous ammonia solution to adjust the bulk medium to pH 8. After sufficient time for aggregation and sedimentation, the precipitate was filtered and combusted. The typical concentration of titanium in the filtrate was below 100 μg/L (∼50 μg), which indicated that the percentage loss of titanium after precipitation was less than 0.1%. The mass recoveries of titanium exceeded 100%, as may be attributed to 2 factors: etching of glass wool by hydrogen fluoride generated in situ, and incomplete combustion of the filter paper or presence of water. These data show that the recovery of titanium in the oxide form with aqueous ammonia solution is an effective approach to recycling titanium. Additional work will be performed to reduce the introduction of impurities so as to avoid buildup of contaminants during iterative uses of this method.
CONCLUSION
In this study, we investigated the feasibility of producing the theranostic radionuclides of scandium on a proton accelerator using a robust and unified process. Results from this work showed that routine production of 43,44,47Sc could be accomplished using enriched TiO2. Importantly, our findings showed that TiO2 targets may tolerate beam currents greater than 30 μA using the target holders designed in this work, that TiO2 can be digested via a 2-step process in 90 min, and that digested titanium may be efficiently recovered via alkali precipitation with aqueous ammonia solution. Future work should investigate the maximum tolerated beam current on TiO2 using these target holders and any impact on downstream chemistry that may be caused by the increased heat deposition in the target material. Additionally, these efforts should seek to better understand and optimize the digestion methods developed in this work in order to further reduce digestion times. Finally, the method to precipitate titanium should be modified to remove sources of silicon. Ultimately, the results in this work showed the potential promise in using enriched titanium targets to produce these matched-pair theranostic radionuclides of scandium.
DISCLOSURE
This work was supported by the Department of Energy Isotope Program under grant DESC0020197. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Is production of the theranostic radionuclides of scandium feasible using enriched TiO2 as a target material?
PERTINENT FINDINGS: The theranostic radionuclides of scandium were produced using naturally isotopic TiO2 on a proton accelerator at energies of up to 24 MeV. These targets were rapidly digested in 90 min, followed by scandium purification using ion-exchange chromatography. This work showed a 94% recovery of scandium radionuclides and demonstrated a simple method to recover digested target material.
IMPLICATIONS FOR PATIENT CARE: The findings add to efforts at identifying easily adoptable methods to produce theranostic radionuclides of scandium, which are increasingly being used in clinical and preclinical studies.
Acknowledgments
We would like to thank the UAB Cyclotron Facility for TR24 irradiations.
Footnotes
Published online Jul. 3, 2020.
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 4, 2020.
- Accepted for publication May 28, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.