


Abstract
As part of our continuous efforts to develop a suitable 18F-labeled PET radioligand with improved characteristics for imaging the N-methyl-d-aspartate receptors (NMDARs) subtype 2B (GluN1/2B), we investigated in the current work ortho-fluorinated (OF) and meta-fluorinated (MF) analogs of 18F-para-fluorinated (PF)-NB1, a 3-benzazepine–based radiofluorinated probe. Methods: OF-NB1 and MF-NB1 were prepared using a multistep synthesis, and their binding affinities toward GluN2B subunits and selectivity over σ1 receptors (σ1Rs) were determined via competitive binding assays. 18F-OF-NB1 was synthesized via copper-mediated radiofluorination and was evaluated in Wistar rats by in vitro autoradiography, PET imaging, ex vivo biodistribution, metabolite experiments, and receptor occupancy studies using CP-101,606, an established GluN2B antagonist. To determine in vivo selectivity, 18F-OF-NB1 was validated in wild-type and σ1R knock-out mice. Translational relevance was assessed in autoradiographic studies using postmortem human brain tissues from healthy individuals and ALS patients, the results of which were corroborated by immunohistochemistry. Results: The binding affinity values for OF-NB1 and MF-NB1 toward the GluN2B subunits were 10.4 ± 4.7 and 590 ± 36 nM, respectively. For σ1R binding, OF-NB1 and MF-NB1 exhibited inhibition constants of 410 and 2,700 nM, respectively. OF-NB1, which outperformed MF-NB1, was radiolabeled with 18F to afford 18F-OF-NB1 in more than 95% radiochemical purity and molar activities of 192 ± 33 GBq/μmol. In autoradiography experiments, 18F-OF-NB1 displayed a heterogeneous and specific binding in GluN2B subunit–rich brain regions such as the cortex, striatum, hypothalamus, and hippocampus. PET imaging studies in Wistar rats showed a similar heterogeneous uptake, and no brain radiometabolites were detected. A dose-dependent blocking effect was observed with CP-101,606 (0.5–15 mg/kg) and resulted in a 50% receptor occupancy of 8.1 μmol/kg. Postmortem autoradiography results revealed lower expression of the GluN2B subunits in ALS brain tissue sections than in healthy controls, in line with immunohistochemistry results. Conclusion: 18F-OF-NB1 is a highly promising PET probe for imaging the GluN2B subunits of the N-methyl-d-aspartate receptor. It possesses utility for receptor occupancy studies and has potential for PET imaging studies in ALS patients and possibly other brain disorders.
N-methyl-d-aspartate receptors (NMDARs) are 1 of 3 glutamate ion channel types prevalent in the central nervous system and are key players in synaptic transmission, plasticity, and higher cognitive functions (1). Once NMDARs, particularly extrasynaptic ones, are overactivated because of uncontrolled glutamatergic stimulation, an apoptotic cascade is triggered rendering l-glutamate a neurotoxin in a pathologic process known as excitotoxicity (2). This deleterious event was found to be involved in neurodegenerative and psychiatric diseases such as Alzheimer disease, vascular dementia, Parkinson disease, depression, stroke, and schizophrenia (3). The discovery of NMDAR subtype 2B (GluN1/2B) as major contributors to glutamate-mediated excitotoxicity and the allosteric ifenprodil-binding pocket situated at the interface of GluN1 and GluN2B channeled drug development strategies toward subunit-selective antagonists (4). These emerging drugs possess activity similar to nonselective NMDARs antagonists but exhibit a favorable safety profile relative to their nonselective predecessors (5). Nonetheless, several promising GluN2B-subunit antagonists, such as CERC-301 (MK-0657), were assessed in clinical trials showing no-to-modest benefit–risk ratio (6). As such, pivotal tools for the efficient evaluation of drug-target engagement and occupancy are of the highest priority (7). Noninvasive PET imaging is such a tool that can be used in preclinical and clinical settings to assess drug-target engagement, receptor occupancy (RO), and therapy monitoring.
Despite more than 2 decades of research, no suitable clinical GluN2B-subunit PET imaging agent currently exists, because of one or more major shortcomings, such as brain radiometabolites; considerable off-target binding, especially to σ1 receptors (σ1Rs); low brain uptake; and brain uptake inconsistent with the known GluN2B expression profile (8–11). Recently, our group successfully developed (R)-11C-Me-NB1, a 3-benzazepin-1-ol–derived GluN2B-subunit PET radiotracer lacking the aforementioned limitations (12) and currently being assessed in a first-in-humans clinical trial. The major inherent limitation of this tracer is the short physical half-life of 11C (20.3 min), which impedes its use in imaging centers without an on-site cyclotron. The development of an 18F (physical half-life, 109.8 min)-radiolabeled GluN2B-subunit PET tracer would offer satellite distribution and higher image resolution (13). Our initial endeavors yielded (R)-18F-OF-Me-NB1, an ortho-fluorinated 18F analog of Me-NB1 (14) that, despite the encouraging preclinical results, displayed a relatively fast brain washout. Nevertheless, 2 characteristics were explicated from this published study. The first is that ortho-fluorination on the phenyl ring critically impacted the affinity of the tracer toward GluN2B subunits whereas para-fluorination led to a loss of affinity. The second evident characteristic was that the (R)-enantiomer of 18F-OF-Me-NB1 was superior to the (S)-enantiomer, as shown by its specific and selective behavior toward GluN2B subunit–containing NMDARs. At the time, it was also reported that 3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-benzazepine-1,7-diol exhibits 18-fold higher GluN2B subunit affinity than its methoxy analog (15). Remarkably, we showed in our previous study that the loss of high binding affinity and selectivity of the para-fluorinated 3-benzazepin-1-ol (PF-Me-NB1) could be restored on cleavage of the methyl ether group (16). More importantly, we found that the (S)- and (R)-enantiomers of PF-NB1 did not differ in their in vitro properties with regard to their binding affinity and selectivity toward GluN2B subunits.
As part of our continuous efforts to develop a 18F-labeled PET radioligand with suitable characteristics for imaging the GluN2B subunits of the NMDAR, we investigated in the current work ortho-fluorinated (OF) and meta-fluorinated (MF) analogs of para-fluorinated (PF)–NB1 (Fig. 1). After the synthesis and in vitro validation of both compounds, OF-NB1 was selected for subsequent radiofluorination and further in vitro and in vivo evaluation. To showcase the utility of this novel tracer, RO studies were performed on rats applying CP-101,606, a GluN2B antagonist. Furthermore, we evaluated in a pilot study a possible in vivo interaction or off-target binding of 18F-OF-NB1 to σ1Rs using the σ1 ligand, fluspidine, and σ1R knock-out (σ1-KO) mice. Finally, for the purpose of clinical translation, we assessed the binding properties of 18F-OF-NB1 on postmortem human brain tissues obtained from amyotrophic lateral sclerosis (ALS) patients using in vitro autoradiography, the results of which were exemplarily confirmed by immunohistochemistry and analyzed by confocal microscopy.

Structures of PF-NB1 and its regioisomers, OF-NB1 and MF-NB1 (16).
MATERIALS AND METHODS
The organic syntheses of the reference compounds and precursors were performed in analogy to our previously published protocols, with minor modifications for the precursor synthesis (Supplemental Figs. 1 and 2; supplemental materials are available at http://jnm.snmjournals.org) (14). Animal experiments were accomplished in accordance with the Swiss Animal Protection Law and the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines after authorization from the local veterinary office of the Canton Zurich in Switzerland. The glass slides for autoradiography and confocal microscopy were prepared at the Institute of Veterinary Pathology, University of Zurich. Wistar rats were acquired from Charles River, whereas CD1 and σ1R-KO mice were acquired from Envigo, and all were retained under standard conditions as previously reported (17). Human ALS and non-ALS brain tissue was obtained from the National Institutes of Health NeuroBioBank and the Harvard Brain Tissue Resource Center. The need for informed consent for brain tissues was waived under local and national law, given that the tissue samples were anonymized. Metabolite and biodistribution studies were conducted as previously described (14). LogD7.4 of 18F-OF-NB1 was determined using the shake-flask method in n-octanol/phosphate-buffered saline (PBS) at pH 7.4.
Chemistry and Binding Affinity Determination
Chemicals and materials were purchased from ABCR, Ametek Scientific, Armar, Merck, Sigma-Aldrich, Acros Organics, and PerkinElmer and were used directly without any further purification. Detection, characterization, and binding affinity experiments were performed in accordance with previously published procedures (16).
Radiochemistry
The 18F-OF-NB1 was radiosynthesized using boronic ester 6 in analogy to a previously published procedure by our group (Supplemental Fig. 3) (16,18). The 18F-fluspidine was radiosynthesized as previously reported (19).
In Vitro Autoradiography
In vitro autoradiography was performed as previously described by our group (14,16). For blocking experiments, 1 μM of GluN2B ligands of either CERC-301 or EVT-101 were used. For off-target binding, 1 μM of 2 σ1R ligands, SA4503 and fluspidine, were used.
Metabolite Study
Wistar rats were injected intravenously with 249–599 MBq (2.74–4.70 nmol/kg) of 18F-OF-NB1. Brain extracts at 15, 30, and 60 min after injection, and blood samples at 5, 15, 30, 45, and 60 min after injection, were acquired, processed, and analyzed by radio–ultraperformance liquid chromatography as previously described by our group (14,16).
Ex Vivo Biodistribution
Eight male Wistar rats (4 baseline and 4 blockade animals) were administered 18F-OF-NB1 (16–22 MBq, 0.31–0.42 nmol/kg) via tail-vein injection, either alone or shortly after a 2 mg/kg injection of eliprodil, and the rats were killed by decapitation under isoflurane anesthesia at 45 min after injection and processed as previously described by our group (14,16).
In Vivo PET and Dose–Response Studies
CD1 and σ1R-KO mice, as well as Wistar rats, were anesthetized with isoflurane and imaged with a PET/CT scanner (Super Argus; Sedecal) upon the tail-vein injection of 18F-OF-NB1 or 18F-fluspidine. Either 28–45 MBq (0.42–8.10 nmol/kg) of the radiotracers were injected into rats or 15–17 MBq (2.54–18.51 nmol/kg) were injected into mice, with a scan time of 90 min. Dose–response and RO studies were accomplished in Wistar rats by tail-vein injection of different doses (0.5, 3.0, 10, and 15 mg/kg) of CP101,606. For the cross-talk studies in mice, the GluN2B blocker (R)-Me-NB1 (2 mg/kg), the σ1 receptor blockers fluspidine (1 mg/kg) and SA4503 (2.5 mg/kg) were used. All blockers were administered 1 min before radiotracer injection. The acquired data were reconstructed as previously reported (17). Time–activity curves were produced by PMOD software (version 3.9; PMOD Technologies) with predefined volumes of interest using an MRI T2 template.
Fluorescence Immunostaining, Confocal Microscopy, and Image Analysis
For fluorescence staining, 10-μm-thick ALS and non-ALS frozen brain tissue sections were washed with PBS and then fixed in 4% paraformaldehyde in PBS for 30 min. Nonspecific adsorption of antibody was prevented by adding bovine serum albumin (2% w/v, 30 min). After the samples had been washed 3 times with PBS, the tissues were incubated with primary antibody anti-NMDAR2B (1/100) for 1 h at room temperature (N59/36 [ab93610]; Abcam). Samples were then washed 3 times with PBS and treated with a secondary antibody (goat anti-mouse Alexa-Fluor 633; Invitrogen), with a dilution of 1:500 in PBS for 1 h at room temperature. Cell nuclei were stained by 4′,6-diamidino-2-phenylindole (2 μg/mL, 10 min). Samples were finally washed 3 times with PBS, mounted in mounting medium, and imaged using a confocal microscope (FV1000; Olympus).
Pixel-by-pixel signal-intensity was analyzed while maintaining identical imaging conditions for samples to be compared. This analysis was performed to compare intraexpression (in different regions of the tissue) and interexpression (between different samples) of GluN2B. The mean intensities from selected parts of an image were measured using ImageJ, and the average intensity of images was plotted for each condition.
Statistical Analysis
An independent 2-tailed Student test of the dataset was used to calculate statistical probability values, assuming a normal distribution.
RESULTS
Chemistry and Binding Affinity
Modifications introduced for the synthesis of boronic ester precursor 6 resulted in an overall yield of 11%, a 7-fold increase compared with the original route. MF-NB1, on the other hand, was obtained in 14% yield. Inhibition constants for OF-NB1 and MF-NB1 toward the GluN2B subunits and σ1Rs were determined using competitive binding assays with 3H-ifenprodil and (+)-3H-pentazocine, respectively, and are summarized in Table 1.
Binding Affinity (Ki) and Selectivity Profile of OF-NB1 and MF-NB1
Radiolabeling
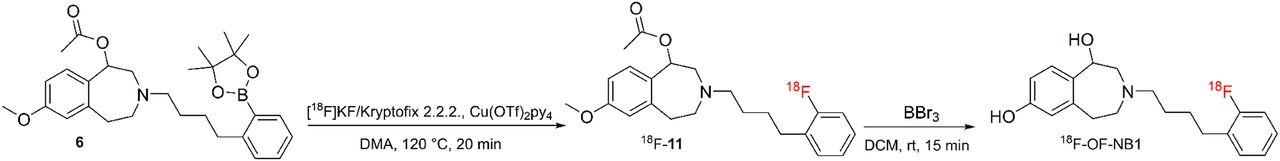
The 18F-OF-NB1 was radiosynthesized using aryl boronic ester precursor 6 in an average total time of 140 min from the end of bombardment (Fig. 2). The isolated radiochemical yield of 18F-OF-NB1 was 5.5% ± 0.1% (n = 6), with radiochemical purity of more than 95%. Molar activity amounted to 192 ± 33 GBq/μmol (n = 6). LogD7.4 was found to be 2.18 ± 0.06 (n = 5), which is an optimal value for passive brain entry (20). The radiochemical yield obtained for 18F-fluspidine was similar to reported values and the purified product was formulated in saline as previously described (19).

Representative radiosynthesis of 18F-OF-NB1. DMA = dimethylacetamide; DCM = dichloromethane. (Kryptofix is a registered trademark of Merck.)
In Vitro Autoradiography
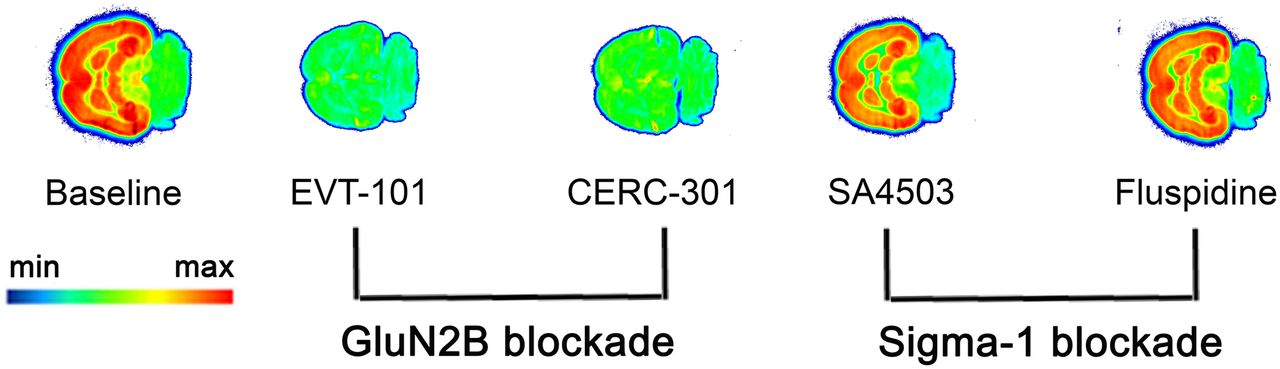
A representative autoradiogram obtained with 18F-OF-NB1 is depicted in Figure 3. 18F-OF-NB1 showed heterogeneous binding with high accumulation in brain areas such as the cortex, striatum, thalamus, and hippocampus, brain regions known to have high densities of the GluN2B subunits (5). The cerebellum, a region with low GluN2B-subunit expression, exhibited negligible radioactivity accumulation. GluN2B antagonists blocked the accumulation of 18F-OF-NB1, whereas σ1R blockers did not compete with 18F-OF-NB1.
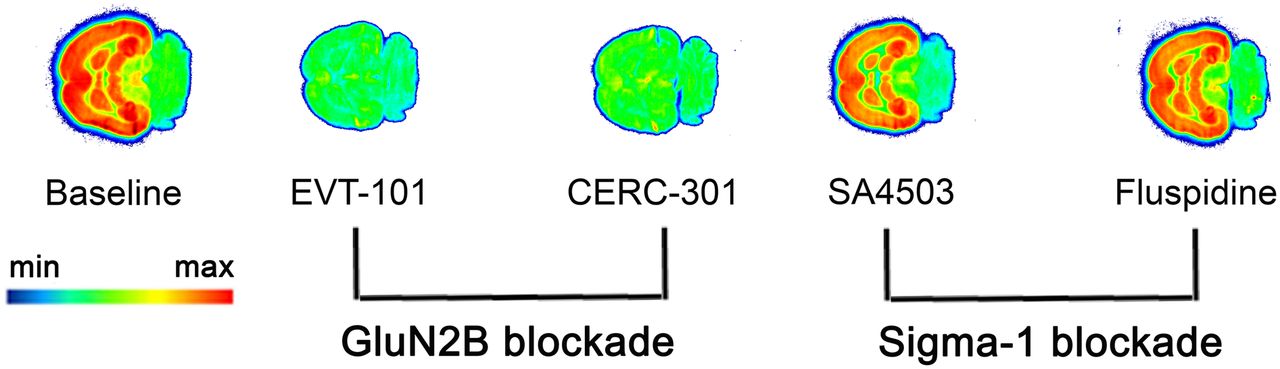
Typical in vitro autoradiogram of 18F-OF-NB1 (3 nM) using rat coronal brain sections. GluN2B blockers and σ1R blockers were used to confirm specificity and selectivity, respectively. Solutions (1 μM) of GluN2B antagonists EVT-101 and CERC-301 were used to determine specificity toward GluN2B subunit, whereas 1 μM solutions of σ1R blockers SA4503 and fluspidine were used to confirm selectivity over σ1Rs.
Dose-Dependency and RO Studies in Wistar Rats
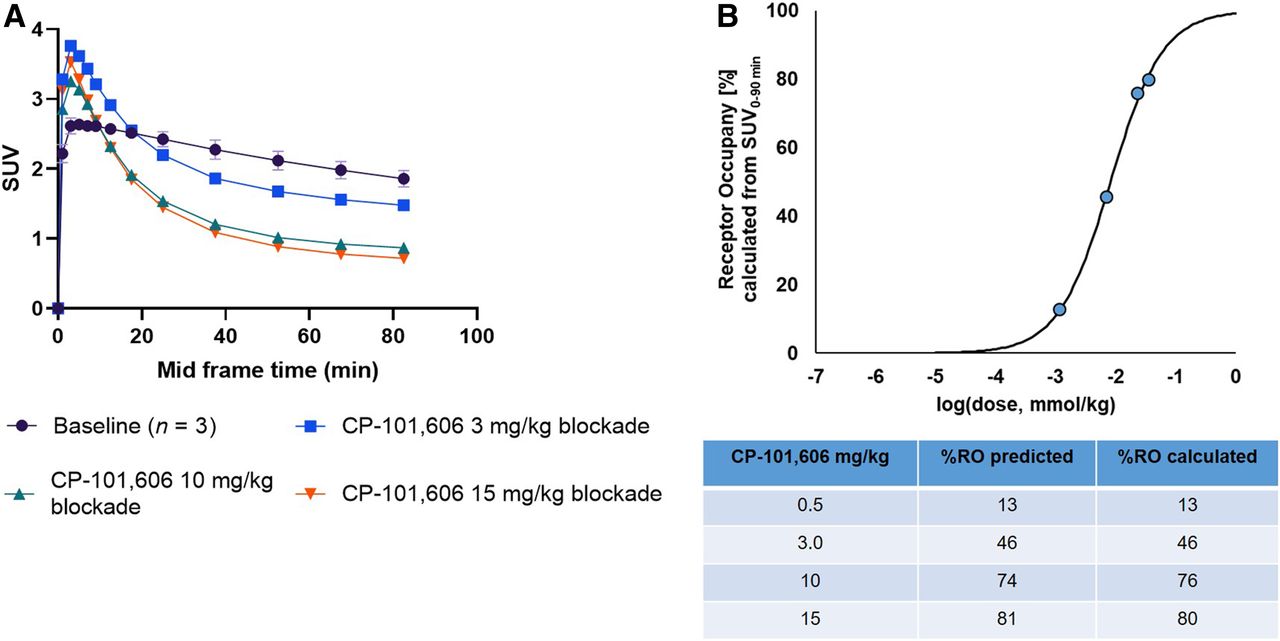
18F-OF-NB1 exhibited excellent blood–brain barrier penetration in line with the measured LogD7.4 value of 2.18. Under baseline conditions, a heterogeneous accumulation in the rat brain, consistent with the GluN2B-subunit expression pattern, was observed (Supplemental Fig. 4). In Figure 4 are shown coronal, sagittal, and axial averaged images of the rat brain under baseline and blockade conditions. Radioactivity uptake was reduced in a dose-dependent manner using the GluN2B subunit antagonist CP-101,606 as reflected by the respective time–activity curves in Figure 5A. The SUVs from the blocking studies were used to calculate the in vivo RO of CP-101,606 (Fig. 5B). A dose of 8.1 μmol/kg was required to achieve 50% RO.

PET images of 18F-OF-NB1 averaged over 30–90 min in rat brain under baseline and blockade (15 mg of CP-101,606 per kilogram) conditions superimposed on MRI T2 template (PMOD). Color bar points to minimum and maximum SUVs (calculated as accumulated radioactivity [Bq] per tissue [g] and divided by injected dose per body weight).
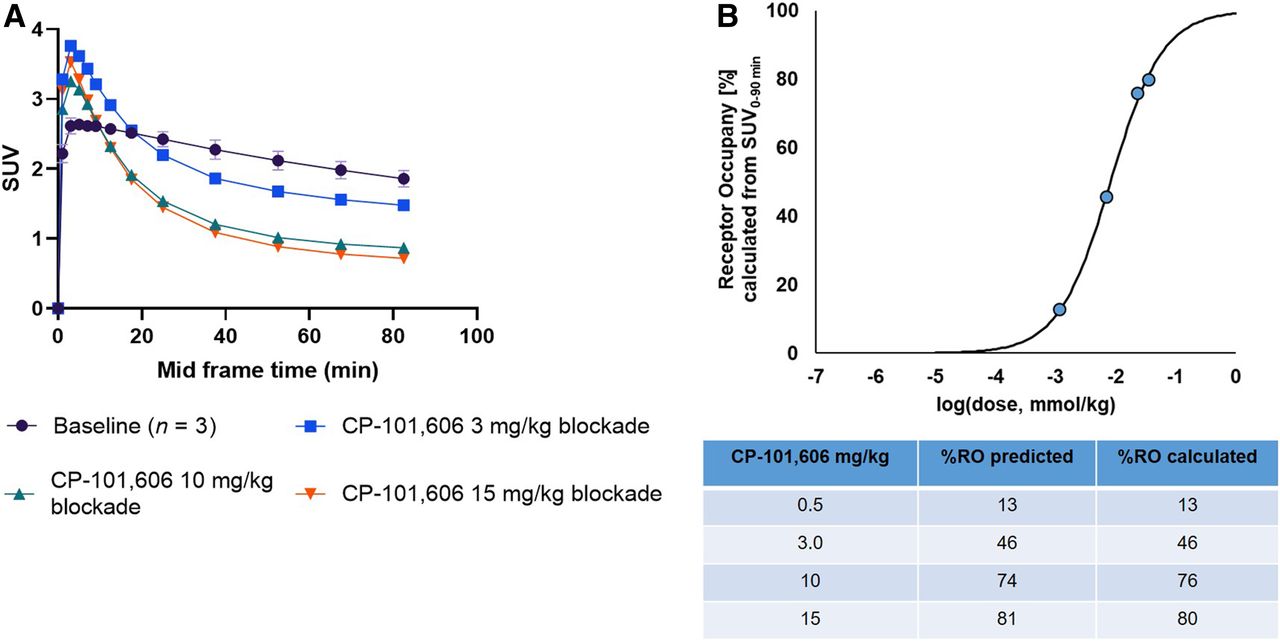
(A) Time–activity curves representing whole-brain accumulation of 18F-OF-NB1 presented as SUVs 0–90 min after tracer injection under baseline conditions, and time–activity curves denoting blockade with GluN2B subunit antagonist CP-101,606 with different doses (3, 10, and 15 mg/kg). (B) RO of GluN2B subunit antagonist CP101,606 using 18F-OF-NB1 in Wistar rats calculated from SUV at 0 min to SUV at 90 min. D50 value of 8.1 μmol/kg was obtained using 18F-OF-NB1. D50 is defined as drug dose required to achieve 50% RO when administered and was calculated as previously outlined (16).
Ex Vivo Metabolite Study
The metabolite analysis revealed only intact parent radiotracer in the brain 60 min after injection, as shown in Supplemental Figure 5.
Ex Vivo Biodistribution Studies
GluN2B subunit–rich brain regions such as the cortex, striatum, thalamus, and hippocampus exhibited a high accumulation of radioactivity compared with regions with poor expression of the GluN2B subunits, that is, the cerebellum. Values for the normalization of GluN2B-rich regions to the cerebellum were as follows: cortex/cerebellum (1.39 ± 0.03), striatum/cerebellum (1.25 ± 0.03), thalamus/cerebellum (1.22 ± 0.06), and hippocampus/cerebellum (1.03 ± 0.02). In the periphery, specific accumulation was highest in the adrenal gland. Nonetheless, previous reports repudiated the presence of GluN2B subunits in the adrenal gland (21). On the other hand, regions such as the brain stem and midbrain showed high and specific accumulation. Biodistribution results are summarized in Supplemental Tables 1 and 2.
PET Imaging in CD1 and σ1-KO Mice
Injection of fluspidine (1 mg/kg) reduced the uptake of 18F-OF-NB1 (Fig. 6A) in both CD1 and σ1R-KO mice. Uptake of 18F-fluspidine, on the other hand, in both CD1 and σ1R-KO mice, was substantially different (Fig. 6B); a rapid washout was observed in the σ1R-KO mice, whereas a slow washout and persistently high time–activity curves were observed in the CD1 mice. Specificity of binding was confirmed using another σ1R ligand, SA4503 (2.5 mg/kg), as a blocker. Furthermore, when a 2 mg/kg dose of (R)-Me-NB1 was used as a blocker in CD1 mice, a blocking effect was observed.

(A) Whole-brain time–activity curves of 18F-OF-NB1 (n = 1 for each condition) in wild-type CD1 and σ1R-KO mice under baseline and blockade conditions with σ1R agonist, fluspidine (1 mg/kg). (B) Whole-brain time–activity curves of 18F-fluspidine (n = 1 for each condition) in CD1 and σ1R-KO mice under baseline and blockade conditions with σ1R agonist SA4503 (2.5 mg/kg) and with GluN2B subunit ligand (R)-Me-NB1 in CD1 mice.
Autoradiography and Confocal Microscopy Using Postmortem Human Brain Tissue
In postmortem cortical brain tissues (BA4 and BA6), we found a lower accumulation of 18F-OF-NB1 in ALS patients than in non-ALS subjects in autoradiographic experiments (Fig. 7A). To confirm that this finding was not confounded by neuronal loss, we further performed fluorescence immunostaining and confocal imaging of the brain cryosections. For the confocal microscopy, the signal intensity of the images was analyzed semiquantitatively and the level of GluN2B subunit expression was normalized to cell nuclei count. Overall, the immunostained images (n = 10) showed a significantly decreased expression of GluN2B and no neuronal loss in brain tissues of ALS patients compared with non-ALS subjects (P < 0.01, Fig. 7B). Furthermore, we found that in areas with a higher cell nucleus density (>150 cells/106 μm2), areas that are most likely cortical regions of the brain, the expression of GluN2B was relatively homogeneous in non-ALS brain sections and significantly higher (P < 0.0001) than in ALS patient brain sections.

(A) Typical in vitro autoradiogram of 18F-OF-NB1 (3 nM) using human postmortem brain sections from ALS and non-ALS subjects. Blocking was achieved using 1 μM solution of GluN2B antagonist CP-101,606. (B) Comparative fluorescence immunostaining and confocal imaging of postmortem human brain cryosections shows relatively lower expression of GluN2B subunits (red) in ALS patient than in non-ALS subject. Images were analyzed and graphically represented, with each dot denoting 1 image. Nuclei are stained with 4′,6-diamidino-2-phenylindole (blue). Scale bar = 100 μm.
DISCUSSION
18F-PF-NB1, a 3-benzazepine-1,7-diol derivative published recently by our group, is by far the best-performing radiofluorinated tracer in view of its favorable in vivo profile in rodents (16). In the current work, we investigated whether shifting the fluorine atom from the para-position in PF-NB1 to the ortho- and meta-positions in the phenyl ring, termed OF-NB1 and MF-NB1, respectively, would have any major impact on the in vitro and in vivo binding properties. Although MF-NB1 showed low affinity and selectivity toward GluN2B subunits, OF-NB1 displayed superior in vitro binding properties and was therefore selected for radiofluorination. PET imaging findings for 18F-OF-NB1 in rats corroborated the in vitro autoradiography results whereby a heterogeneous and high radioactivity uptake was observed in GluN2B-rich regions. The administered drug dose required to achieve 50% RO (D50), obtained from the CP-101,606 RO studies, was similar to the values observed in previous studies. The time–activity curves were also similar to those of 18F-PF-NB1 (12,16). 18F-OF-NB1 established its superiority over 18F-PF-NB1 in ex vivo biodistribution studies, in which case higher target-to-cerebellum accumulation ratios were achieved. However, 18F-OF-NB1 showed specific binding in regions of low GluN2B expression, such as the midbrain, brain stem, and cerebellum, in line with previous reports (12,14,16,17). Whether this is a GluN2B-related specific binding or an off-target binding remains to be investigated. The administration of CP101,606 caused an initial surge in brain uptake of 18F-OF-NB1, which may be explained by the increased availability of unbound tracer arising from blockade of peripheral GluN2B-bearing NMDA receptors.
Using GluN2B KO mice is an elegant method to investigate the potential in vivo interaction or off-target binding of 18F-OF-NB1 toward σ1Rs; however, homozygous GluN2B KO mice are not viable (22). Alternatively, σ1R-KO mice, which survive for a relatively long period, can be used. On the basis of our previously reported cross-talk experiments, we hypothesized that the administration of a σ1R ligand would alter the binding of 18F-OF-NB1 in wild-type mice but not in σ1R-KO mice, given the absence of σ1R modulation (17). To modulate σ1Rs, we selected fluspidine since the 18F-labeled version is an established PET radioligand for imaging σ1Rs (19). As anticipated, injection of a 1 mg/kg dose of fluspidine in CD1 mice reduced uptake of 18F-OF-NB1. Surprisingly, a similar blocking effect was observed in the σ1R-KO mice which could not be attributed to σ1R modulation (Fig. 6A). Possible explanations could be that off-target binding of fluspidine to GluN2B subunits occurs or that σ1R-KO mice are not entirely devoid of σ1Rs. To validate the σ1R-KO mice, we administered 18F-fluspidine into wild-type CD1 and σ1R-KO mice. A fast washout of 18F-fluspidine was observed in the brain of σ1R-KO mice, indicating the absence of σ1Rs and off-target binding of 18F-fluspidine (Fig. 6B). To probe the off-target potential of the 3-benzazepine series, we applied (R)-Me-NB1, a selective GluN2B subunit ligand (12). Unexpectedly, a 2 mg/kg dose of (R)-Me-NB1 showed a substantial blocking effect comparable to a 2.5 mg/kg dose of SA4503, a σ1R ligand. From these studies, we concluded that fluspidine in concentrations of 1 mg/kg might exhibit off-target binding toward GluN2B subunit–containing NMDARs and that the observed effects are not necessarily an exclusive consequence of cross-talk between σ1Rs and GluN2B-containing NMDARs. Similar blocking effects with SA4503 were observed in a recent study on another GluN2B radioligand, 11C-(S)-NR2B-Me (23). The off-target effect is also valid for some GluN2B ligands, such as (R)-Me-NB1, for which off-target binding toward σ1Rs becomes more prominent at high concentrations. For future cross-talk imaging experiments with different σ1R agonists, one should consider the use of a wild-type CD1/σ1R-KO mouse pair, as done in this study, to exclude the direct effect of the σ1R agonists on GluN2B-containing NMDARs. Additionally, we suggest that future σ1R in vitro binding experiments should not focus solely on σ1/σ2R selectivity but should also take into account the off-target binding affinity to GluN2B subunits.
Given our ultimate goal of clinical translation, we explored the expression level of GluN2B subunits in postmortem cortical tissues of ALS patients and healthy controls using in vitro autoradiography. Glutamatergic excitotoxicity is described as one of the contributing factors to the susceptibility of the motor neurons to neurodegeneration in ALS (24). Supporting evidence suggested changes in the expression of GluN2B subunit–containing NMDARs in mouse and rat models of ALS (25,26), and therefore, for our investigations, we selected ALS as a disease model for neurodegeneration. In particular, 2 subcortical regions (motor and frontal, BA4 and BA6) were selected for this experiment. We found a decreased accumulation of the tracer in tissues from ALS patients compared with healthy volunteers. This decreased accumulation was further corroborated with immunohistochemistry as shown by confocal microscopy, suggesting that GluN2B subunit levels can be measured in ALS patients undergoing GluN2B-related treatments.
CONCLUSION
18F-OF-NB1 has been developed as a front-runner of our first-generation radiofluorinated ligand, (R)-18F-OF-Me-NB1. It showed favorable in vitro and in vivo attributes in terms of GluN2B subunit specificity, selectivity over σ1Rs, metabolism, and kinetics. Its utility for drug development was demonstrated in an RO study with CP-101,606, wherein a dose dependency of radioligand uptake was observed. Off-target binding of GluN2B ligands to σ1Rs and vice versa is a potential confounder when developing drugs targeting these receptors or studying the featured in vivo cross-talk between both receptors. Finally, in vitro autoradiography using ALS and healthy postmortem brain tissues revealed a decreased accumulation of 18F-OF-NB1 in GluN2B-rich regions in ALS patients when compared with healthy controls. This finding possibly indicates a substantial GluN2B subunit downregulation, which was confirmed by immunohistochemistry. Overall, the results suggest that 18F-OF-NB1 is a promising GluN2B radioligand with utility for RO studies and PET imaging studies in patients with ALS and potentially also for other neurodegenerative diseases not investigated in this study.
DISCLOSURE
This project was supported by grants 310030E_160403/1 and 310030E_182872/1 from the Swiss National Science Foundation to Simon Ametamey. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can we selectively image GluN2B subunit–containing NMDARs with 18F-OF-NB1 in vitro and in vivo, and can GluN2B-targeted imaging serve as a diagnostic marker for ALS?
PERTINENT FINDINGS: PET imaging using 18F-OF-NB1 in Wistar rats showed heterogeneous uptake in GluN2B-rich brain regions that could be blocked in a dose-dependent manner by the GluN2B antagonist CP-101,606, and autoradiography experiments using postmortem cortical brain tissue revealed lower GluN2B subunits in ALS patients than in healthy controls, as was in line with immunohistochemistry results.
IMPLICATIONS FOR PATIENT CARE: 18F-OF-NB1 has the potential to detect GluN2B subunits in patients with neurodegenerative diseases, especially in ALS patients, and has utility for RO studies in humans.
Acknowledgments
We thank the NIH NeuroBioBank and the Harvard Brain Tissue Resource Center for supplying ALS and healthy postmortem human brain tissue. Dr. Jose Miguel Vela is acknowledged for providing our group with the σ1R-KO mice.
Footnotes
Published online Jul. 31, 2020.
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 7, 2020.
- Accepted for publication June 26, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Hippocampal mGluR5 levels are comparable in Alzheimers and control brains, and divergently influenced by amyloid and tau in control brain
- Evaluation of (rac)-, (R)-, and (S)-18F-OF-NB1 for Imaging GluN2B Subunit-Containing N-Methyl-D-Aspartate Receptors in Nonhuman Primates
- First-in-Humans Brain PET Imaging of the GluN2B-Containing N-methyl-D-aspartate Receptor with (R)-11C-Me-NB1