


Visual Abstract

Abstract
Despite 2 decades of research, no N-methyl-d-aspartate (NMDA) glutamate receptor (GluN) subtype 2B (GluN1/2B) radioligand is yet clinically validated. Previously, we reported on (rac)-18F-OF-NB1 as a promising GluN1/2B PET probe in rodents and its successful application for the visualization of GluN2B-containing NMDA receptors in postmortem brain tissues of patients with amyotrophic lateral sclerosis. In the current work, we report on the in vivo characterization of (rac)-, (R)-, and (S)-18F-OF-NB1 in nonhuman primates. Methods: PET scans were performed on rhesus monkeys. Plasma profiling was used to obtain the arterial input function. Regional brain time–activity curves were generated and fitted with the 1- and 2-tissue-compartment models and the multilinear analysis 1 method, and the corresponding regional volumes of distribution were calculated. Blocking studies with the GluN1/2B ligand Co 101244 (0.25 mg/kg) were performed for the enantiopure radiotracers. Receptor occupancy, nonspecific volume of distribution, and regional binding potential (BPND) were obtained. Potential off-target binding toward σ1 receptors was assessed for (S)-18F-OF-NB1 using the σ1 receptor ligand FTC-146. Results: Free plasma fraction was moderate, ranging from 12% to 16%. All radiotracers showed high and heterogeneous brain uptake, with the highest levels in the cortex. (R)-18F-OF-NB1 showed the highest uptake and slowest washout kinetics of all tracers. The 1-tissue-compartment model and multilinear analysis 1 method fitted the regional time–activity curves well for all tracers and produced reliable regional volumes of distribution, which were higher for (R)- than (S)-18F-OF-NB1. Receptor occupancy by Co 101244 was 85% and 96% for (S)-18F-OF-NB1 and (R)-18F-OF-NB1, respectively. Pretreatment with FTC-146 at both a low (0.027 mg/kg) and high (0.125 mg/kg) dose led to a similar reduction (48% and 49%, respectively) in specific binding of (S)-18F-OF-NB1. Further, pretreatment with both Co 101244 and FTC-146 did not result in a further reduction in specific binding compared with Co 101244 alone in the same monkey (82% vs. 81%, respectively). Regional BPND values ranged from 1.3 in the semiovale to 3.4 in the cingulate cortex for (S)-18F-OF-NB1. Conclusion: Both (R)- and (S)-18F-OF-NB1 exhibited high binding specificity to GluN2B subunit–containing NMDA receptors. The fast washout kinetics, good regional BPND values, and high plasma free fraction render (S)-18F-OF-NB1 an attractive radiotracer for clinical translation.
Glutamatergic N-methyl-d-aspartate receptors (NMDARs) are heterotetrameric ion channels ubiquitously expressed in the mammalian brain. The receptor complex is assembled from distinct subunits, namely N-methyl-d-aspartate (NMDA) glutamate receptor (GluN) subtypes 1a–1h (GluN1(a–h)), GluN subtypes 2A–2D (GluN2(A–D)), and GluN subtypes 3A and 3B, where each GluN2 subtype exhibits a unique spatiotemporal expression and function (1). NMDARs are known to be the linchpin for synaptic plasticity and transmission as well as higher cognitive functions. Notably, synaptic NMDARs are promotors of cell survival whereas extrasynaptic GluN2B subunit–enriched NMDARs are mediators of cell death. The GluN1/2B receptors are associated with several neuropathologies, such as Alzheimer’s disease, Parkinson’s disease, depression, schizophrenia, and stroke, among others (2–5). As such, the development of GluN1/2B antagonists has been the prime focus of therapeutic development efforts over the past 2 decades, with 19 therapeutic patent applications published in the past 5 years alone (6,7). Although these subtype-selective antagonists possess pharmacologic action similar to conventional NMDA ion channel blockers, they exhibit a much more favorable neurologic safety profile, as their selectivity is thought to preserve the physiologic functions of NMDARs but minimize potential undesired pharmacologic effects (4,8). To date, several GluN1/2B antagonists, such as CERC-301, have been evaluated in clinical trials with no-to-modest clinical benefits, whereas others are still in development (7,9). PET imaging is a powerful modality that can accelerate drug development—for example, to be used in target engagement and drug occupancy studies—and serve as a credible tool in patient selection and monitoring (10,11). Despite the long-standing interest, efforts to develop a GluN1/2B-specific PET radioligand have largely been unsuccessful. Challenges include poor brain uptake, homogeneous distribution, and the existence of brain radiometabolites, as well as binding to other brain receptors, most notably σ1 receptors (σ1Rs) (12).
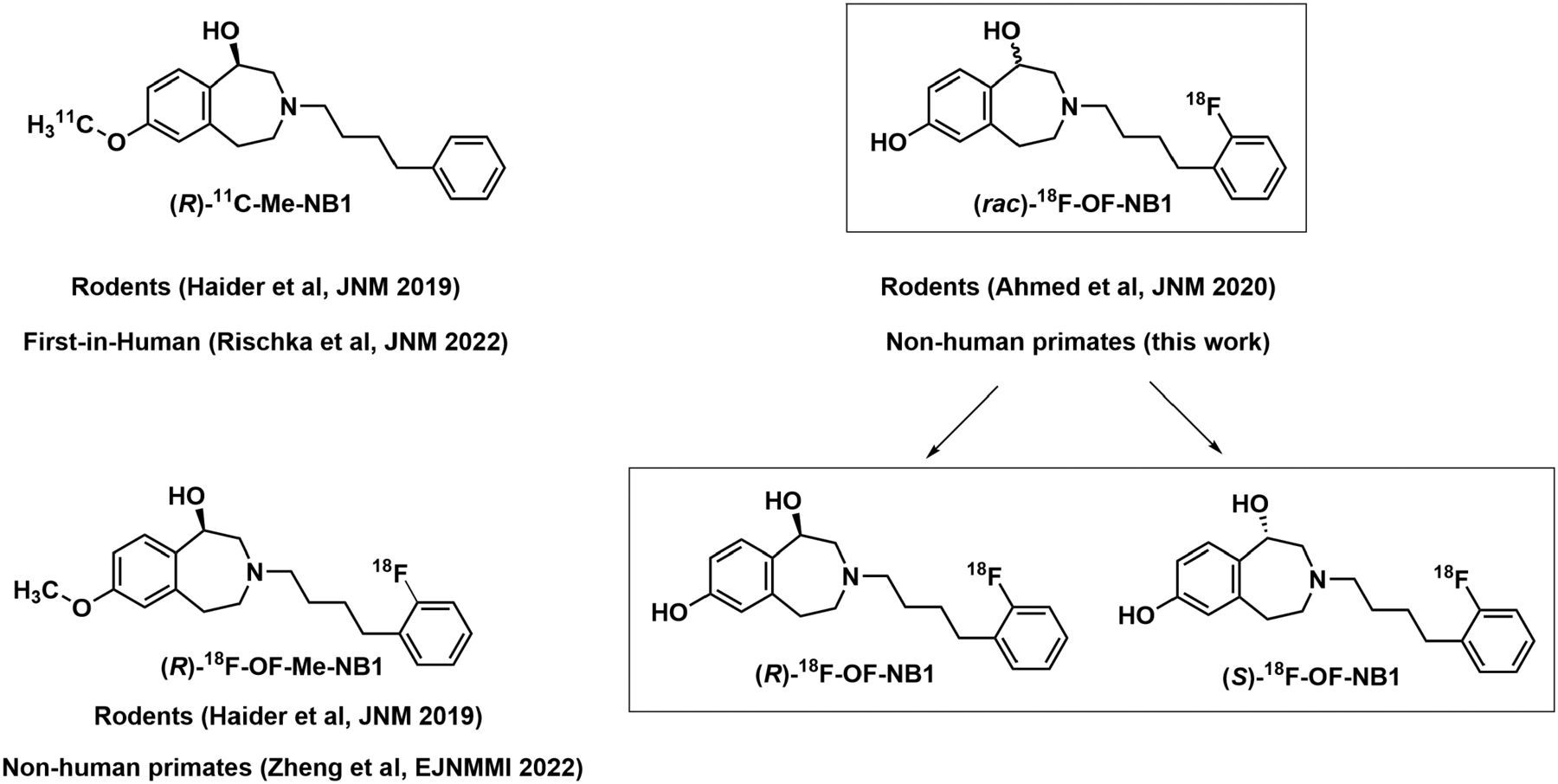
The 3-benzazepine structural class of compounds has recently emerged as potential PET radioligand candidates for the imaging of GluN1/2B (Fig. 1) (13,14). Recently, Ametamey’s group successfully developed the 3-benzazepin-1-ol derivative, (R)-11C-Me-NB1, for imaging GluN1/2B in humans (15–17). To overcome the limitation of the short physical half-life of 11C-labeled radioligands, we evaluated several radiofluorinated derivatives in animals (18–20). (Rac)-18F-OF-NB1 (inhibition constant Ki (GluN1/2B), 10.4 ± 4.7 nM) emerged as one of the most promising candidates for evaluation in nonhuman primates (19). In the current work and for the purpose of clinical translation, we evaluated the pharmacokinetic and imaging properties of (rac)-, (S)-, and (R)-18F-OF-NB1 in nonhuman primates, and performed a comprehensive assessment of (S)-18F-OF-NB1 regarding its binding specificity to GluN1/2B receptors and selectivity over σ1Rs in vivo.

Structures of (R)-11C-Me-NB1, the lead radiotracer recently evaluated in clinical trial; (R)-18F-OF-Me-NB1, the first radiofluorinated derivative of (R)-11C-Me-NB1 evaluated in rats; and (rac)-, (R)-, and (S)-18F-OF-NB1 evaluated in the current work.
MATERIALS AND METHODS
Chemistry
Synthesis of the reference compound (rac)-OF-NB1 and the corresponding aryl boronic ester precursors followed previously published procedures, as did their chiral high-performance liquid chromatography (HPLC) separation (18,19).
Radiochemistry
18F-fluoride was produced via the 18O(p,n)18F nuclear reaction using H218O (Huayi Isotopes) in a 16.5-MeV GE Healthcare PETtrace cyclotron. After bombardment, the activity was transferred to a shielded hot cell and trapped on an anionic-exchange resin cartridge (Chromafix PS-HCO3; Macherey-Nagel). Trapped 18F− was eluted into a sealed 5-mL borosilicate glass reaction vial using a 2-mL solution of K2C2O4 (1.0 mg/mL in water), K2CO3 (0.1 mg/mL in water), and Kryptofix 222 (Merck) (6.3 mg/mL in acetonitrile). The solution was azeotropically dried for 5 min at 110°C under nitrogen, followed by drying twice using 0.4 mL of acetonitrile each time. Afterward, the vial was purged with 20 mL of air before the addition of 0.4 mL of the boronic ester precursor solution (6 mg of precursor and 12 mg of Cu(OTf)2(Py)4 in anhydrous dimethylacetamide). The reaction mixture was stirred at 110°C for 20 min, diluted with 10 mL of deionized water, and passed through a Waters C18 SepPak cartridge. The cartridge was washed with 10 mL of deionized water. The trapped activity was eluted from the SepPak with 1 mL of ethanol into a second reaction vial. The solvent was dried at 90°C under a nitrogen stream. Dichloromethane and anhydrous BBr3 were added and left to react for 15 min at room temperature. The solvent was removed by gentle nitrogen blowing, and the crude product was redissolved in the HPLC mobile phase (25% acetonitrile/75% 0.1 M ammonium formate with 0.5% acetic acid, pH 4.2). The solution was injected into a semipreparative HPLC system equipped with an Agilent Eclipse XDB-C18 (9.4 × 250 mm, 5 μm) column eluting with the above mobile phase at a flow rate of 5 mL/min. The radioactive peak from 15 to 16 min was collected and diluted with 50 mL of water. The solution was passed through a Waters C18 SepPak cartridge. The product was eluted from the SepPak with 1 mL of ethanol and dried under gentle nitrogen blowing. The product was redissolved with the second HPLC mobile phase (32% acetonitrile/68% water with 0.05% triethylamine) and injected into the chiral semipreparative HPLC system equipped with a Regis reflect I-Amylose A column (250 × 10 mm, 5 μm) eluting with the aforementioned mobile phase at a flow rate of 5 mL/min. The radioactive peaks corresponding to (R)-18F-OF-NB1 (eluting from 23 to 25 min) and (S)-18F-OF-NB1 (eluting from 27 to 29 min) were collected in 2 separate bottles and diluted with 50 mL of water each. The solutions were passed through Waters C18 SepPak cartridges separately. Each SepPak was washed with 1 mM HCl (10 mL), dried with air, and then eluted with 1 mL of U.S. Pharmacopeia–grade ethanol, followed by 3 mL of U.S. Pharmacopeia–grade saline, into a collection vial. The solution was then passed through a 0.22-μm Millex-GV filter (Millipore) into a 10 mL dose vial precharged with 7 mL of U.S. Pharmacopeia–grade saline for formulation.
Measurement of Lipophilicity (log D7.4)
The lipophilicity (log D7.4) of (S)-18F-OF-NB1 was determined according to previously published procedures (21,22). The log D7.4 was calculated as the ratio between the concentrations of decay-corrected radioactivity in 1-octanol and pH 7.4 phosphate-buffered saline (Dulbecco).
PET Imaging in Rhesus Monkeys
All experimental procedures were approved by the Yale University Institutional Animal Care and Use Committee. In total, 11 dynamic PET scans (120 min each) were performed on 3 rhesus monkeys (Macaca mulatta, 2 males and 1 female) on the Focus 220 scanners (Siemens Medical Solutions).
Baseline scans were acquired for (rac)-, (R)-, and (S)-18F-OF-NB1, whereas blockade scans using the GluN1/2B antagonist Co 101244 (0.25 mg/kg; half-maximal inhibition concentration (IC50), 4 ± 1 nM for GluN1/2B) were performed for the enantiopure radiotracers (23). Further, blockade scans using either the σ1R antagonist FTC-146 (2 doses; 0.027 and 0.125 mg/kg; Ki, 0.0025 nM for σ1R) alone or GluN1/2B (0.25 mg/kg dose of Co 101244) followed by σ1R blockade (0.125 mg/kg dose of FTC-146) were accomplished using (S)-18F-OF-NB1 (24). Procedures for PET scanning, image processing, and image analysis are detailed in the supplemental materials (available at http://jnm.snmjournals.org).
Arterial Input Function Measurement and Metabolite Analysis
Plasma activity and parent fraction were determined in accordance with previously published procedures (25,26). Radioactivity was measured in whole blood and plasma from arterial samples taken at various time points after radiotracer injection using cross-calibrated γ-counters (Wizard 1480/2480; Perkin Elmer). The radiometabolic profile was investigated by HPLC analysis using a column-switching method with plasma samples from 0, 5, 15, 30, 60, 90, and 120 min after radiotracer injection (27). The parent fraction was calculated as the ratio of the radioactivity in parent compound–containing fractions to the total radioactivity measured. Finally, the arterial input function was calculated as the product of total plasma concentrations and the interpolated parent fraction at each time point.
Measurement of Radiotracer Free Fraction in Plasma (fp)
Ultrafiltration was the method of choice for measuring the unbound radiotracer in plasma fp (28). The fp measurement was performed in triplicates per scan and was defined as the ratio of the radioactivity concentration in the filtrate to the total activity in plasma.
Kinetic Modeling
The regional time–activity curves and metabolite-corrected arterial input functions were analyzed with the 1- and 2-tissue compartment models, as well as the multilinear analysis 1 method (starting time, 30 min) to calculate the kinetic parameters (28). The regional volume of distribution (VT, mL/cm3) from each model was derived and compared. Target occupancy and the nondisplaceable VT (VND) in the brain were derived from the Lassen plot, generated using the baseline regional VT values and the VT difference between baseline and blockade scans (29). Regional binding potential (BPND) was calculated as BPND = (VT − VND)/VND using the VND value derived from the Lassen plot for the GluN1/2B blocking scan.
RESULTS
Chemistry
The overall chemical yields of the reference compounds (rac)-, (S)-, and (R)-OF-NB1 and the respective boronic ester precursors were comparable to those published previously (18,19).
Radiochemistry
The radiosynthetic strategy is depicted in Figure 2. Despite starting from enantiopure precursors, racemization occurred during the deprotection step, requiring additional chiral purification. The final activities obtained were 1,480 ± 185 MBq (n = 5) for (rac)-18F-OF-NB1 and 203.7 ± 81.4 MBq (n = 10) for (S)- and (R)-18F-OF-NB1. The radiotracers were prepared in more than 99% radiochemical purity. (S)- and (R)-18F-OF-NB1 were both obtained in more than 98% enantiomeric purity. Molar activity was 143.5 ± 127.3 GBq/μmol (n = 10) at the end of synthesis. The total synthesis time for (S)- and (R)-18F-OF-NB1 was 200 min on average.

Radiosynthesis of (rac)-, (R)-, and (S)-18F-OF-NB1. DMA = dimethylacetamide; rt = room temperature.
Measurement of log D7.4
The measured log D7.4 of (S)-18F-OF-NB1 was 2.05 ± 0.08 (n = 4), similar to the log D7.4 of (rac)-18F-OF-NB1, and in the optimal range for brain penetration (19,30).
PET Imaging Experiments on Rhesus Monkeys
The amount of injected radioactivity and associated mass were 147.1 ± 47.4 MBq and 1.05 ± 0.70 μg, respectively (n = 10). The parent fractions at 30 min after injection were 33% (n = 2), 32% (n = 2), and 57% (n = 1) for the baseline scans of (rac)-, (S)-, and (R)-18F-OF-NB1, respectively (Fig. 3). For (S)- and (R)-18F-OF-NB1, the parent fractions decreased to 26% (n = 2) and 27%, respectively, after pretreatment of the animals with the GluN1/2B antagonist Co 101244, whereas preblocking with the σ1R antagonist FTC-146, whether at a low or a high dose, did not change the parent fraction of (S)-18F-OF-NB1 (32%). The fp was 0.12 ± 0.01 for (rac)-18F-OF-NB1 (n = 2), 0.16 ± 0.01 for (S)-18F-OF-NB1 (n = 8), and 0.13 for (R)-18F-OF-NB1 (n = 1).

Parent fraction remaining of (rac)-, (R)-, and (S)-18F-OF-NB at different time points under baseline and blockade conditions. For GluN1/2B blockade, dose of 0.25 mg/kg of Co 101244 was used. For σ1R blockade, either low dose (0.027 mg/kg) or high dose (0.125 mg/kg) of FTC-146 was used.
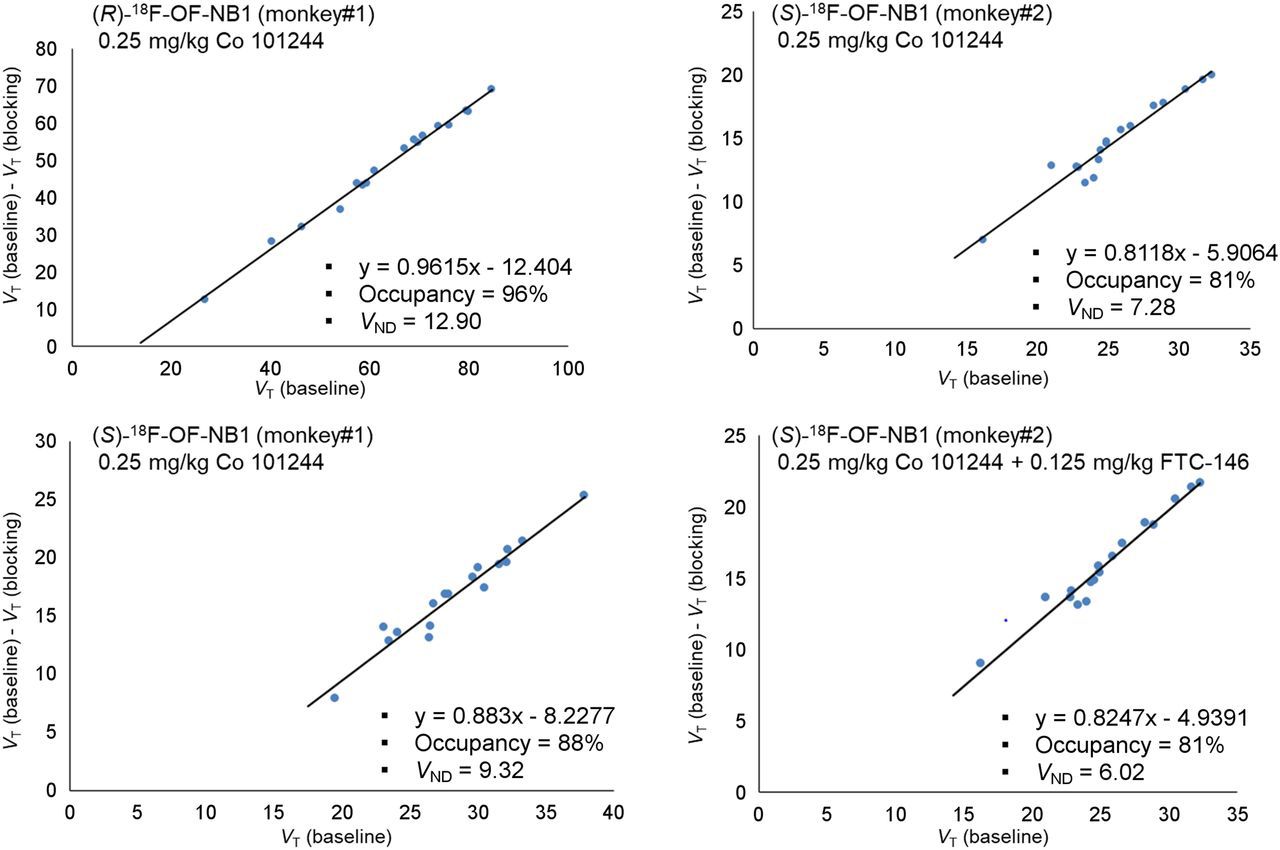
In the monkey brain, all tracers showed a heterogeneous distribution, with uptake high in the cortex, putamen, and cerebellum; moderate in the hippocampus and thalamus; and lowest in the centrum semiovale (Fig. 4). Brain uptake of (S)-18F-OF-NB1 peaked at an SUV of 4–5 within 20 min after injection, followed by a relatively fast clearance. The (R)-enantiomer, on the other hand, displayed slow brain clearance, with uptake increasing throughout the 120-min scan. Brain uptake levels and tissue clearance of (rac)-18F-OF-NB1 were between those of the 2 enantiomers (Supplemental Fig. 1). Pretreatment with the GluN1/2B antagonist Co 101244 (0.25 mg/kg) reduced radioactivity levels in all regions to nearly those in the semiovale for both enantiomers at the end of the scans. Receptor occupancy by a 0.25 mg/kg dose of Co 101244 was estimated at 81% and 88% across the brain in 2 separate monkeys for (S)-18F-OF-NB1 and 96% for (R)-18F-OF-NB1 (Fig. 5). Pretreatment with a σ1R antagonist reduced the uptake of (S)-18F-OF-NB1 across all brain regions. A low dose (0.027 mg/kg) and a high dose (0.125 mg/kg) of FTC-146 reduced the specific binding of (S)-18F-OF-NB1 by 48% and 49%, respectively. Double blocking with sequential administration of both Co 101244 (0.25 mg/kg) and high-dose FTC-146 (0.125 mg/kg) in 1 monkey at 25 and 15 min before the radiotracer injection, respectively, did not further reduce brain uptake of (S)-18F-OF-NB1 when compared with blocking with Co 101244 alone (82% vs. 81%).

(Top) Representative PET SUV images (summed from 45 to 60 min) and time–activity curves in selected brain regions from baseline, GluN1/2B blockade (0.25 mg/kg dose of Co 101244), σ1R blockade (0.125 mg/kg dose of FTC-146), and sequential GluN1/2B/σ1R blockade (0.25 mg/kg dose of Co 101244 + 0.125 mg/kg dose FTC-146) scans of (S)-18F-OF-NB1 obtained in the same rhesus monkey (monkey 2). (Bottom) PET SUV images and time–activity curves from baseline and GluN1/2B blockade (0.25 mg/kg dose of Co 101244) scans of (R)-18F-OF-NB1 (monkey 1).

Lassen occupancy plots of (R)- and (S)-18F-OF-NB1 after pretreatment with either GluN1/2B ligand Co 101244 (0.25 mg/kg) only or with addition of σ1R ligand FTC-146 (0.125 mg/kg). Each point represents unique brain region.
The time–activity curves were well fitted with both the 1-tissue-compartment model and the multilinear analysis 1 method for reliable VT estimates, with a relative SE of less than 5% (Supplemental Tables 1 and 2). On average, VT values were similar for (rac)- and (R)-18F-OF-NB1 and were lower for (S)- 8F-OF-NB1. Regional VT values ranged from 17 to 39 mL/cm3 for (S)-18F-OF-NB1 in the selected brain regions (Table 1). From the GluN1/2B blocking studies, VND was found to be 9.3 mL/cm3 for (S)-18F-OF-NB1 in monkey 1 and 7.3 mL/cm3 in monkey 2. From these VND values, regional BPND values were calculated and ranged from 1.2 (semiovale) to 3.4 (cingulate cortex) for (S)-18F-OF-NB1 (Table 1).
One-Tissue-Compartment Model VT and BPND Values in Selected Brain Regions Under Baseline and Blockade Conditions
DISCUSSION
Given the involvement of GluN1/2B receptors in various neuropathologies, great effort has been taken to develop therapeutic and PET imaging agents, and several radioligands are currently in development. (R)-11C-Me-NB1 was recently evaluated in healthy volunteers and shown to have excellent brain uptake and kinetics (17). These results warrant the development of a radiofluorinated PET tracer that would allow wider use. We previously synthesized and tested several ligands, and (rac)-18F-OF-NB1 emerged as one of the most promising candidates for clinical translation, given its excellent properties in rodents (19). In the current work, we evaluated the performance characteristics of (rac)-18F-OF-NB1 and its enantiopure (S)- and (R)-18F-OF-NB1 with respect to their brain uptake, kinetics, and metabolism, to select the most promising candidate for clinical translation. To our knowledge, there have been no reports of clinical trials on the evaluation of a radiofluorinated probe for PET imaging of GluN1/2B in humans.
The radiosynthesis of 18F-OF-NB1 proceeded in 2 steps: copper-catalyzed radiofluorination of the boronic ester precursor, followed by simultaneous cleavage of the 2 hydroxyl protecting groups (19). Despite starting with enantiopure precursors, we could not obtain the respective enantiopure (S)- and (R)-18F-OF-NB1 without racemization during the second deprotecting step. As such, the radiosynthetic strategy was adapted to include a chiral HPLC purification step to provide enantiopure radiotracers at the end of the synthesis (Supplemental Fig. 2). This allowed the production of both enantiomers from a single radiosynthetic run. Metabolite analysis showed a higher parent fraction in the plasma for (R)-18F-OF-NB1 than for the (S)-enantiomer. This phenomenon can be explained by the fact that different enantiomers can bind to enzymes with different affinities and therefore exhibit different metabolic rates (31). Further, all forms of 18F-OF-NB1 presented good fp values (0.12–0.16), offering the opportunity for high-precision measurement and subsequently improving the accuracy of quantitative PET measurements and calculation of VT/fp, a potentially valuable outcome measure in clinical trials.
The brain uptake and distribution of (rac)-18F-OF-NB1 corroborated results in rodents (19). Kinetic analysis, however, is not suited for racemic mixtures unless both enantiomers exhibit identical plasma clearance. The 2 enantiopure radiotracers displayed brain distribution and kinetics that are in line with the results in rodents. (R)- and (S)-18F-OF-NB1 showed high uptake in GluN1/2B-rich brain regions, with the highest uptake in the cingulate cortex. The binding of both enantiopure tracers was displaceable across all brain regions, indicating the lack of a reference region for kinetic modeling analysis. Regional time–activity curves of all 3 tracers, on the other hand, were well fitted by the 1-tissue-compartment model. The 2-tissue-compartment model also produced reasonable model fits, but the SE of VT values was high and nonphysiologic values were generated in several gray matter regions. The multilinear analysis 1 method also provided good fits and VT values comparable to those derived from the 1-tissue-compartment model (32). Regional VT estimates were higher for (R)-18F-OF-NB1 than for the (S)-enantiomer. When plotting VT values of (R)- and (S)-18F-OF-NB1 against each other to generate the Guo plot (33), the slope of this curve indicated 4- to 5-fold higher in vivo affinity for (R)-18F-OF-NB1 than for (S)-18F-OF-NB1 (Fig. 6).

Guo plots (33) comparing regional VT values of (S)-18F-OF-NB1 (y-axis) and (R)-18F-OF-NB1 (x-axis) in same monkey (left) and in 2 different monkeys (right). y-intercept is > 0, signifying higher BPND values of tracer on x-axis than on y-axis. Slope indicates relative in vivo binding affinity of 2 tracers, where (R)-18F-OF-NB1 exhibits 4- to 5-fold higher binding affinity than (S)-enantiomer.
To investigate the in vivo binding specificity of both enantiomers to GluN1/2B, blocking experiments with the GluN1/2B antagonist Co 101244 (0.25 mg/kg) were performed and a Lassen plot analysis for each enantiomer was undertaken. Both enantiomers showed high specific binding, with an estimated occupancy of 96% (n = 1) across the brain by Co 101244 (0.25 mg/kg) when measured with (R)-18F-OF-NB1, and 85% (n = 2) with (S)-18F-OF-NB1. Furthermore, both enantiomers exhibited good regional BPND values in the range of 2–3, highlighting their clinical translation potential in terms of specific binding (Table 1). The high affinity of (R)-18F-OF-NB1 came at the expense of a less desirable pharmacokinetic profile due to its extremely slow washout from the brain, which precluded its further development, since a very long scan might be required in humans. Pretreatment with a low dose (0.027 mg/kg) or a high dose (0.125 mg/kg) of the σ1R antagonist FTC-146 led to partial blocking, with a 49% and 48% reduction in the specific binding of (S)-18F-OF-NB1, respectively, as calculated from the Lassen plots, and no clear dose–blockade level relationship. To address this potential off-target binding component of (S)-18F-OF-NB1, a sequential blockade experiment was conducted in which Co 101244 (0.25 mg/kg) was injected first, followed by the σ1R antagonist FTC-146 (0.125 mg/kg). Blocking with a 0.25 mg/kg dose of Co 101244 alone resulted in 81% occupancy, and virtually no change in occupancy was observed when FTC-146 was administered afterward. The absence of a further reduction in binding after treatment with FTC-146 suggests that (S)-18F-OF-NB1 binds specifically to the GluN1/2B receptors and is selective over σ1Rs. The partial blockade of (S)-18F-OF-NB1 binding by FTC-146 when administered alone can be attributed to σ1R-NMDAR cross-talk stemming from the inherent chaperone nature of σ1Rs and their ability to regulate the NMDARs (34,35). Another plausible explanation might be off-target binding of FTC-146, as the σ1R bears a particular binding site that is structurally similar to the ifenprodil-binding site of the GluN1/2B receptor, and thus σ1R ligands are known for their off-target binding at the GluN1/2B. For example, we have reported potential off-target binding by the σ1R ligand fluspidine (19). Nonetheless, further studies are warranted to clarify the nature of potential off-target binding by these novel benzazepine-based radioligands.
CONCLUSION
We successfully synthesized and evaluated (rac)-, (S)-, and (R)-18F-OF-NB1 in rhesus monkeys. The high GluN1/2B-specific binding of (S)-18F-OF-NB1 (gray matter BPND values in the range of 1.6–3.4) and its attractive pharmacokinetic profile render it a promising candidate for clinical translation. Clinical PET imaging of GluN1/2B could expedite the development of GluN1/2B therapeutics through target engagement and occupancy studies. Furthermore, PET imaging of diseases in which the GluN2B subunit–containing NMDARs are implicated could potentially improve the diagnosis and treatment monitoring of these diseases.
DISCLOSURE
This study was funded by the National Institute of Health (grant U01MH107803) and also was partly supported by the Swiss National Science Foundation (grants 310030E-160403/1 and 310030E-182872/1). Hazem Ahmed, Simon Ametamey, and Ahmed Haider hold shares in Nemosia A.G. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can we selectively image GluN2B subunit–containing NMDARs in nonhuman primates and identify a suitable radiofluorinated PET probe for clinical translation?
PERTINENT FINDINGS: (S)-18F-OF-NB1 emerged as an attractive candidate for clinical translation, as it displayed high specific binding to GluN1/2B receptors and a favorable pharmacokinetic profile.
IMPLICATIONS FOR PATIENT CARE: A GluN1/2B PET tracer suitable for use in humans will facilitate the investigation and diagnosis of neuropsychiatric diseases, as well as the development of therapeutics targeting GluN2B-containing NMDARs.
ACKNOWLEDGMENTS
We thank the staff at the Yale PET Center for their expert technical support.
Footnotes
Published online Jun. 16, 2022.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 7, 2022.
- Revision received April 7, 2022.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.