


Abstract
We recently introduced the potent gastrin-releasing peptide receptor (GRPR) antagonist 68Ga-SB3 (68Ga-DOTA-p-aminomethylaniline-diglycolic acid-DPhe-Gln-Trp-Ala-Val-Gly-His-Leu-NHEt), showing excellent tumor localizing efficacy in animal models and in patients. By replacement of the C-terminal Leu13-Met14-NH2 dipeptide of SB3 by Sta13-Leu14-NH2, the novel GRPR antagonist NeoBOMB1 was generated and labeled with different radiometals for theranostic use. We herein report on the biologic profile of resulting 67/68Ga-, 111In-, and 177Lu-NeoBOMB1 radioligands in GRPR-expressing cells and mouse models. The first evidence of prostate cancer lesion visualization in men using 68Ga-NeoBOMB1 and PET/CT is also presented. Methods: NeoBOMB1 was radiolabeled with 67/68Ga, 111In, and 177Lu according to published protocols. The respective metalated species natGa-, natIn-, and natLu-NeoBOMB1 were also synthesized and used in competition binding experiments against [125I-Tyr4]BBN in GRPR-positive PC-3 cell membranes. Internalization of 67Ga-, 111In-, and 177Lu-NeoBOMB1 radioligands was studied in PC-3 cells at 37°C, and their metabolic stability in peripheral mouse blood was determined by high-performance liquid chromatography analysis of blood samples. Biodistribution was performed by injecting a 67Ga-, 111In-, or 177Lu-NeoBOMB1 bolus (74, 74, or 370 kBq, respectively, 100 μL, 10 pmol total peptide ± 40 nmol Tyr4-BBN: for in vivo GRPR blockade) in severe combined immunodeficiency mice bearing PC-3 xenografts. PET/CT images with 68Ga-NeoBOMB1 were acquired in prostate cancer patients. Results: NeoBOMB1 and natGa-, natIn-, and natLu-NeoBOMB1 bound to GRPR with high affinity (half maximal inhibitory concentration, 1–2 nM). 67Ga-, 111In-, and 177Lu-NeoBOMB1 specifically and strongly bound on the cell membrane of PC-3 cells displaying low internalization, as expected for receptor antagonists. They showed excellent metabolic stability in peripheral mouse blood (>95% intact at 5 min after injection). After injection in mice, all 3 (67Ga-, 111In-, and 177Lu-NeoBOMB1) showed comparably high and GRPR-specific uptake in the PC-3 xenografts (e.g., 30.6 ± 3.9, 28.6 ± 6.0, and >35 percentage injected dose per gram at 4 h after injection, respectively), clearing from background predominantly via the kidneys. During a translational study in prostate cancer patients, 68Ga-NeoBOMB1 rapidly localized in pathologic lesions, achieving high-contrast imaging. Conclusion: The GRPR antagonist radioligands 67Ga-, 111In-, and 177Lu-NeoBOMB1, independent of the radiometal applied, have shown comparable behavior in prostate cancer models, in favor of future theranostic use in GRPR-positive cancer patients. Such translational prospects were further supported by the successful visualization of prostate cancer lesions in men using 68Ga-NeoBOMB1 and PET/CT.
- molecular tumor imaging
- PET
- SPECT
- radionuclide therapy
- theranostic applications
- prostate cancer
- gastrin releasing peptide receptor antagonist
The high-density expression of gastrin-releasing peptide receptors (GRPRs) in frequently occurring human cancers, such as prostate cancer and mammary carcinoma, compared with their lack of expression in surrounding healthy tissue provides valid opportunities for molecular imaging and internal radiotherapy with peptide radioligands (1–6). The first-generation radiopeptides developed for such purposes were GRPR agonists derived from C-terminal fragments of the amphibian tetradecapeptide bombesin (BBN) (7–12) or the respective human 27mer peptide GRP (13–15). The use of resulting radioligands in humans was soon linked to undesirable effects after agonist-induced GRPR activation (16). This finding limited the translational prospects of radioagonists despite their ability to internalize in cancer cells, a property considered at first essential for good diagnostic sensitivity and therapeutic efficacy.
This notion has drastically changed with the advent of the second-generation GRPR radiotracers, which were instead based on GRPR antagonists (17,18). Several studies have shown that noninternalizing radioantagonists can successfully target and be sufficiently retained in GRPR-expressing cancer lesions while rapidly clearing from physiologic organs in both animal models and humans. The superior pharmacokinetics of GRPR radioantagonists over their agonist counterparts in combination with their higher inherent biosafety has shifted current research toward the design of new improved GRPR radioantagonist candidates with clear potential for clinical translation (19–23).
Following this rationale, we have initially developed 99mTc-demobesin 1 (99mTc-DB1), a GRPR antagonist radioligand for SPECT imaging, labeled with 99mTc and containing the [DPhe6,Leu-NHEt13,des-Met14]BBN(6-14) peptide fragment (17,18). To allow labeling with other clinically relevant radiometals, we then replaced the acyclic tetraamine chelator of DB1 by the universal chelator DOTA. The resulting peptide-conjugate SB3 was labeled with 68Ga and evaluated as a PET tracer first in animals and eventually in patients with prostate and breast cancer (23). Interestingly, the respective 111In- and 177Lu-labeled radioligands, in addition to showing less affinity for GRPR, turned out to be less stable in the biologic milieu. As a result, GRPR targeting of xenografts in animal models in the case of 111In- and 177Lu-SB3 was found inferior to 68Ga-SB3, restricting the theranostic application options of SB3 in the clinic (24).
Truncation of C-terminal Met14-NH2 and alkylation of the Leu13NH2 amide is only one of many C-terminus modifications in the BBN(6-14) motif, shown to convey antagonistic properties to resulting receptor ligands. For example, replacement of the C-terminal Leu13-Met14-NH2 dipeptide by Sta13-Leu14-NH2 and coupling of DOTA has recently fostered the development of promising GRPR radioantagonists (20,21,25). We have shown though that such a GRPR radioantagonist, [68Ga/177Lu]JMV4168, can reach its full theranostic potential only after injection of phosphoramidon (22). The latter, by in situ inhibition of the catabolic action of neutral endopeptidase, increased the stability of [68Ga/177Lu]JMV4168 in peripheral blood, leading to notably enhanced tumor uptake. This study has highlighted the importance of in vivo stability for the success of GRPR-supported tumor targeting.
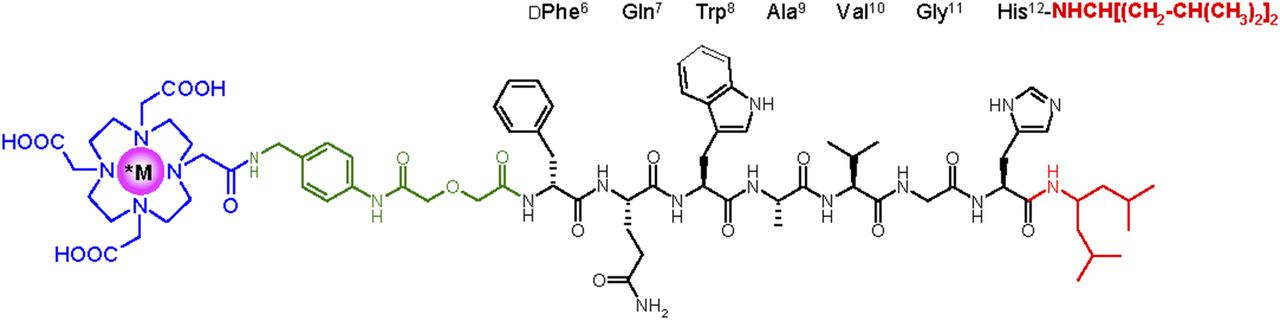
In the quest of radioligands with higher stability, our attention was attracted by the improved stability, and potency, reported for the well-characterized GRPR antagonist Ac-His-Trp-Ala-Val-Gly-His-NH-CH[CH2-CH(CH3)2]2 (26). By replacing the C-terminal -His12-Leu13NHEt of SB3 by -His12-NH-CH[CH2-CH(CH3)2]2, we produced NeoBOMB1, which we labeled with 68Ga (for PET), 111In (for SPECT), and 177Lu (for radionuclide therapy). The effect of radiometal on the biologic properties of forming radioligands (Fig. 1), especially on GRPR affinity and in vivo stability, was thoroughly investigated in cell and animal models to assess the theranostic prospects of NeoBOMB1. First translational evidence of the 68Ga-NeoBOMB1 efficacy as a PET tracer was acquired in prostate cancer patients using PET/CT.
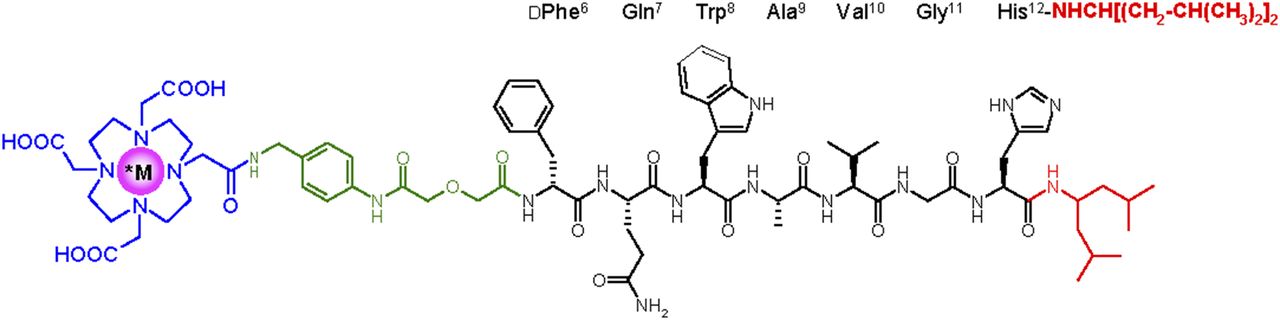
Chemical structure of *M-NeoBOMB1, *M = 67/68Ga, 111In, and 177Lu.
MATERIALS AND METHODS
Ligands and Radioligands
NeoBOMB1 was provided free by NCSR “Demokritos,” and Tyr4-BBN was purchased from PSL GmbH. Radiolabeling of NeoBOMB1 with 67/68Ga, 111In, and 177Lu and subsequent quality control were performed as detailed in the supplemental material (available at http://jnm.snmjournals.org). Metalation of NeoBOMB1 with natGa, natIn, and natLu used in in vitro assays is also described therein, along with radioiodination of Tyr4-BBN with 125I.
In Vitro Assays
Human androgen-independent prostate adenocarcinoma PC-3 cells expressing the GRPR (LGC Promochem) cultured as previously reported were used in biologic assays (10,27). Competition binding experiments were conducted by incubation of the [125I-Tyr4]BBN radioligand (50 pM, ∼40,000 cpm per assay tube) with increasing concentrations of either test peptide (NeoBOMB1, natGa-NeoBOMB1, natIn-NeoBOMB1, or natLu-NeoBOMB1) or reference (Tyr4-BBN) in PC-3 cell membrane homogenates at 22°C for 1 h, as previously described (21,23). Filter samples were measured for radioactivity in an automatic well-type γ-counter (NaI(Tl) crystal, Canberra Packard Auto-γ-5000 series instrument). Half maximal inhibitory concentration (IC50) values were determined by nonlinear regression according to a 1-site model applying the PRISM 2 program (GraphPad Software) and represent mean ± SD from 3 independent experiments performed in triplicate.
For cell-binding assays, confluent PC-3 cells were seeded in 6-well plates (∼1.2 × 106 cells per well) 24 h before the experiment. Test peptide (67Ga-NeoBOMB1, 111In-NeoBOMB1, or 177Lu-NeoBOMB1 [250 fmol total peptide in 150 μL of 0.5% bovine serum albumin/phosphate-buffered saline]) was added alone (total) or in the presence of 1 μM Tyr4-BBN (nonspecific), and the experiment was performed following a published protocol (21,23). Results were calculated as percentage specific and nonspecific internalized plus membrane-bound of total added activity applying the Microsoft Excel program and represent the mean ± SD of 4 experiments performed in triplicate.
Radioligand Stability and Biodistribution in PC-3 Xenograft–Bearing Mice
For metabolic studies in mice, each radioligand (67Ga-NeoBOMB1, 111In-NeoBOMB1, or 177Lu-NeoBOMB1) was injected as a 100-μL bolus (3 nmol NeoBOMB1, associated with 11.1–22.2 MBq of 67Ga or 111In and with 111 MBq of 177Lu) in the tail vein of healthy male Swiss albino mice (30 ± 5 g; NCSR “Demokritos” Animal House Facility). Mice were euthanized and blood (0.5–1 mL) collected at 5 min and, additionally for 67Ga-NeoBOMB1 and 177Lu-NeoBOMB1, at 30 min after injection was analyzed by high-performance liquid chromatography, as previously described (21,23). The Symmetry Shield RP18 (5 μm, 3.9 × 20 mm) column was eluted at a flow rate of 1.0 mL/min with the following gradient: 100% A to 90% A in 10 min and from 90% A to 60% for the next 60 min; A = 0.1% aqueous trifluoroacetic acid (v/v) and B = MeCN.
A bolus (150 μL) containing a suspension of about 1.2 × 107 freshly harvested human PC-3 cells in saline was subcutaneously injected in the flanks of female severe combined immunodeficiency mice (15 ± 3 g, 6 wk of age on arrival day; NCSR “Demokritos” Animal House Facility), and 3–4 wk later the mice developed well-palpable tumors at the inoculation site (80–150 mg). On the day of the experiment, a [67Ga/111In/177Lu]NeoBOMB1 bolus (10 pmol NeoBOMB1, associated with up to 74 kBq of 67Ga or 111In, or up to 370 kBq of 177Lu, in 100 μL of vehicle: saline/EtOH 9/1 v/v) was injected in the tail vein of mice. Biodistribution for the 1-, 4-, 24-, and 72-h (for 177Lu only) postinjection time intervals was conducted as previously described (21,23); for in vivo GRPR blockade, animals of an additional 4-h group were coinjected with Tyr4-BBN (40 nmol). Biodistribution data were calculated as percentage injected dose per gram of tissue (%ID/g) using the Microsoft Excel program and with the aid of suitable standards of the injected dose and represent mean ± SD, n = 4. Statistical analysis was conducted using the unpaired 2-tailed Student t test; P values of less than 0.05 were considered statistically significant.
All animal experiments were performed in compliance with European and national regulations and after approval of protocols by national authorities.
Patients: Ethical and Regulatory Issues
Four patients with prostate adenocarcinoma who were referred by their urologists underwent 68Ga-NeoBOMB1 PET/CT at the Theranostics Research Center, Zentralklinik Bad Berka, Germany. The study protocol was approved by the Institutional Review Board, and all subjects signed a substantive written informed consent form. All patients were studied before by conventional imaging modalities (e.g., MRI, 18F-choline PET, and 99mTc-methylene diphosphonate bone scanning). The decision to perform 68Ga-NeoBOMB1 PET/CT was based on the opinion of the referring urologists as the best choice for the patients’ respective clinical conditions. 68Ga-NeoBOMB1 was administered in compliance with the German Medicinal Products Act (section 13, subsection 2b), the 1964 Declaration of Helsinki, and the responsible regulatory body (Government of Thuringia) and under the compassionate-use clause of the German Medicinal Products Act (Federal Institute for Drugs and Medical Devices, “Compassionate use” programs, http://www.bfarm.de/EN/Drugs/licensing/clinicalTrials/compUse/_node). This pilot study was performed in accordance with the regulations of the German Federal Agency for Radiation Protection.
68Ga-NeoBOMB1 PET/CT Imaging
A dual-modality PET/CT tomograph (Biograph Duo; Siemens Medical Solutions) was used in the study. Patients were prepared and injected with 60–339 (median, 199.5, adapting the injected activity to the patient body weight) MBq of 68Ga-NeoBOMB1 before data acquisition, as detailed in the supplemental materials. Information on the applied imaging protocol, data acquisition, and reconstruction methods and image analysis are also reported therein.
RESULTS
In Vitro Studies
As shown in Figure 2A, NeoBOMB1 was able to displace [125I-Tyr4]BBN from GRPR sites in PC-3 membranes in a monophasic and dose-dependent manner. The calculated IC50 value of 1.49 ± 0.07 nM was found comparable to that of Tyr4-BBN reference (IC50 = 1.33 ± 0.09 nM). Interestingly, incorporation of gallium, indium, or lutetium by the DOTA chelator did not negatively affect receptor affinity, with the corresponding IC50 values of 1.17 ± 0.06 nM found for natGa-NeoBOMB1, 1.28 ± 0.14 nM for natIn-NeoBOMB1, and 1.38 ± 0.09 nM for natLu-NeoBOMB1. Likewise, high and specific binding was observed for 67Ga-NeoBOMB1 (47.8% ± 0.5%), 111In-NeoBOMB1 (43.5% ± 0.4%), and 177Lu-NeoBOMB1 (44.2% ± 0.5%) in PC-3 cells after 1-h incubation at 37°C (nonspecific values < 3%) (Fig. 2B). In all cases, most radioactivity was associated with the cell membrane, with a smaller portion (≈10%) found within the cells, as expected for radioligands based on GRPR antagonists.
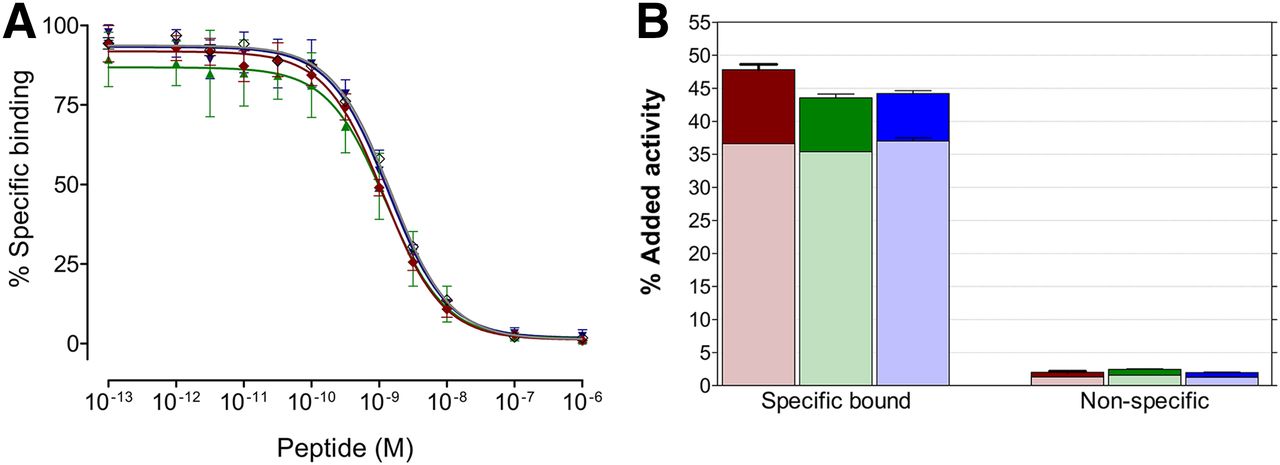
(A) Displacement of [125I-Tyr4]BBN from GRPR sites in PC-3 cell membranes by increasing concentrations of NeoBOMB1 (gray curve) (IC50 = 1.49 ± 0.07 nM); natGa-NeoBOMB1 (red curve) (IC50 = 1.17 ± 0.06 nM); natIn-NeoBOMB1 (green curve) (IC50 = 1.28 ± 0.14 nM); natLu-NeoBOMB1 (blue curve) (IC50 = 1.38 ± 0.09 nM); and reference, Tyr4-BBN (curve not shown) (IC50 = 1.33 ± 0.09 nM). Results represent mean IC50 values ± SD, n = 3. (B) Internalized plus cell membrane–bound fractions in PC-3 cells shown for  , 67Ga-NeoBOMB1;
, 67Ga-NeoBOMB1;  , 111In-NeoBOMB1; and
, 111In-NeoBOMB1; and  , 177Lu-NeoBOMB1, respectively (1-h incubation at 37°C); nonspecific values were determined in presence of 1 μM BBN. Results represent average IC50 values ± SD, n = 4.
, 177Lu-NeoBOMB1, respectively (1-h incubation at 37°C); nonspecific values were determined in presence of 1 μM BBN. Results represent average IC50 values ± SD, n = 4.
Radioligand In Vivo Stability: Biodistribution in PC-3 Tumor–Bearing Mice
As shown by the representative radiochromatograms in Figures 3A, 3B, and 3C, respectively, 67Ga-NeoBOMB1, 111In-NeoBOMB1, and 177Lu-NeoBOMB1 remained more than 95% intact in peripheral mouse blood at 5 min after injection. Furthermore, 67Ga-NeoBOMB1 and 177Lu-NeoBOMB1 were found more than 90% and 80% intact, respectively, in mouse circulation when further tested at 30 min after injection (Supplemental Fig. 3).
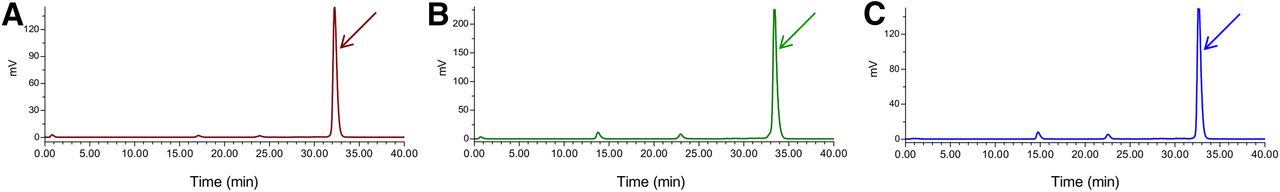
Representative radiochromatograms of blood samples collected 5 min after injection of 67Ga-NeoBOMB1 (A), 111In-NeoBOMB1 (B), and 177Lu-NeoBOMB1 (C) in mice, showing more than 95% of radioligand detected intact; retention time of injected analog was determined by coinjection of radiopeptide in high-performance liquid chromatography column and is indicated by arrow.
As shown in Figures 4A, 4B, and 4C, respectively, 67Ga-NeoBOMB1, 111In-NeoBOMB1, and 177Lu-NeoBOMB1 efficiently localized in the PC-3 xenografts in severe combined immunodeficiency mice (30.7 ± 3.9 %ID/g, 28.6 ± 6.0 %ID/g, and 42.4 ± 5.0 %ID/g, respectively, at 4 h after injection). High uptake was observed also in mouse pancreas, a GRPR-rich organ. Coinjection with excess Tyr4-BBN, a receptor agonist, was found to be partially effective to confirm specificity of uptake in the tumor and GRPR-expressing organs, as previously reported for GRPR antagonist–based radioligands (17,23). On the other hand, coinjection with excess NeoBOMB1 was more than 90% effective to block GRPR sites in vivo (28). All analogs showed a fast blood clearance and were excreted predominantly via the kidneys; 67Ga-NeoBOMB1 showed somewhat elevated blood values at the initial time intervals as compared with its 111In and 177Lu counterparts. There was high tumor retention for 67Ga-NeoBOMB1 (serving as a 68Ga-NeoBOMB1 surrogate) between 1 and 4 h after injection and for 111In-NeoBOMB1 between 4 and 24 h after injection, both matching the respective radionuclide half-life. On the other hand, the uptake of 177Lu-NeoBOMB1 in the PC-3 xenografts, although declining between 4 and 24 h, still remained at impressively high levels at 24 h after injection (>25 %ID/g at 24 h after injection). At later time points, tumor uptake declined further (≈5 %ID/g at 72 h after injection) but surpassed that in all other organs, including the GRPR-positive pancreas (≈2 %ID/g at 72 h after injection).

Biodistribution data of 67Ga-NeoBOMB1 ( 1 and
1 and  4 h after injection) (A), 111In-NeoBOMB1 (
4 h after injection) (A), 111In-NeoBOMB1 ( 4 and
4 and  24 h after injection) (B), and 177Lu-NeoBOMB1 (
24 h after injection) (B), and 177Lu-NeoBOMB1 ( 4,
4,  24, and
24, and  72 h after injection) (C) in PC-3 tumor–bearing severe combined immunodeficiency mice expressed as %ID/g and representing mean ± SD, n = 4;
72 h after injection) (C) in PC-3 tumor–bearing severe combined immunodeficiency mice expressed as %ID/g and representing mean ± SD, n = 4;  in vivo GRPR blockade at 4 h after injection. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = PC-3 xenograft.
in vivo GRPR blockade at 4 h after injection. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = PC-3 xenograft.
PET/CT Results of 68Ga-NeoBOMB1 in Prostate Cancer Patients
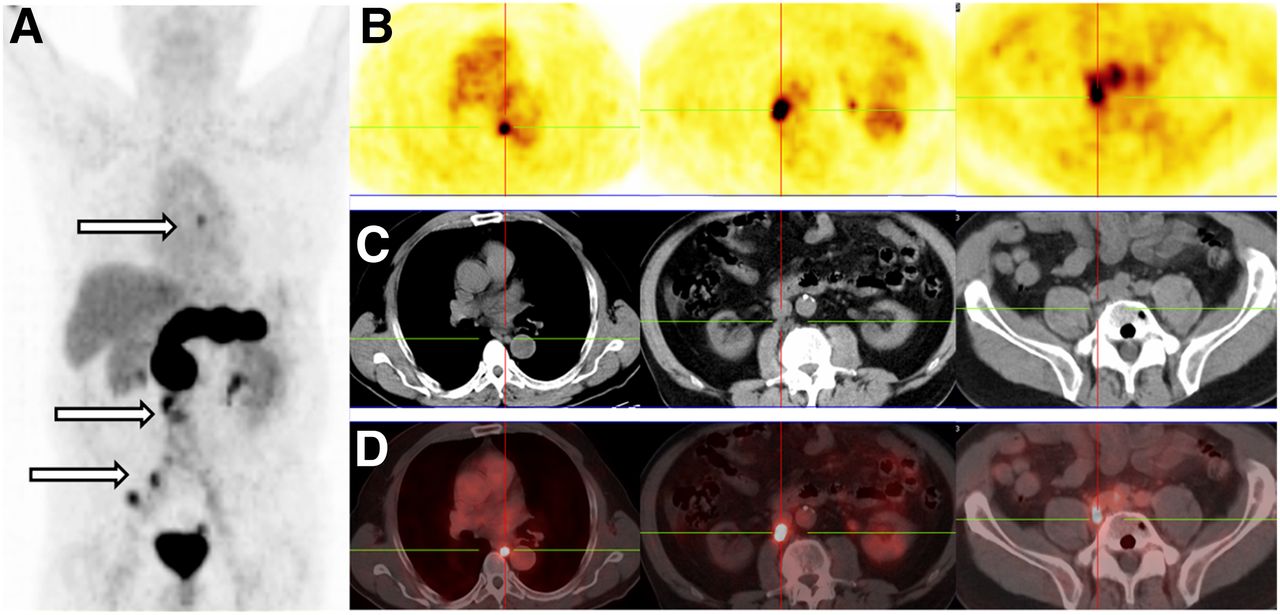
Four prostate cancer patients from an ongoing proof-of-principle study who underwent 68Ga-NeoBOMB1 PET/CT and with a complete dataset are presented herein first. The tracer was well tolerated by all subjects, and no side effect was registered from the administration of 68Ga-NeoBOMB1. A representative 68Ga-NeoBOMB1 PET/CT scan of a 69-y-old patient with primary bilateral prostate adenocarcinoma (initially classified cT1 [G3] cNx cMx; Gleason score, 8 [4 + 4]) and a prostate-specific antigen (PSA) of 6.33 ng/mL is presented in Figure 5, showing strong tracer uptake in the right and middle prostate lobes suggestive of multifocal prostate carcinoma. In Figure 6, multiple mediastinal, paraesophageal, abdominal, and pelvic lymph node metastases are strongly positive during 68Ga-NeoBOMB1 PET/CT in a 65-y-old patient. The patient was diagnosed 9 y before with prostate adenocarcinoma (classification pT3b [G3] pN1c M0; Gleason score, 7) and subsequently underwent radical prostatovesiculectomy with pelvic lymphadenectomy, intensity-modulated radiotherapy, and androgen-deprivation therapy. Because of an increasing PSA, restaging with 68Ga-NeoBOMB1 PET/CT was indicated (PSA at 21.77 ng/mL on the date of scan). Two additional representative 68Ga-NeoBOMB1 PET/CT scans are included and are further commented in Supplemental Figures 4 and 5.

Detection of primary tumor (arrow and crossbars) in a 69-y-old patient with prostate adenocarcinoma (bilateral) (Gleason score, 8 [4 + 4] and PSA on day of scan, 6.33 ng/m). 68Ga-NeoBOMB1 PET/CT fusion coronal (A), PET transverse (B), corresponding CT transverse (C), and fusion PET/CT (D) images are shown. Tracer uptake in right and middle prostate lobes is suggestive of multifocal prostate carcinoma.
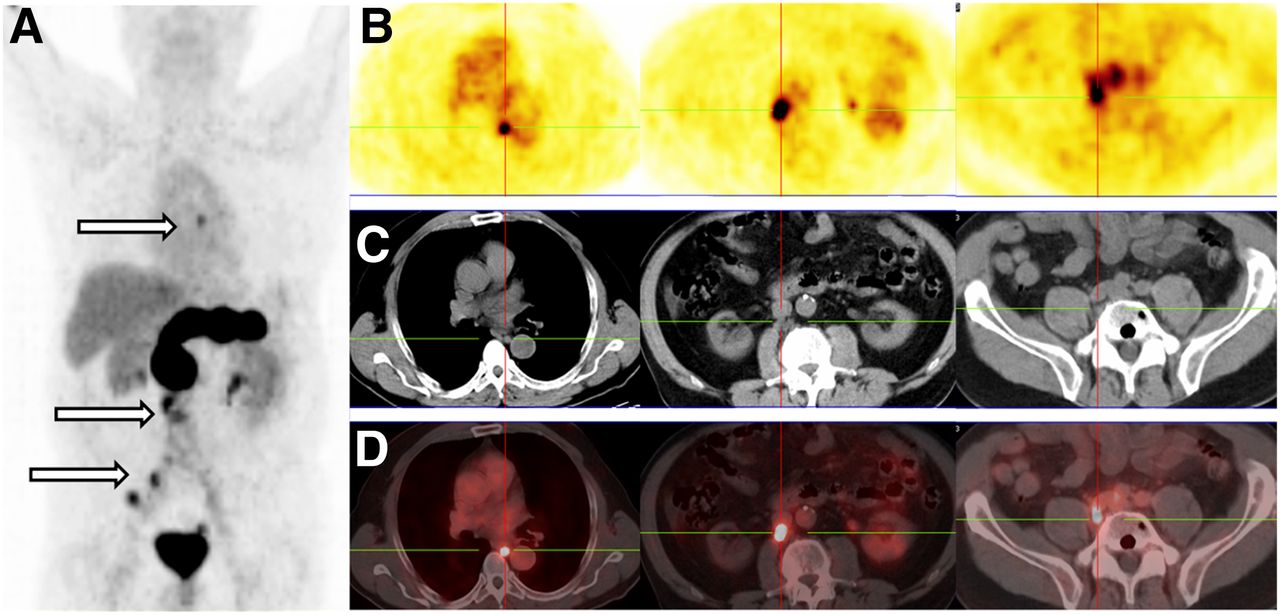
Patient with prostate adenocarcinoma, postradical prostatovesiculectomy with pelvic lymphadenectomy, intensity-modulated radiotherapy, and androgen-deprivation therapy (PSA on day of scan, 21.77 ng/mL). 68Ga-NeoBOMB1 PET maximum-intensity projection (A), serial PET transverse (B), corresponding CT transverse (C), and fusion PET/CT (D) images are shown. Multiple mediastinal, abdominal, paraesophageal, and pelvic lymph node metastases are indicated by arrows and crossbars.
DISCUSSION
In recent years, radiolabeled analogs of the frog tetradecapeptide BBN and its C-terminal BBN(6-14) fragment have been studied at length as potential candidates in the diagnosis and treatment of GRPR-positive human cancer, especially prostate and breast cancer (7,8). In principle, this approach can be effective in targeting both primary and metastatic disease as long as a high density of GRPR expression in malignant lesions is preserved (1–4). According to the latest preclinical and clinical studies, radiolabeled GRPR antagonists often outperform their agonist counterparts (17,19,23). Furthermore, GRPR antagonists are deemed safer for human use than agonists. The latter induce GRPR activation after intravenous administration in humans, leading to strong adverse effects (16).
Previous experience in the management of neuroendocrine tumors with radiolabeled somatostatin analogs has highlighted the unparalleled strength of nuclear medicine to provide a theranostic approach in cancer (29–31). Ideally, a theranostic pair, represented by a peptide-conjugate that can be labeled with either a diagnostic (111In/67Ga for SPECT, or 68Ga for PET) or a therapeutic radionuclide (177Lu), is used for disease diagnosis, localization, staging, patient stratification, dosimetry, radiotherapy, and also therapeutic response monitoring (32). Both diagnostic and therapeutic radiopeptide should effectively target the GRPR-expressing lesions in vivo and, therefore, need to display similarly high receptor affinity and in vivo stability.
Following this rationale, we have recently introduced 68Ga-SB3 for PET imaging of prostate and breast cancer, effectively localizing in GRPR-positive lesions in mouse models and in patients (23). However, the respective therapeutic 177Lu-SB3 showed a receptor affinity inferior to and was less stable in vivo than its diagnostic counterpart 68Ga-SB3. Consequently, lower tumor uptake was achieved in mice, a handicap that could be reversed by in situ neutral endopeptidase inhibition with phosphoramidon (24). However, this promising approach, recently shown to improve the theranostic potential of other GRPR radioantagonists as well, has not yet been translated in humans (22).
In the present work, we introduce [68Ga/111In/177Lu]NeoBOMB1 based on a previously well-characterized GRPR antagonist (26) and showing excellent qualities for PET or SPECT imaging as well as radionuclide therapy. First, NeoBOMB1 displayed single-digit nanomolar affinity for the human GRPR, even after incorporation of any of gallium, indium, or lutetium (Fig. 2A), thus appearing more suitable for theranostic applications than SB3. In line with affinity findings, the respective 67Ga-NeoBOMB1, 111In-NeoBOMB1, and 177Lu-NeoBOMB1 radioligands specifically bound to the cell membrane of PC-3 cells with comparably high efficacy and, as expected for receptor radioantagonists, showed minimal internalization efficacy. Of great interest is the fact, that all 3 radioligands were found more than 95% intact in peripheral mouse blood at 5 min after injection (Fig. 3) but also when tested at later time points (67Ga-NeoBOMB1 > 90% intact and 177Lu-NeoBOMB1 80% intact at 30 min after injection; Supplemental Fig. 3), highlighting again the promising theranostic prospects of NeoBOMB1 as compared with SB3.
Taken together, these properties of receptor affinity, cell-binding efficacy, and metabolic stability seem to have led to excellent uptake of 67Ga-NeoBOMB1, 111In-NeoBOMB1, and 177Lu-NeoBOMB1 in the PC-3 tumors xenografted in mice (Fig. 4), further supporting the theranostic prospects of NeoBOMB1. Minor differences in biodistribution profiles can be attributed to differences in coordination chemistries with DOTA across radiometals, which, despite identical in vitro properties, slightly affected pharmacokinetic parameters in our mouse models. The high pancreatic uptake of the radioligands, although a result of high receptor affinity and in vivo stability, is rather undesirable for radiotherapy with 177Lu-NeoBOMB1. Nevertheless, it seems to decline more rapidly than the tumor uptake in our mouse model. By administering different NeoBOMB1 peptide doses in mice, it has been possible to impressively improve tumor-to-pancreas ratios and overall pharmacokinetics (28). On the other hand, interspecies differences of biologic profiles of GRPR-directed radioligands between mice and men have been reported (33,34). Therefore, translational studies are warranted to establish the diagnostic (and also therapeutic) value of new GRPR radioantagonists.
For this purpose, 68Ga-NeoBOMB1 has been tested in a first-in-man study in 4 prostate cancer patients applying PET/CT. First, the excellent tolerability of NeoBOMB1 in men is a valuable asset for future GRPR-targeted radionuclide therapy options. Furthermore, the new tracer strongly localized in primary prostate-confined disease but also in multimetastatic foci during disease spread, as seen in the representative scans in Figures 5 and 6, respectively. Impressively, 68Ga-NeoBOMB1 uptake was evident in a liver micrometastasis not visualized on the arterial phase of CT but later confirmed on follow-up angiography (Supplemental Fig. 4). 68Ga-NeoBOMB1 was able to detect lymph node metastases down to 5 mm in size, normally considered negative on conventional imaging, overall exhibiting a high diagnostic sensitivity. Of particular interest is the localization ability of 68Ga-NeoBOMB1 in osseous metastases (Supplemental Fig. 5), a finding requiring further investigation in a bigger cohort of patients. Previous reports have demonstrated the high upregulation of GRPR in well-differentiated prostate carcinoma, with the expression density declining in more advanced stages of the disease (1–3). Indeed, excellent outcome in therapy-naïve patients has recently been established with 68Ga-SB3 and PET/CT (35,36). However, existing experience with 68Ga-SB3 (23) and the BBN-based 99mTc-DB4 tracer (37) indicated good tracer localization in several, but not all, bone metastases of prostate cancer.
On the other hand, prostate-specific membrane antigen represents an attractive target for small-size inhibitors in patients with advanced and less-differentiated disease, but less in early stages of prostate cancer (38). The clinical implications of prostate-specific membrane antigen and GRPR expression at different disease stages are not fully understood thus far, and further clinical studies are warranted. In a recent direct clinical comparison of a prostate-specific membrane antigen– and GRPR-directed 68Ga-labeled probe in 7 prostate cancer patients, the authors suggested that personalizing biomarker assessments are to be conceived in the future for patients with prostate cancer (39).
CONCLUSION
Theranostic approaches for GRPR-expressing human tumors, such as prostate and breast cancer, are becoming realistic with the advent of new-generation radiopeptides based on GRPR antagonists. In addition to higher biosafety, the new GRPR radioantagonists most often exhibit potent targeting of tumor lesions and attractive pharmacokinetics in animal models and patients. We herein introduce NeoBOMB1 and the [67/68Ga/111In/177Lu]NeoBOMB1 radiopeptides, showing a remarkably preserved GRPR affinity and cell binding, high in vivo stability, and an ability to target prostate cancer xenografts in mice independent of radiometal used. These features highlight the theranostic prospects of NeoBOMB1. First-in-man results with 68Ga-NeoBOMB1 PET/CT in prostate cancer patients revealed the translational strength of the new tracer. Ongoing preclinical and clinical studies aim to investigate the true theranostic value of NeoBOMB1 in nuclear oncology.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Aug. 4, 2016.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication May 28, 2016.
- Accepted for publication July 13, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Additive Value of [68Ga]Ga-RM26 PET/CT to [68Ga]Ga-PSMA-617 PET/CT in Detecting Pelvic Lymph Node Metastasis in Prostate Cancer: A Prospective, Single-Center, Phase II Study
- Preclinical Investigation of [212Pb]Pb-DOTAM-GRPR1 for Peptide Receptor Radionuclide Therapy in a Prostate Tumor Model
- Prospective Comparison of 68Ga-NeoB and 68Ga-PSMA-R2 PET/MRI in Patients with Biochemically Recurrent Prostate Cancer
- Comparison of 68Ga-PSMA-617 PET/CT and 68Ga-RM2 PET/CT in Patients with Localized Prostate Cancer Who Are Candidates for Radical Prostatectomy: A Prospective, Single-Arm, Single-Center, Phase II Study
- A Radiotracer for Molecular Imaging and Therapy of Gastrin-Releasing Peptide Receptor-Positive Prostate Cancer
- GRPr Antagonist 68Ga-SB3 PET/CT Imaging of Primary Prostate Cancer in Therapy-Naive Patients
- MITIGATE-NeoBOMB1, a Phase I/IIa Study to Evaluate Safety, Pharmacokinetics, and Preliminary Imaging of 68Ga-NeoBOMB1, a Gastrin-Releasing Peptide Receptor Antagonist, in GIST Patients
- Imaging the Distribution of Gastrin-Releasing Peptide Receptors in Cancer
- Primary Preclinical and Clinical Evaluation of 68Ga-DOTA-TMVP1 as a Novel VEGFR-3 PET Imaging Radiotracer in Gynecological Cancer
- Development and Characterization of a Novel, High-Affinity, Specific, Radiolabeled Ligand for BRS-3 Receptors
- PET Using a GRPR Antagonist 68Ga-RM26 in Healthy Volunteers and Prostate Cancer Patients
- Prospective Evaluation of 68Ga-RM2 PET/MRI in Patients with Biochemical Recurrence of Prostate Cancer and Negative Findings on Conventional Imaging
- Will GRPR Compete with PSMA as a Target in Prostate Cancer?
- Dual-Integrin {alpha}v{beta}3- and Gastrin-Releasing Peptide Receptor-Targeting PET Radiotracer (68Ga-BBN-RGD)