


Abstract
We analyzed and compared 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-[methyl-11C]thymine (11C-FMAU), 3′-deoxy-3′-[18F]fluorothymidine (18F-FLT) and 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5-[76Br]bromouracil (76Br-BFU) with respect to tissue uptake, DNA incorporation, and excretion modulation in rats. The goal of the investigation was to evaluate the efficiency of the 3 nucleoside tracers as potential tracers for measuring proliferation. Methods: Sprague-Dawley rats were divided into 3 groups and administered 5 MBq 11C-FMAU, 1 MBq 18F-FLT, or 2 MBq 76Br-BFU. For each tracer, a subgroup was also administered 6 mg/kg cimetidine. The rats in the 11C-FMAU group were killed at 5, 20, 40, 60, and 80 min after injection; the rats in the 18F-FLT group were killed at 80 min and 2 and 4 h; and the rats in the 76Br-BFU group were killed at 5, 20, 40, and 80 min and 2, 4, 6, and 24 h. Samples of blood, liver, kidney, spleen, and intestine were taken, and the radioactivity was measured. DNA separation was made in the samples of spleen, and the radioactivity in the DNA fraction was measured. Results: Maximal uptake of radioactivity was seen in the spleen and intestine, organs with active DNA synthesis. The highest relative radioactivity uptake was at 60 min in the 11C-FMAU groups and at 4 h in the 18F-FLT group. In the 76Br-BFU group, the uptake increased gradually during the observation period, and uptake of radioactivity increased markedly in rats receiving cimetidine. Cimetidine did not affect radioactivity uptake in the 11C-FMAU or 18F-FLT groups. The fraction of radioactivity in DNA was 78% in spleen at 60 min in the 11C-FMAU group, 80% at 60 min and 97% at 4 h in the 76Br-BFU group. The DNA-incorporation was only 2% in the 18F-FLT group. Conclusion: 76Br-BFU predominantly incorporates into DNA and has great potential as a PET tracer for the assessment of proliferation in vivo. 11C-FMAU also may have potential as a proliferation marker, but the observation time is limited. 18F-FLT does not incorporate into DNA and is therefore not a direct marker of proliferation.
PET is a powerful method for in vivo characterization of tumor biochemistry and has been applied extensively in clinical practice for diagnosing and grading malignancies. Most studies have been conducted using 18F-FDG and 11C-methionine (1–5). The uptake of these tracers in tumor tissue has been shown to correlate with malignancy grades within the same tumor type, but the mechanism of uptake is only indirectly related to cell division, which reduces their applicability for proliferation assessment. In addition, 18F-FDG is not a highly selective tracer for tumor imaging, because many types of nontumor cells (e.g., granulocytes) show high tracer uptake. Inflammatory tissue might be difficult to separate from tumor tissue (6).
PET studies with 11C-thymidine have suggested that thymidine can be used for tumor imaging, because it is incorporated into DNA and provides a measure of cell proliferation (7–9). The short half-life of 11C, the rapid metabolism of thymidine, and the relatively slow incorporation into DNA make it, however, less suitable as a proliferation marker (10–12). Many researchers have investigated the potential use of radiolabeled nucleosides or their analogues. The goal of these studies was to develop radionuclide imaging agents or agents to be used for radiation treatment. Thus, attempts have been made with 124I-iododeoxyuridine (124I-IUdR), 125I-IUdR, 77Br-bromodeoxyuridine (77Br-BrdU), 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-[methyl-11C]thymine (11C-FMAU), and 3′-deoxy-3′-[18F]fluorothymidine (18F-FLT), some of which have been suggested as potential PET tracers to characterize malignant tumors. The studies indicated that it is possible to determine cell proliferation potential in vivo using these labeled nucleosides or their analogues (13–18).
76Br is a positron-emitting radionuclide with a half-life of 16 h and is suitable for PET. 76Br-BrdU, an analog of thymidine, was investigated by several groups (19–23). Although 76Br-BrdU is incorporated into DNA, the results did not support it as a valid radiotracer for clinical practice. A large fraction of 76Br-bromide is rapidly generated through metabolism of the tracer, and this 76Br-bromide remains in the extracellular space and disturbs the contrast between proliferating and nonproliferating tissues.
1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5-[76Br]bro-mouracil (76Br-BFU) has a slight modification with respect to 76Br-BrdU, substituting a hydrogen with a fluorine atom in the 2′ position of the sugar moiety, which makes it considerably more stable to degradation. Preclinical evaluations of 76Br-BFU have suggested that it should be a valid tracer for proliferation (24). With the same fluorine modification to the original thymidine molecule, we synthesized 11C-FMAU, with 11C-labeling in the methyl group of thymine. A range of trials is ongoing with 18F-FLT, an 18F-labeled nucleoside analog that is supposedly not incorporated into DNA but taken up and phosphorylated and thereby trapped in tissue (17). The uptake of 18F-FLT is very high in proliferating tissues and gives excellent images of several malignancy types.
In the present study, comparisons of 11C-FMAU, 18F-FLT, and 76Br-BFU with respect to tissue uptake, DNA incorporation, and excretion modulation in healthy rats and tumor-bearing mice were performed. The aim of the study was to evaluate the efficiency of these compounds as potential tracers for measuring proliferation. Because cimetidine has previously shown a marked effect on elimination of nucleosides, this mode was included in the comparisons.
MATERIALS AND METHODS
Animals
A total of 200 male Sprague-Dawley rats weighing 350–450 g were used in the studies. A tumor model was made with 30 male C3H mice weighing 32–42 g. NFSA, a spontaneous fibrosarcoma syngeneic to C3Hf/Kam mice, was transplanted subcutaneously into the right back of each mouse by injecting tumor pieces generated according to the method previously described by Milas et al. (25). Two weeks after the transplantation, mice with tumors of about 1 cm in diameter were studied.
The animals were housed under standard laboratory conditions at 20°C and 50% humidity under a 12-h light/dark cycle. They were given free access to laboratory animal food and water before and during the experiments. The studies with research animals were approved by the Animal Ethics Committee of Uppsala University.
Radiotracers
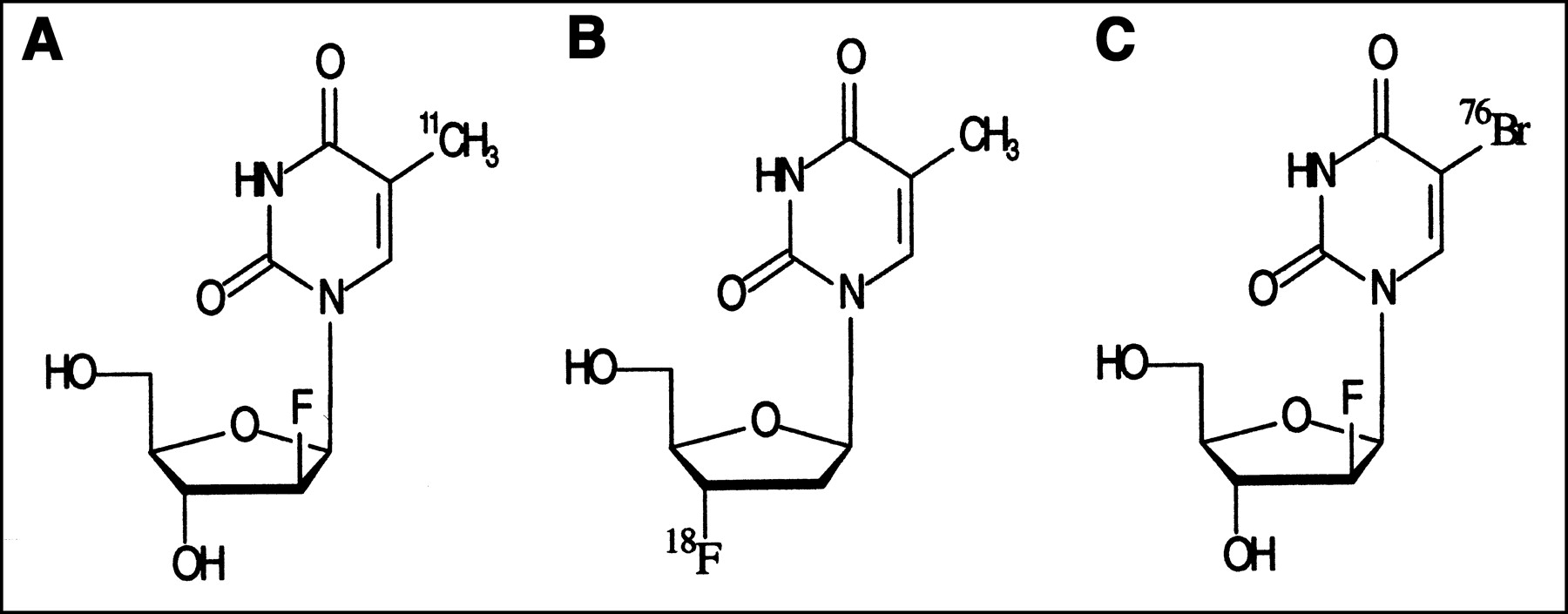
The structures of the tracers used are shown in Figure 1.

Structures of 11C-FMAU, 18F-FLT, and 76Br-BFU.
11C-FMAU.
11C-carbon dioxide was produced by the Scanditronix MC-17 cyclotron at the Uppsala University PET Center (UUPC), with the 14N(p,α)11C reaction using a gas target containing nitrogen (Nitrogen 6.0; AGA, Stockholm, Sweden) and 0.05% oxygen (Oxygen 6.0; AGA) bombarded with 17 MeV protons. 11C-methyl iodide was synthesized according to the published procedure (26) and was used in a Stille cross-coupling reaction (27) with the same trimethylstannyl nucleoside used for the synthesis of 76Br-BFU. 11C-methyl-iodide was trapped in a vial containing 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5-(trimethylstannyl)uracil (0.5 mg), tris(dibenzylidenacetone)dipalladium (0.9 mg), and tri(o-tolyl)-phosphine (1.2 mg) in DMF (300 μL) at room temperature. The mixture was heated at 130°C for 5 min and then diluted with 1,200 μL distilled water before injection into a semipreparative LC column (Ultrasphere ODS C18 5 μm, 10 × 250 mm; Beckman Coulter, Inc., Fullerton, CA). The mobile phase used was 17 mmol/L acetic acid + 1 mmol/L ascorbic acid and ethanol at a flow of 7.0 mL/min. A gradient of 5%–10% ethanol during 5 min was used. The fraction eluting at 8.5 min was collected and pH was adjusted to 4.5. Analytic LC was used to determine the identity and radiochemical purity of the product (Ultrasphere ODS C18 5-μm column, 4.6 × 250 mm; Beckman Coulter, Inc.) using water and ethanol as the mobile phase at a flow of 2.0 mL/min using a gradient from 5% to 8% ethanol during 8 min. The retention time of the product was 6.3 min, and the radiochemical purity was shown to be >95%. The product coeluted with the authentic reference compound. The synthesis was completed at 35 min after the end of bombardment, and the decay-corrected radiochemical yield was 10% of the original 11C-methyl iodide. Typically, 400–800 MBq 11C-FMAU was obtained from 20–30 GBq of 11C-methyl iodide.
18F-FLT.
18F-FLT was synthesized according to the method of Grierson and Shields (28). 18F-fluoride was produced using the MC-17 cyclotron (Scanditronix, Uppsala, Sweden) at the UUPC, using the 18O(p,n)18F nuclear reaction by irradiation of a water target containing 18O-enriched water. The target water with 18F-fluoride was added to a solution of potassium carbonate (0.7 mg, 5.0 μmol) and Kryptofix 2.2.2 (13 mg, 35 μmol). The water was removed by azeotropic distillation with acetonitrile (4 × 1 mL) under helium flow at 90°C.
1-(2-deoxy-3-O-(4-nitrobenzenesulfonyl)-5-O-(4,4′-dime-thoxy-trytyl)-β-d-threo-penta-furanosyl)-3-(2,4-dimethoxybenzyl) thymine (5.1 mg, 5.8 μmol) in 600 μL acetonitrile was added to the same vessel. The reaction mixture was heated at 100°C for 10 min, then passed through neutral alumina Sep-Pak (Waters, Milford, MA). Solvent was removed under helium flow at 100°C.Ceric ammonium nitrate (16 mg, 30 μmol) in 4:1:1 acetonitrile/ethanol/water (240 μL) was added and heated at 100°C for 3 min. The mixture was quenched with 4% aqueous NaHCO3 and passed through a microfilter (0.2 μm) and a neutral alumina Sep-Pak. The product was purified by preparative high-performance liquid chromatography (HPLC) on an Ultrasphere C18 column (10 × 250 mm; Beckman Coulter, Inc.). Ten percent ethanol was used as the mobile phase at a flow rate of 4 mL/min. The product 18F-FLT eluted at about 12 min. Radiochemical purity and identity were assessed using HPLC and an analytic Discovery C18 (4.6 × 250 mm; SUPELCO, St Louis, MO) with 7% acetonitrile/93% water as mobile phase and a flow rate of 1 mL/min.
76Br-BFU.
76Br-Br− was produced at the Scanditronix MC-17 cyclotron at the UUPC using the 76Se(p,n)76Br− nuclear reaction by irradiation of a 76Se-Se-enriched (96.5% enrichment) Cu2Se target. The 76Br-Br− was separated from the Cu2Se pellet using a thermal diffusion procedure (29). The precursor 5-trimetylstannyl-2′-fluoro-2′-deoxyuridine was synthesized from 5-iodo-2′-fluoro-2′-deoxyuridine according to a published procedure (30,31). The 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5 76Br-bromouracil 76Br-BFU) was prepared from the precursor 5-trimetylstannyl-2′-fluoro-2′-deoxyuridine by an electrophilic substitution reaction using chloramine-T as an oxidizing agent. The product was isolated in about 80% radiochemical yield and >99% radiochemical purity within 45 min from the start of synthesis (24).
Other Chemicals
14C-thymidine was bought from Dupont (Stockholm, Sweden). Cimetidine (Acinil; A/S GEA, Fredriksburg, Denmark) was purchased from the Uppsala University hospital pharmacy and hydroxyurea was purchased from Sigma (St. Louis, MO). DNAzol reagent was obtained from Life Technologies Co. (Grand Island, NY).
Experiments with 11C-FMAU
Radioactivity Distribution.
Twelve groups of rats, with 4–6 rats in each group, were included in the experiments. In 5 untreated groups, the animals received injections of 5 MBq 11C-FMAU in the tail vein. In 5 treated groups, the animals were administered 6 mg/kg cimetidine mixed with 11C-FMAU. The animals were killed by CO2 inhalation at 5, 20, 40, 60, or 80 min after radioactivity administration, and each time point included 1 cimetidine-treated group and 1 untreated group. In the remaining 2 treated groups, each animal was given 200 mg hydroxyurea intravenously 30 min before radioactivity administration, and the rats were killed at 20 or 60 min after tracer administration. Samples of liver, kidney, spleen, intestine, and blood were rapidly removed, and radioactivity in the samples was measured in a γ-counter (built in-house). The weight of the samples was recorded. Standardized uptake values (SUVs) were calculated as radioactivity concentration in tissue divided by the ratio of total administered radioactivity and the animal’s body weight. Ratios of the SUVs of the organs and blood were calculated as further standardization.
DNA Separation.
Twenty rats were divided into 3 cimetidine-treated and 3 untreated groups. Each rat received 100 MBq 11C-FMAU. The same dose of 11C-FMAU and cimetidine at 6 mg/kg were coinjected into the rats in the treated groups. The rats were killed at 20, 40, or 60 min after the injections, and each time point included 1 untreated group and 1 treated group, with 3–5 rats at each time point. Samples of 200 mg spleen and 200 mg intestine were taken and 2.2 mL DNAzol (DNA isolation reagent) were added to each sample. The tissues then were homogenized using a polytron homogenizer. The homogenate was centrifuged at 2,500 rpm for 6 min to remove fragments of tissues that were not homogenized, and 1 mL supernatant was taken to measure the radioactivity, representative for the concentration of radioactivity in the tissue. The radioactivity was measured in a γ-counter. Then 0.5 mL 99.5% ethanol were added to the supernatant to precipitate the DNA fraction. After DNA precipitation, with centrifugation at 15,000 rpm at 4°C for 10 min, the supernatant was removed and the pellet representing DNA was washed once more with 0.5 mL 99.5% ethanol. The radioactivity in DNA was measured, and the DNA-incorporated radioactivity was expressed as percentage of total radioactivity in the tissue.
Experiments with 18F-FLT
Nine groups of rats were included in the experiments. Each group consisted of 5–7 rats. Three groups received only 18F-FLT in the tail vein. Three groups were cimetidine treated and received 1 MBq 18F-FLT and 6 mg/kg cimetidine at the same time. In the remaining 3 groups, each rat was injected with 200 mg hydroxyurea 30 min before the 18F-FLT injection. The animals from each of the different groups were sacrificed at 80 min or 2 or 4 h after the radioactivity administration. Organ radioactivity was measured, and the tissue radioactivity concentration was calculated in the same way as in the 11C-FMAU experiments.
DNA separation was performed on 8 rats, 4 treated and 4 untreated, killed 2 h after radioactivity injection. In the untreated group, the rats received 10 MBq 18F-FLT. The cimetidine-treated rats received 6 mg/kg cimetidine, coinjected with the radioactivity. The procedure of DNA separation and presentation of fraction of DNA-incorporated radioactivity was conducted as in the 11C-FMAU experiments.
Experiments with 76Br-BFU
In the 76Br-BFU experiments, rats were divided into 14 groups, 5 untreated and 9 cimetidine treated, with 4–6 rats in each group. 76Br-BFU (5 MBq) was administered to each animal, and these rats were sacrificed at 5, 20, 40, 60, or 80 min. The same dose of 5 MBq 76Br-BFU plus 6 mg/kg cimetidine was injected into each rat in the treated groups, and these rats were killed at 5, 20, 40, 60, or 80 min and 2, 4, 6 or 24 h after tracer administration. The radioactivity measurements and calculations of radioactivity concentration in the organs were made as described previously for the 11C-FMAU experiments.
Samples of spleen and intestine were taken from the untreated groups at 20-, 40-, and 60-min time points and from the treated groups at 20, 40, and 60 min and 2, 4, 6, and 24 h and analyzed with respect to the DNA fractions. The measurements for the fraction of radioactivity incorporated into DNA and calculations for presentation of fraction of radioactivity in the DNA were performed as in the 11C-FMAU experiments.
Urinalysis
Ten groups of rats, 5 cimetidine-treated and 5 untreated groups with 2–4 rats in each group, were included in the experiments. Rats in 2 untreated groups received either 5 MBq 11C-FMAU or 5 MBq 76Br-BFU. The remaining group of untreated rats received 1 MBq 18F-FLT. In the treated groups, each of these tracers was coinjected with 6 mg/kg cimetidine. The rats were placed separately in individual plastic boxes with tissue paper in the bottom for urine collection. In the 11C-FMAU and 76Br-BFU groups, the rats were killed by CO2 inhalation at 5 or 80 min after radioactivity administration. The rats in the 18F-FLT groups were killed at 80 min after radioactivity administration. All urine samples from the tissue paper and the rats’ bladders were collected for measurement of total radioactivity in urine. The radioactivity of each sample was calculated as a percentage of the total radioactivity injected, and comparisons were made between treated and untreated groups at the same time points.
Experiments in Tumor Model
Twenty-four tumor-bearing mice were used in the tumor model experiments. They were divided into 2 groups, an untreated and a cimetidine-treated group. All mice were injected intraperitoneally with a combination of 0.5 MBq 11C-FMAU and 37 KBq 14C-thymidine. In the treated group, 10 mg/kg cimetidine were mixed and coinjected with the radioactivity. The mice were killed by cervical dislocation at 20, 40, 60, or 80 min after the injection, including 3 mice from each of the 2 groups and from each time point. Samples of heart and tumor were taken, and 11C radioactivity was measured in a γ-counter. 14C radioactivity was measured 3 d later in a scintillation counter. SUVs were calculated as previously described.
DNA extraction was performed on specimens of tumor from 6 mice. These mice were injected with 5 MBq 11C-FMAU and 37 KBq 14C-thymidine. In 3 of these mice, 10 mg/kg cimetidine were mixed with the radioactivity before the injection. All mice were sacrificed at 60 min after the radioactivity administration. 11C and 14C radioactivities in the DNA were measured separately. The DNA-incorporated radioactivity was calculated as a percentage of the total sample radioactivity.
Data Analysis
All results were analyzed by ANOVA using the StatView 4.0 system (Abacus Concepts, Inc., Berkeley, CA). The data are presented as mean ± SD.
RESULTS
Experiments with 11C-FMAU
Radioactivity Distribution.
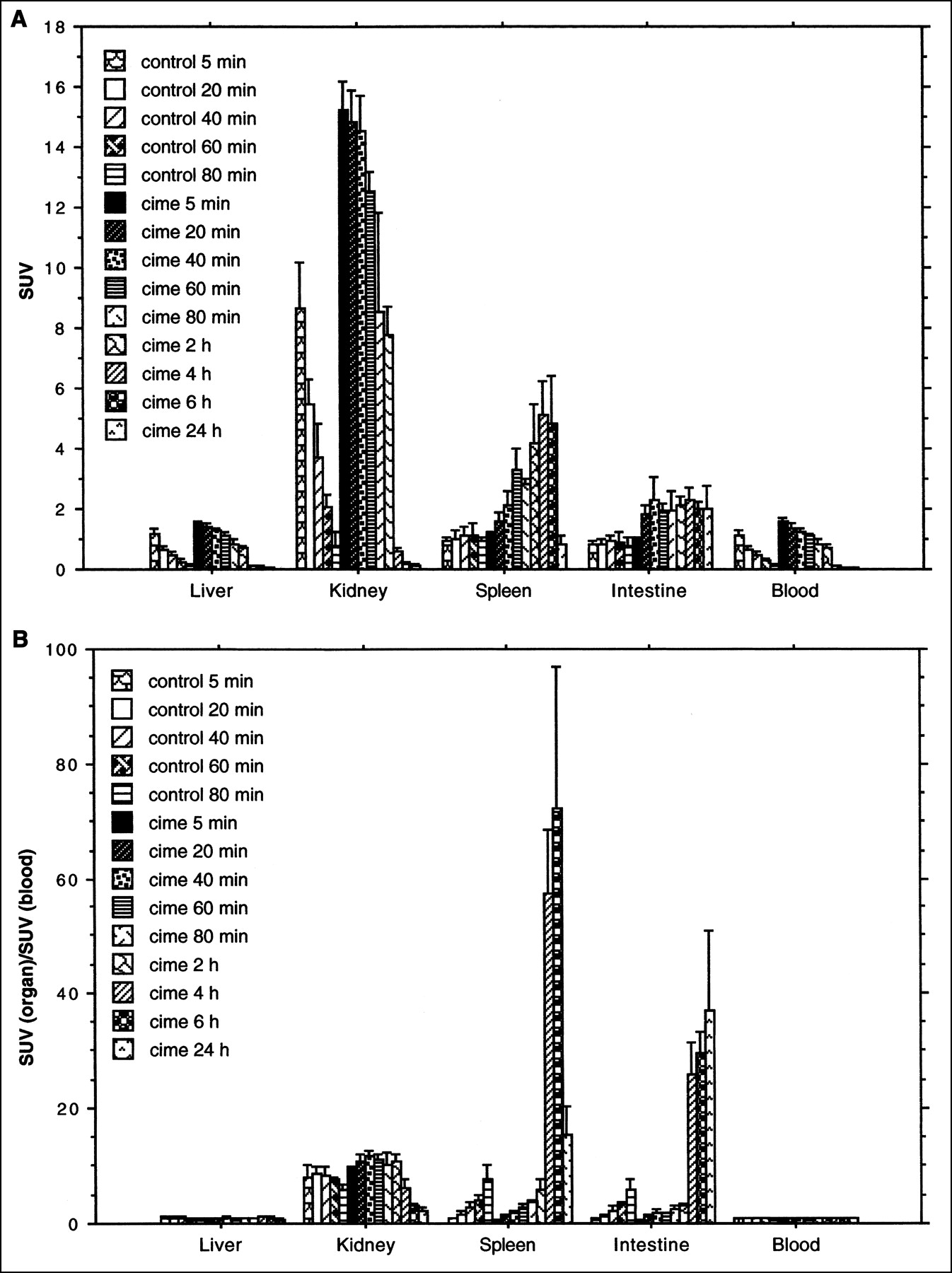
The 11C-FMAU uptake values in the spleen and intestine increased with time after tracer administration in both untreated and cimetidine-treated groups. Comparing the uptake values between the treated and untreated groups at the same time points, a relatively higher uptake was found in the treated group. In the spleen, the highest uptake was observed at the 60-min time point in the untreated group (SUV = 3.6, SD = 1.4) and at the 80-min time point in the treated group (SUV = 5.0, SD = 1.2). In the intestine, the highest uptake was found at the 80-min time point in both groups (SUV = 2.0, SD = 0.4, in the untreated group; SUV = 2.9, SD = 0.1, in the treated group). The uptake values in the liver, kidney, and blood decreased with time in the untreated but not in the treated group (Fig. 2A). A clear temporal pattern also was demonstrated when the radioactivity uptake in the spleen and intestine were represented as a ratio to blood radioactivity (Fig. 2B). In the rats treated with hydroxyurea, the radioactivity at the 20-min time point was decreased about 55% in the spleen and 34% in the intestine compared with untreated rats; at the 60-min time point, the radioactivity was 71% lower in the spleen and 47% lower in the intestine.
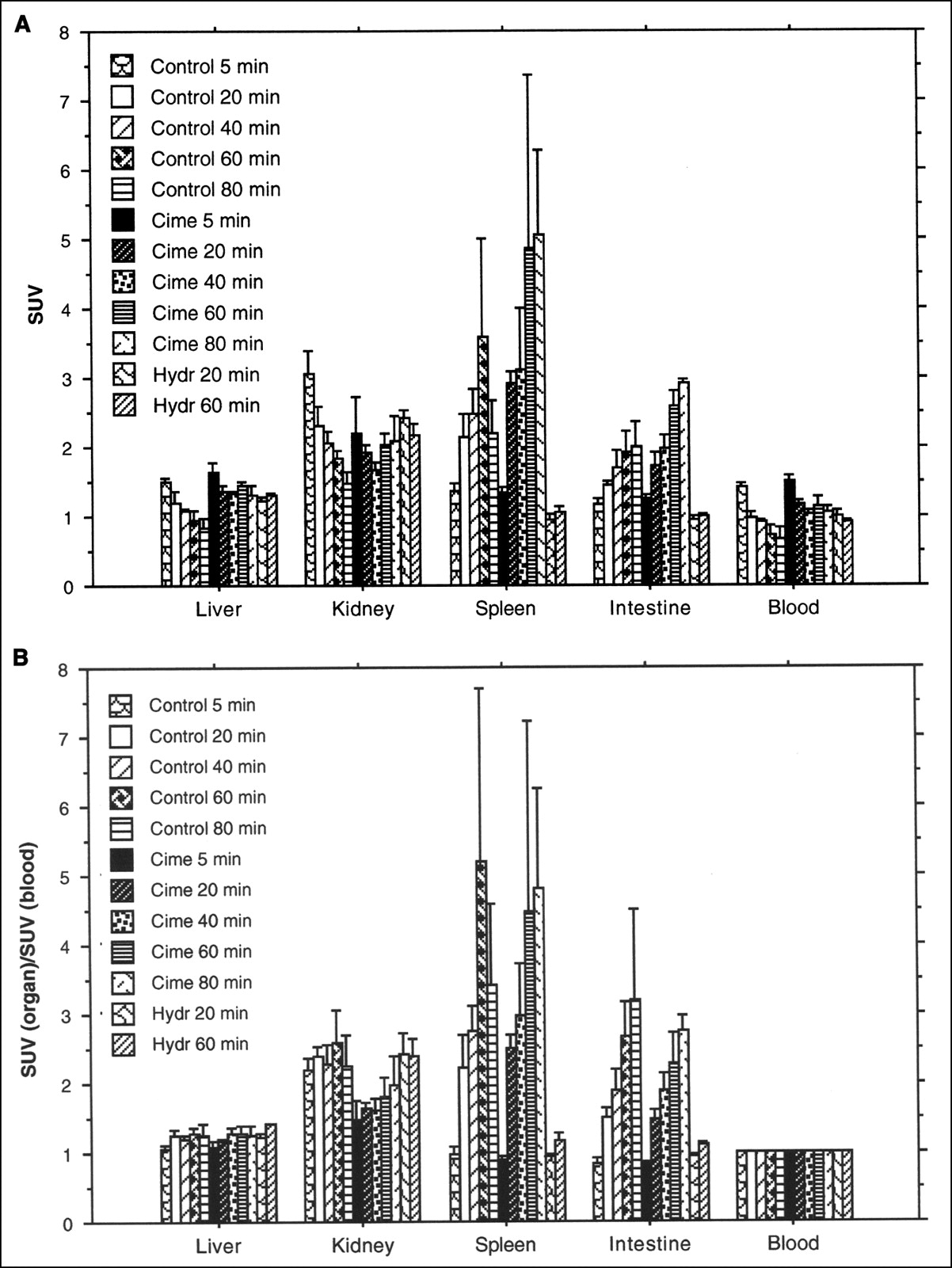
(A) Uptake of 11C-FMAU, expressed as SUV (mean ± SD) in rat organs. Animals were killed after 5, 20, 40, 60, or 80 min. Different animals were administered cimetidine together with 11C-FMAU or hydroxyurea 30 min before administration of radioactivity. (B) Uptake of 11C-FMAU, expressed as ratio to blood radioactivity concentration (mean ± SD) in rat organs. Animals were killed after 5, 20, 40, 60, or 80 min. Different animals were administered cimetidine with 11C-FMAU or hydroxyurea 30 min before administration of radioactivity.
DNA Separation.
Analyses of the radioactivity fraction incorporated into DNA showed a tendency toward increased radioactivity in both spleen and intestine with extended observation time. In both tissues, the highest value of radioactivity in DNA was seen at the 60-min time point. In the spleen, the radioactivity fractions were 78.3% (SD = 2.1) in the untreated group and 70.4% (SD = 8.8) in the treated group. In the intestine, the radioactivity fractions were 56.9% (SD = 8.2) in the untreated group and 56.7% (SD = 2.9) in the cimetidine-treated group (Fig. 3). At the same time points there were no significant differences between the treated and untreated groups.
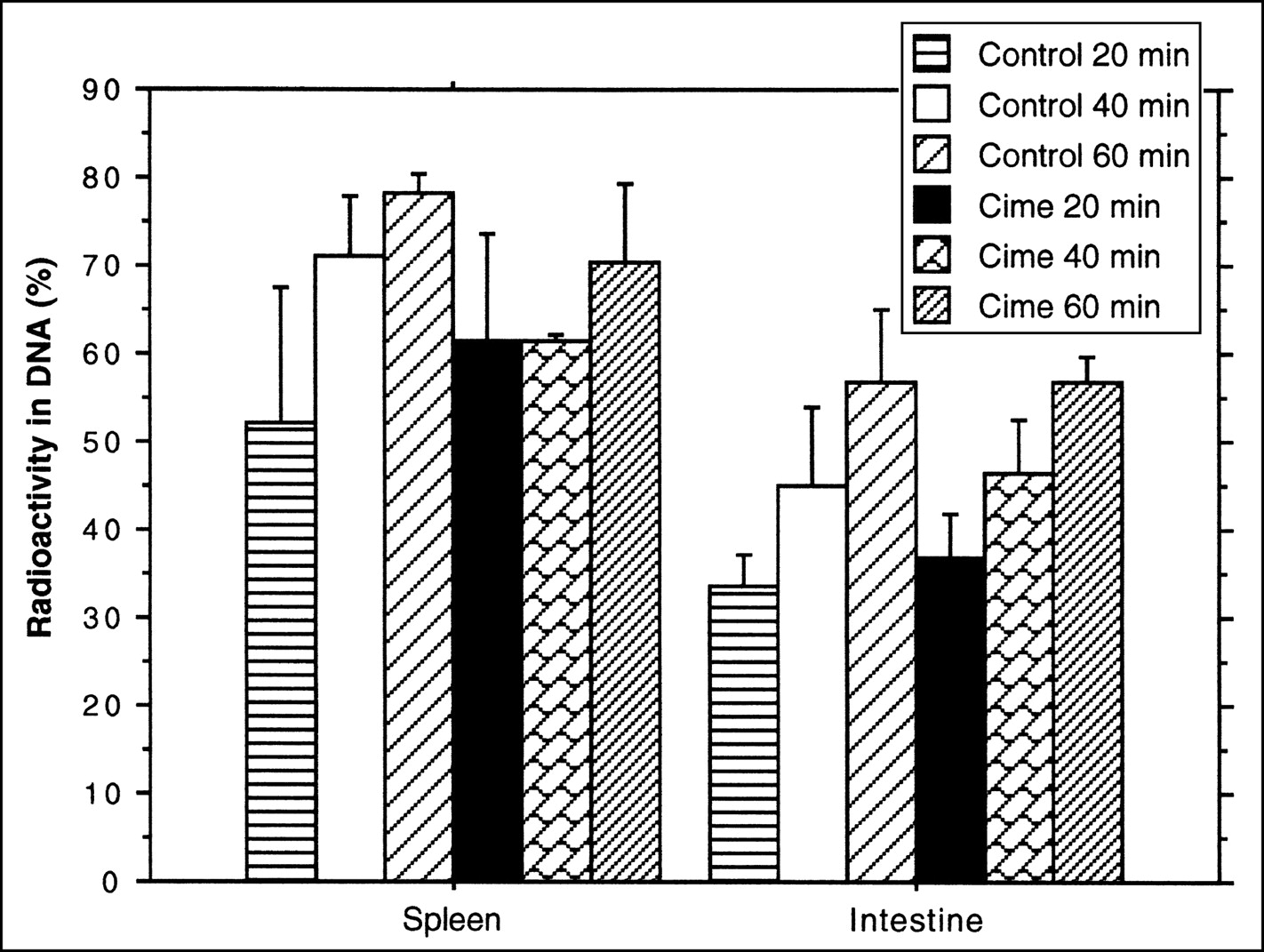
Fraction of 11C-FMAU radioactivity, expressed as percentage of total tissue radioactivity (mean ± SD), recovered in DNA fraction in rat organs. Different animals were administered cimetidine with 11C-FMAU.
Experiments with 18F-FLT
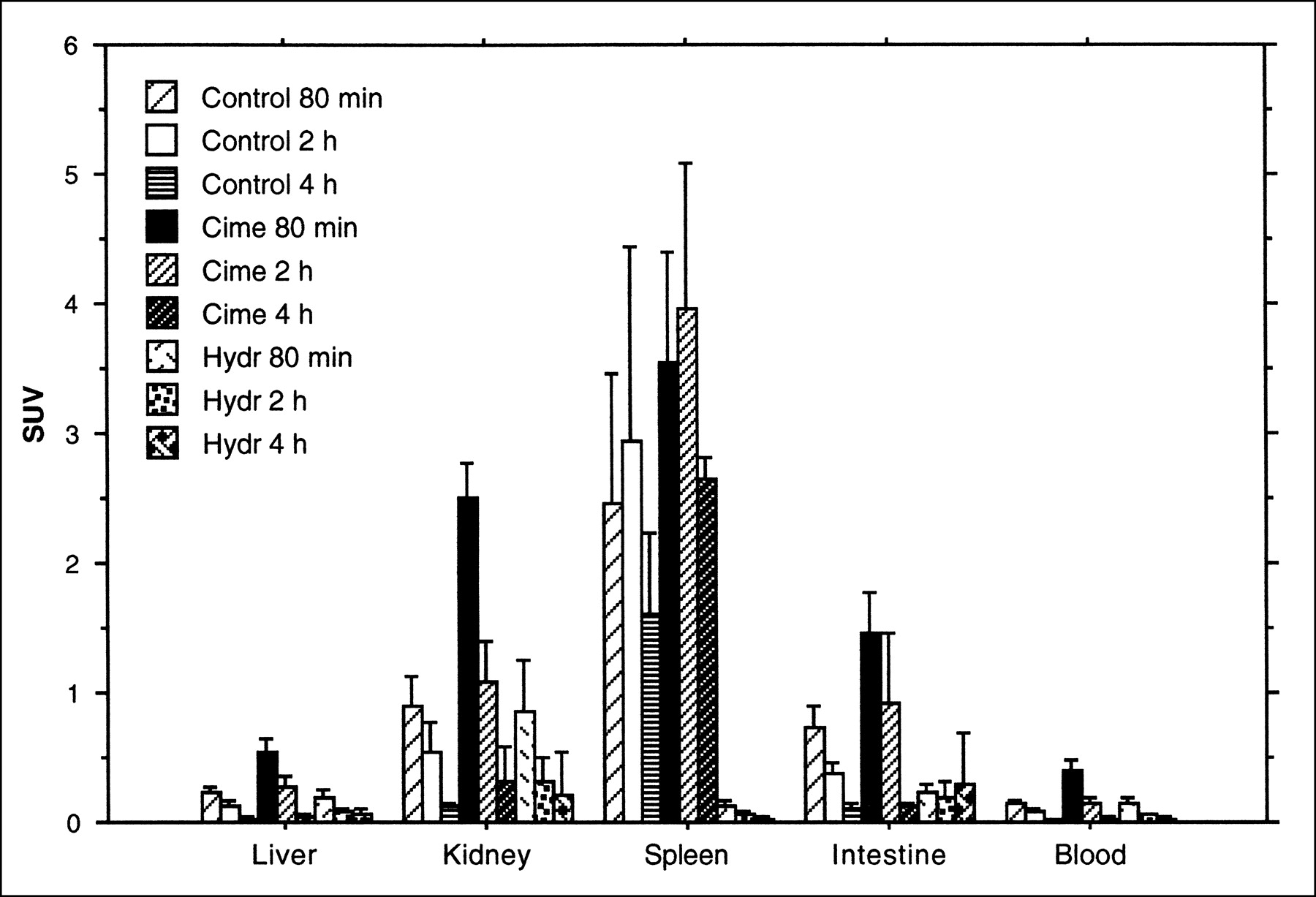
In these experiments, the highest 18F-FLT uptake was found in the spleen, followed by the kidney and intestine. In the spleen, the highest uptake value was observed at the 2-h time point. In the kidney and intestine, the highest uptake values were seen at the 80-min time point and decreased with extended observation time (Fig. 4). When compared at the same time points, the uptake values of the cimetidine-treated group were all higher than the untreated group. In the hydroxyurea-treated group, a pronounced decrease of uptake was found in the spleen.
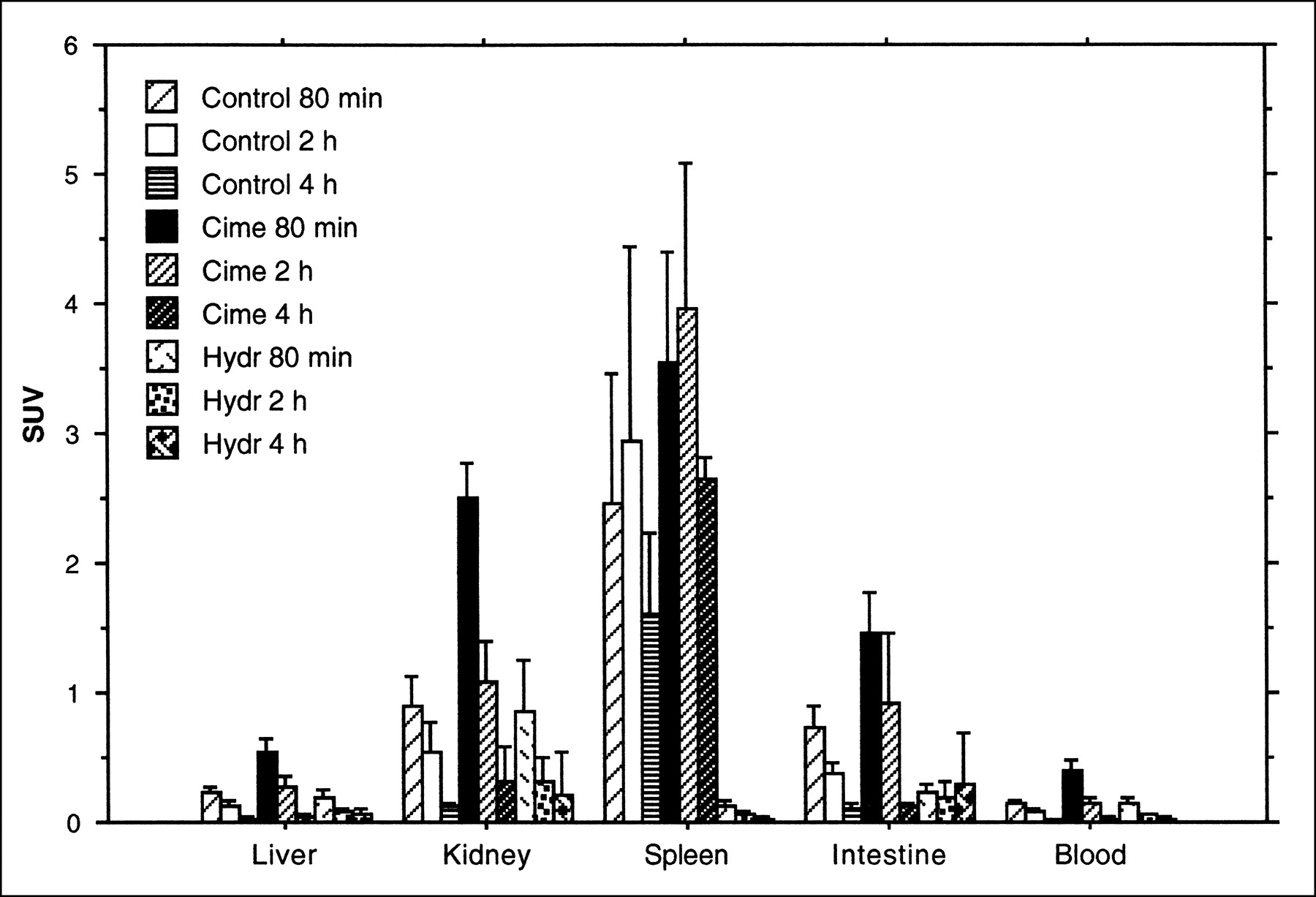
Uptake of 18F-FLT, expressed as SUV (mean ± SD) in rat organs. Animals were killed after 80 min or 2 or 4 h. Different animals were administered cimetidine with 18F-FLT or hydroxyurea 30 min before administration of radioactivity.
The percentage of the radioactivity incorporated into the DNA fraction was <2% in both spleen and intestine. There was no difference between the treated and untreated groups.
Experiments with 76Br-BFU
Figure 5A shows that in the rats injected with 76Br-BFU the highest uptake was found in the kidney at the 5-min point (SUV = 8.7, SD = 1.5). During the 80-min observation time, the radioactivity concentration in the kidney, liver, and blood decreased with time. In the spleen and intestine, however, the uptake maintained a constant level and the SUVs were between 0.8 and 1.0 during the entire observation period. In the rats treated with cimetidine, the radioactivity concentrations in the liver, kidney, and blood were much higher than in the untreated group, and these concentrations also decreased with time. After 4 h, the SUVs of these organs were all <0.5. In the spleen, radioactivity concentration increased with time. In the intestine, the radioactivity concentration remained at a relatively stable level during the observation period. When the radioactivity in an organ was represented as a fraction of blood radioactivity, significantly higher uptake was found in the spleen (P < 0.0001) and intestine (P < 0.0001) than in other organs, such as the liver, at 4 h after the radioactivity was administered (Fig. 5B).
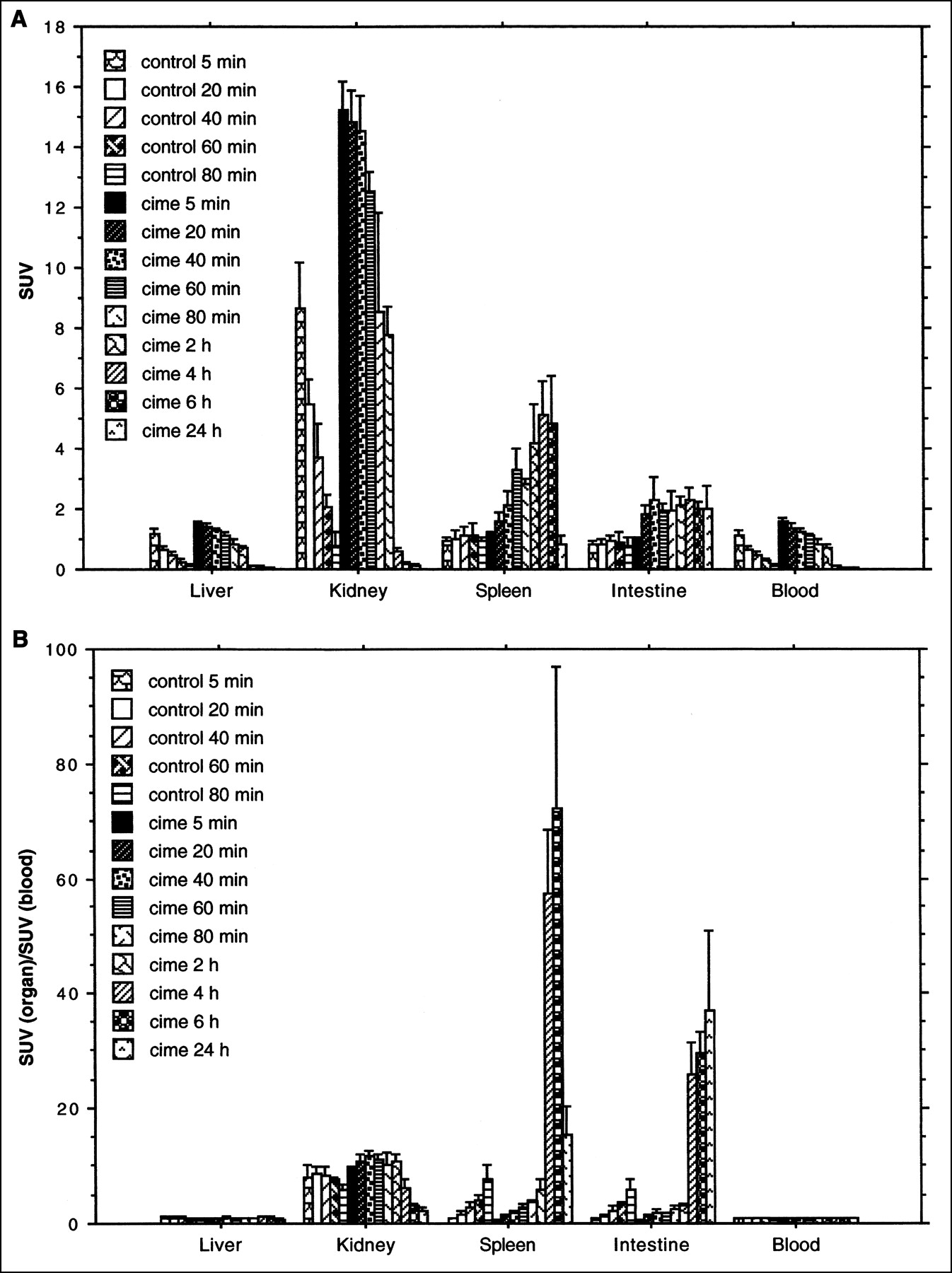
(A) Uptake of 76Br-BFU, expressed as SUV (mean ± SD), in rat organs. Animals were killed after 5, 20, 40, 60, or 80 min. Different animals were administered cimetidine with 76Br-BFU and were killed after 5, 20, 40, 60, or 80 min and 2, 4, 6, or 24 h. (B) Uptake of 76Br-BFU, expressed as ratio to blood radioactivity concentration (mean ± SD) in rat organs. Animals were killed after 5, 20, 40, 60, or 80 min. Different animals were administered cimetidine with 76Br-BFU and were sacrificed after 5, 20, 40, 60, or 80 min and 2, 4, 6, or 24 h.
The results of the DNA separation showed that the fraction of radioactivity incorporated into DNA increased with observation time. One hour after the radioactivity injection, the fraction of radioactivity in DNA in the spleen was 80.5% (SD = 3.8) in the untreated group and 77.9% (SD = 3.5) in the treated group. In the intestine, the values were 58.2% (SD = 10.5) in the untreated group and 54.3% (SD = 7.6) in the treated group. Four hours after the radioactivity injection, the radioactivity fractions exceeded 97% in the spleen and 93% in the intestine and became constant with time (Fig. 6).
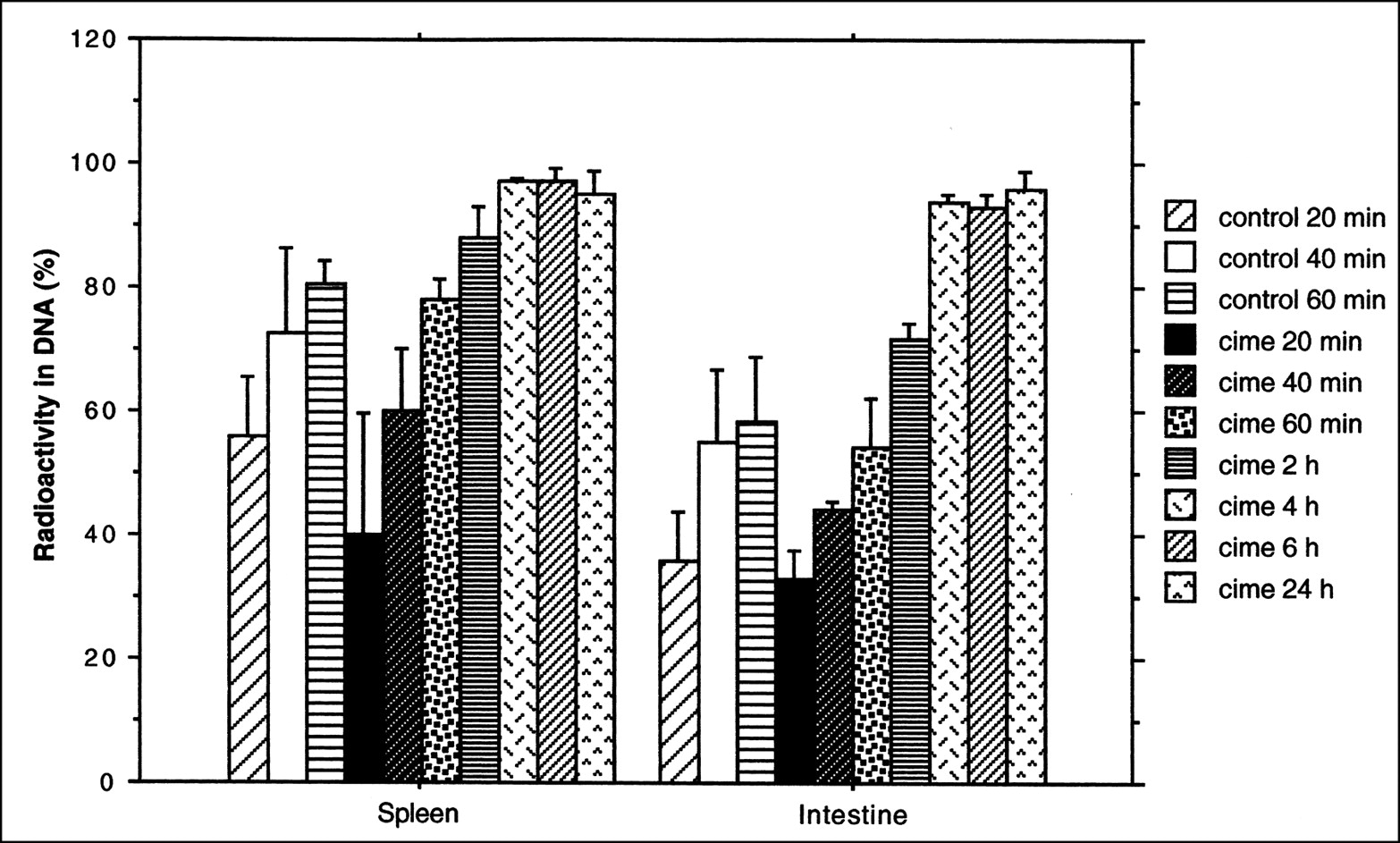
Fraction of 76Br-BFU radioactivity, expressed as percentage of total tissue radioactivity (mean ± SD), recovered in DNA fraction in rat organs. Different animals were administered cimetidine with 76Br-BFU.
Urinalysis
In the rats treated with cimetidine, the elimination of total administered radioactivity was reduced with all 3 tracers. At the 5-min time point, the elimination rate of 11C-FMAU, defined as the fraction of given radioactivity appearing in urine, was reduced about 80%. At the same time point, the elimination rate was reduced about 96% for 76Br-BFU. At the 80-min time point, the rate was reduced about 91% for 11C-FMAU, about 74% for 76Br-BFU, and about 36% for 18F-FLT (Table 1).
Elimination of Radioactivity from Rat Kidney*
Experiments with Tumor Model
The highest uptake of 11C-FMAU in the heart in the untreated group was observed at the 20-min time point (SUV = 1.35, SD = 0.15). The radioactivity decreased after this time point. In the tumor tissue, the highest uptake was found at the 40-min time point (SUV = 1.03, SD = 0.21). In the cimetidine-treated group, the radioactivity concentration in the heart was stable during the observation time, with SUVs about 1.4. In the tumor tissue, the uptake was significantly lower than in the heart at the 20-min time point (SUV = 0.75, SD = 0.38) but had a tendency to increase with time. At the 60-min time point, the uptake was higher than in the heart (SUV = 1.63, SD = 0.24).
In the 14C-thymidine measurement, the highest uptake in the tumor tissue was found at the 40-min time point in the untreated group (SUV = 1.39, SD = 0.27). In the heart, the uptake was much lower than in tumor (SUV = 0.31, SD = 0.04). In the cimetidine-treated group, the highest uptake was observed at the 80-min time point in the tumor (SUV = 1.20, SD = 0.59), whereas the radioactivity concentration in the heart was as low as in the untreated group.
The results of the DNA separation showed that the fraction of 11C-FMAU incorporated into DNA was 28.2% (SD = 13.3) in the untreated group and 45.2% (SD = 7.5) in the treated group. The fraction of 14C-thymidine incorporated into DNA was 41.7% (SD = 20.4) in the untreated group and 58.0% (SD = 24.0) in the treated group.
DISCUSSION
There is a strong need for a PET method that would indicate proliferation potential. Such a method would have great value in grading the malignancy of tumors and would be important in the early assessment of antitumoral effects by established anticancer drugs as well as new drugs under development. DNA synthesis and renewal of the genetic material is a cornerstone in proliferation. Classic in vitro assays have used radiolabeled thymidine with recording of the DNA-incorporated radioactivity. In tumor cells cultured as a monolayer, the growth rate is so exaggerated that the DNA-incorporated radioactivity dominates. Under in vivo conditions, however, with more limited growth rates as a result of contact inhibition and other factors, the fraction of the total radioactivity found in DNA can be relatively small, so that nonincorporated thymidine and metabolites thereof can be substantial. This fact has severely restricted the use of 11C-thymidine for in vivo characterization of proliferation potential using PET. A further problem is that thymidine is so rapidly metabolized that tissue exposure to intact nucleoside is too brief. In addition, labeled metabolites may contribute significantly to tissue radioactivity.
The most promising nucleoside analogues developed recently retain nucleoside transport and phosphorylation as thymidine but are protected from systemic metabolism by the introduction of fluorine in the sugar moiety. We have previously reported (24) on the use of 76Br-BFU for proliferation measurements and shown that the major part of the tissue radioactivity in proliferating tissues is constituted by DNA-incorporated radioactivity. As a side issue, we observed that the very rapid elimination through the kidneys could be retarded by pretreatment with cimetidine. The mechanism was postulated to be cimetidine inhibition of the cation transporter.
Very successful imaging of tumors has been achieved with another nucleoside, 18F-FLT. It is believed that this molecule does not incorporate into DNA, yet its uptake and retention in tissue are proportional to proliferation potential.
In the present work, we compared the magnitude of uptake of these tracers with the inclusion of a third, 11C-FMAU, to understand their respective prospects as proliferation markers. As part of this comparison, we wished to assess their sensitivity to cimetidine treatment and evaluate the degree of DNA incorporation.
When 76Br-BFU was administered alone, the radioactivity concentration in the organs without active DNA synthesis, such as the liver and kidney, decreased in parallel with the decreased concentration of the tracer in blood (Fig. 5A). In organs with active DNA synthesis, such as the spleen and intestines, however, the radioactivity concentration maintained relatively constant levels, although the radioactivity concentration in the blood decreased with time. As a consequence, the tissue-to-blood ratio increased with time for these organs. With cimetidine treatment and follow-up times of 4 h or more, tissue-to-blood ratios of 30–60 were obtained.
Soon after the radioactivity administration, a peak concentration level was observed in the kidneys, a level that decreased quickly with time. Together with the urinalysis, these facts indicate that 76Br-BFU is rapidly eliminated through the kidney into urine. Cimetidine, as an inhibitor of the organic cation secretory system, may block secretion of the some nucleosides (32,33). After treatment with cimetidine, the 76Br-BFU elimination in the kidneys is markedly hindered, leading to an increased level of its concentration in the blood. This results in a general increase in the concentration of 76Br-BFU in all organs, including the spleen and intestines. At time points later than 4 h, however, a pronounced decrease of the radioactivity was noted in organs without active DNA synthesis. This might indicate a limited duration of the cimetidine effect.
As previously reported, a very large fraction of the radioactivity was seen in the DNA fraction at time points later than 1 h after tracer administration.
The results obtained with 76Br-BFU are very encouraging and suggest that this tracer could be used in vivo in patients with tumors. The main obstacles are that 76Br is more cumbersome to produce and that this radionuclide generates a proportionally high radiation dose. To counteract this, the amount of radioactivity that can be administered to a patient must be reduced, leading to noisy images.
In the group given 11C-FMAU alone, uptake of the radioactivity in the organs measured showed patterns similar to those of 76Br-BFU, except that it decreased more slowly with time. Because the radioactivity concentration in the blood maintained higher levels than with76Br-BFU, a higher uptake in the organs with active DNA synthesis (such as the spleen and intestines) could be expected. In spite of the short follow-up times allowed by 11C, a significant portion of the radioactivity was found in DNA. Compared with the other 2 tracers, the contrast between proliferating and nonproliferating tissues is lower, perhaps because the time allowed for systemic elimination is shorter.
When hydroxyurea was used, the 11C-FMAU uptake in the spleen and intestines decreased to a level close to that in the organs with minimal DNA synthesis. This can be explained by the fact that hydroxyurea may block DNA synthesis (34–36).
Cimetidine administration with 11C-FMAU did not show the same dramatic effect on radioactivity uptake as observed with 76Br-BFU. Analysis of the urine radioactivity indicates that the rate of 11C-FMAU secretion in the kidney is significantly lower than that of the 76Br-BFU at the 2 observation time points. However, cimetidine significantly reduced the urinary elimination. We have previously suggested that 76Br-BFU is eliminated in the kidneys through a cation transporter that can be inhibited by cimetidine. We suggest that the same mechanism operates on 11C-FMAU but with lower efficiency.
The results of 11C-FMAU distribution in the organs and the DNA analyses showed that both the radioactivity uptake and the fraction of DNA-incorporated radioactivity in the organs with active DNA synthesis had a tendency to increase with time. However, because of the short half-life of 11C, a longer observation time is not feasible.
The studies performed in the mouse tumor model showed a higher uptake of 11C-FMAU in the tumor than in heart in the mice treated with cimetidine, but the contrast was still relatively low. It is possible that the DNA synthesis rate in tumors is in fact lower than that observed in intestines and spleen. Other factors may also contribute to the low tracer uptake, such as selectivity and expression of nucleotide transporters and kinase activity. The relative low uptake of 14C-thymidine, a known marker of tumor proliferation (37), in the same tumor model may also be explained by this hypothesis.
In the experiment with 18F-FLT, the highest radioactivity uptake values were seen in the spleen and intestines, but the DNA analysis showed that the tracer was not incorporated into DNA. The radioactivity in the intestines decreased over time, in parallel with the blood radioactivity. This supports the observation that 18F-FLT is not introduced into DNA in this organ. Shields et al. (17) showed a high accumulation in dog, but no analysis of DNA incorporation was performed. There may be species differences in this respect. We believe that our results are representative with respect to DNA incorporation, but the magnitude of uptake could be species dependent. The radioactivity uptake in the spleen and intestines decreased significantly after treatment with hydroxyurea, but the mechanism is unclear. It is possible that hydroxyurea inhibits the activity of kinases, or reduces the activity of the nucleoside transporter in the cell membrane, either directly or indirectly as a consequence of inhibition of DNA synthesis.
The radioactivity concentration in the kidney and urinalysis showed that 18F-FLT was eliminated quickly via the kidneys. However, cimetidine could only partially inhibit its secretion, indicating that there are other mechanisms for 18F-FLT elimination than the known organic cation secretory system in the kidney.
CONCLUSION
76Br-BFU is predominantly incorporated into DNA and has great potential as a PET tracer for assessment of proliferation in vivo. Cimetidine may be used to prevent the rapid secretion of the tracer via the kidneys and thereby increase the uptake of 76Br-BFU in organs with active DNA synthesis. 11C-FMAU also may have potential as a proliferation marker but, becaues of its short half-life, allows a limited time for observation. 18F-FLT is not incorporated into DNA and cannot directly measure proliferation. However, it could be used as an indirect indicator of proliferation potential if it is proven that the uptake of this nucleoside via nucleoside transporter and phosphorylation parallels that of thymidine analogues that are incorporated into DNA. Cimetidine does not significantly affect the uptake of 11C-FMAU or 18F-FLT in the organs.
Acknowledgments
This work was in part supported by grants from the Swedish Cancer Society and the Erik, Karin, and Gösta Selanders Foundation.
Footnotes
Received Dec. 18, 2001; revision accepted July 26, 2002.
For correspondence or reprints contact: Mats Bergström, PhD, PET Center, Uppsala University Hospital, 751 85 Uppsala, Sweden.
E-mail: Mats.Bergstrom{at}pet.uu.se
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Different Modes of Transport for 3H-Thymidine, 3H-FLT, and 3H-FMAU in Proliferating and Nonproliferating Human Tumor Cells
- Imaging of Cell Proliferation: Status and Prospects
- Molecular Imaging of Proliferation in Malignant Lymphoma.
- Evaluation of 76Br-FBAU as a PET Reporter Probe for HSV1-tk Gene Expression Imaging Using Mouse Models of Human Glioma
- Biodistribution and Radiation Dosimetry Estimates of 1-(2'-Deoxy-2'-18F-Fluoro-1-{beta}-D-Arabinofuranosyl)-5-Bromouracil: PET Imaging Studies in Dogs
- Potential of PET in oncology and radiotherapy
- Imaging DNA Synthesis In Vivo with 18F-FMAU and PET
- Monitoring Antiproliferative Responses to Kinase Inhibitor Therapy in Mice with 3'-Deoxy-3'-18F-Fluorothymidine PET
- Molecular Targeting with Radionuclides: State of the Science
- PET Imaging with 18F-FLT and Thymidine Analogs: Promise and Pitfalls