


Visual Abstract
Abstract
Reactive astrocytes play a key role in the pathogenesis of various neurodegenerative diseases. Monoamine oxidase-B (MAO-B) is one of the promising targets for the imaging of astrogliosis in the human brain. A novel selective and reversible MAO-B tracer, (S)-(2-methylpyrid-5-yl)-6-[(3-18F-fluoro-2-hydroxy)propoxy]quinoline (18F-SMBT-1), was successfully developed via lead optimization from the first-generation tau PET tracer 18F-THK-5351. Methods: SMBT-1 was radiolabeled with 18F using the corresponding precursor. The binding affinity of radiolabeled compounds to MAO-B was assessed using saturation and competitive binding assays. The binding selectivity of 18F-SMBT-1 to MAO-B was evaluated by autoradiography of frozen human brain tissues. The pharmacokinetics and metabolism were assessed in normal mice after intravenous administration of 18F-SMBT-1. A 14-d toxicity study after the intravenous administration of 18F-SMBT-1 was performed using rats and mice. Results: In vitro binding assays demonstrated a high binding affinity of 18F-SMBT-1 to MAO-B (dissociation constant, 3.7 nM). In contrast, it showed low binding affinity to MAO-A and protein aggregates such as amyloid-β and tau fibrils. Autoradiographic analysis showed higher amounts of 18F-SMBT-1 binding in the Alzheimer disease brain sections than in the control brain sections. 18F-SMBT-1 binding was completely displaced with the reversible MAO-B inhibitor lazabemide, demonstrating the high selectivity of 18F-SMBT-1 for MAO-B. Furthermore, 18F-SMBT-1 showed a high uptake by brain, rapid washout, and no radiolabeled metabolites in the brain of normal mice. 18F-SMBT-1 showed no significant binding to various receptors, ion channels, or transporters, and no toxic effects related to its administration were observed in mice and rats. Conclusion: 18F-SMBT-1 is a promising and selective MAO-B PET tracer candidate, which would be useful for quantitative monitoring of astrogliosis in the human brain.
Reactive astrocytosis is the astrocyte response observed in various neurologic disorders. It is characterized by distinct morphologic alternations such as enlarged cell bodies and overexpression of glial fibrillary acidic protein, vimentin, and nestin (1). In neurodegenerative conditions, astrocytes turn reactive and secrete neurotoxic inflammatory cytokines (1). Reactive astrocytes also overexpress monoamine oxidase-B (MAO-B) in the outer membrane of the mitochondria. MAO-B, a major enzyme that metabolizes dopamine and histamine, is widely distributed in the human brain. Higher levels of MAO-B expression are physiologically observed in the basal forebrain, substantia nigra, basal ganglia, thalamus, and hippocampal uncus relative to the cerebellar cortex (2). MAO-B levels in whole brain regions also increase linearly in an age-dependent manner during normal aging processes (2,3). Elevated MAO-B levels in autopsy-confirmed Alzheimer disease (AD) brains were observed during in vitro binding studies with selective MAO-B radioligands, such as 3H-l-deprenyl and 3H-lazabemide (4–6). In addition, MAO-B elevation was also observed in the postmortem brains of parkinsonian syndromes, progressive supranuclear palsy, and multiple-system atrophy (7). This finding indicates that MAO-B could be an attractive target for visualizing reactive astrocytes in vivo during neuroinflammatory processes.
Much effort has been made to develop novel MAO-B PET tracers based on the chemical structure of selective MAO-B inhibitors (8,9). 11C-l-deprenyl-D2 is the most commonly used PET tracer for imaging MAO-B in the human brain (Fig. 1). However, quantifying 11C-l-deprenyl-D2 binding is difficult because of its irreversible binding. 11C-SL25.1188, a reversible MAO-B PET tracer, was developed and used in human studies (10). The short half-life of 11C (20 min) limits the clinical utility of these PET tracers. Therefore, several 18F-labeled PET tracers have been developed for imaging MAO-B (11–14).

Chemical structures of MAO-B tracers tested in humans, 18F-THK-5351 and 18F-SMBT-1.
18F-THK-5351 was originally designed to detect neurofibrillary tangles in vivo and was found to bind MAO-B with high affinity (15,16). Clinical 18F-THK-5351 PET studies demonstrated high tracer retention in sites susceptible to astrogliosis in various neurodegenerative conditions (17–26). The nonselective binding of THK-5351 to MAO-B and tau limits its clinical utility as a biomarker. Lead optimization for generating a selective MAO-B binding tracer led to the development of (S)-(2-methylpyrid-5-yl)-6-[(3-18F-fluoro-2-hydroxy)propoxy]quinoline (18F-SMBT-1) (Fig. 1), a novel radiolabeled tracer compound. Here, we report the preclinical binding, pharmacokinetics, and metabolic properties of 18F-SMBT-1 and its derivatives.
MATERIALS AND METHODS
Synthesis of 18F-SMBT-1 and Its Derivatives
Methods for the synthesis and characterization of the tracer compounds are described in the supplemental data (Supplemental Schemes 1–4; supplemental materials are available at http://jnm.snmjournals.org).
Radiochemistry
18F-fluoride, produced by the 18O(p,n)18F reaction on enriched 18O-H2O with a cyclotron, was separated from the irradiated target with a Sep-Pak Light Accell Plus QMA cartridge (Waters), which was washed in advance with K2CO3 followed by water. The trapped 18F-fluoride was eluted with a solution of Kryptofix 222 (8 mg; Merck), K2CO3 (1.5 mg), acetonitrile (0.45 mL), and water (0.13 mL). The eluted solution was then used for subsequent radiofluorination. The solution was evaporated to dryness by azeotropic distillation with a helium flow (300 mL/min), heating at 110°C, and stirring. After drying, the tosylate precursor (S)-(2-methylpyrid-5-yl)-6-[[2-(tetrahydro-2H-pyran-2-yloxy)-3-tosyloxy]propoxy]quinoline (THK-5475; 2 mg) dissolved in dimethylsulfoxide (0.45 mL) was transferred and stirred at 110°C for 10 min. Next, hydrochloride aqueous solution (2 M, 0.2 mL) was added to the reaction solution and stirred at 110°C for an additional 3 min for deprotection of the hydroxy group. The reaction was then quenched with potassium acetate aqueous solution (0.2 M, 4 mL), followed by solid-phase extraction. The crude mixture was passed through the activated Sep-Pak tC18 Plus (Waters) and then washed with water. Radioactive products remaining in the solid phase were eluted with 70% ethanol (0.7 mL) and diluted with water (0.12 mL) and then subjected to semipreparative high-performance liquid chromatography (column, Inertsil ODS-4 (GL Sciences, Inc.; mobile phase, 20 mmol/L NaH2PO4/acetonitrile [67/33 for 18F-SMBT-1]; flow rate, 5.0 mL/min). A detailed account of the optimized procedure is provided in Supplemental Table 1. 18F-SMBT-1 was also produced by 1-pot microscale radiosynthesis (27). 18F-THK-5351 was prepared as described previously (15). 18F-fluoroethyl harmine was prepared from the corresponding precursor using the microscale radiosynthesis method, as previously described (27). 125I-Ro 43-0463 was prepared as previously described (11). 18F-SMBT-1, 18F-THK-5351, 18F-fluoroethyl harmine, and 125I-Ro 43-0463 were obtained in greater than 95% radiochemical purity after high-performance liquid chromatography purification. The averages of decay-corrected radiochemical yields and molar activity at the end of 18F-SMBT-1 synthesis were 39% and 414 GBq/μmol, respectively.
In Vitro Binding Studies
3H-THK-5351 (molar activity, 2.96 TBq/mmol; radiochemical purity, 98.9%) was custom-labeled by Sekisui Medical Inc. 3H-Pittsburgh compound B (molar activity, 2.96 TBq/mmol; radiochemical purity, 99%) was obtained from American Radiolabeled Chemicals. 3H-MK-6240 (molar activity, 0.83 TBq/mmol; radiochemical purity, 99%) was obtained from ViTrax. Competitive binding assays were performed as previously described (28). 3H-Pittsburgh compound B (1 nM) and 3H-MK-6240 (1.5 nM) were used as radioligands for amyloid and tau aggregates, respectively. An in vitro saturation binding assay was also performed using 18F-labeled compounds as previously described (29). 18F-fluoroethyl harmine and recombinant MAO-A (M7316; Sigma-Aldrich) were used for an in vitro competitive binding assay against MAO-A. The off-rate was determined as previously described (30). All assays were performed at room temperature in quadruplicate, and the off-rate was determined by GraphPad Prism, version 7.0. Correlation analysis between tracer binding and MAO-B activity was performed as previously described (16).
In Vitro Autoradiography
The Ethics Committee of the Tohoku University Graduate School of Medicine approved this study, and all subjects provided written informed consent. Postmortem brain sections from control subjects and AD patients were acquired from Tohoku University Brain Bank. In vitro autoradiography was performed as previously described (16). To account for MAO-B binding in the brain tissues, the reaction was incubated in the presence of the MAO-B inhibitor lazabemide (1 μM). After postfixation in 4% paraformaldehyde for 30 min, adjacent frozen sections were immunostained with anti-MAO-B (1:400; Sigma-Aldrich), antitau (AT8; Innogenetics), and anti-β-amyloid (6F/3D; Dako) antibodies.
Biodistribution Study in Normal Mice
All animal experimental protocols were approved by the Laboratory Animal Care Committee of Tohoku University. The biodistribution study was performed as previously described (29). The radiation dose and mass dose for humans were estimated on the basis of the biodistribution data from mice (31).
Metabolite Analysis
Male Institute of Cancer Research (ICR) mice (6 wk old) were killed by decapitation under anesthesia at 2, 10, and 30 min after intravenous administration of 18F-SMBT-1 (19.4 MBq), and the brain and cardiac blood were collected. Extraction and metabolite analysis were performed as previously described (32). To identify the major metabolite of 18F-SMBT-1, in vitro enzyme assays were performed using human sulfotransferase 1E1 (Cypex Ltd.) as described previously (28).
Receptor Binding Assays
Receptor binding screen assays were performed by Sekisui Medical Inc. Binding inhibition (%) was determined by competitive radioligand assays against 60 common neurotransmitter receptors, ion channels, and transporters, as previously described (29).
Animal Toxicity Studies
Acute toxicity studies were performed on Sprague–Dawley rats and ICR mice. A single intravenous dose of 18F-SMBT-1 was administered by LSI Medience Inc., as previously described (29).
RESULTS
In Vitro Competitive Binding Studies
We performed an in vitro competitive binding assay using 3H-THK-5351 to measure the binding affinity of various compounds against recombinant MAO-B (Supplemental Table 2). Arylquinoline derivatives including THK-5105 showed high binding affinity to MAO-B. The binding affinity of THK-5105 to MAO-B was substantially reduced after substitution of the hydroxy group in the (3-fluoro-2-hydroxy)propoxyl group by hydrogen. The chirality of compounds was also associated with the binding affinity to MAO-B. For example, R-enantiomer (THK-5451) showed lower affinity to MAO-B than S-enantiomer (THK-5351). Previous analysis of the structure–activity relationship of 2-arylquinoline derivatives showed that the 2 amino groups on the pyridine ring were essential for binding to tau aggregates (Ryuichi Harada, unpublished data, November 2013). We therefore investigated the substituent on the pyridine ring at the 2-position to ensure high selectivity of compounds for MAO-B over tau aggregates. Although the hydrogen substituent on the pyridine ring (SMBT-0) reduced the binding affinity to MAO-B, the methyl substituent (SMBT-1) maintained a high binding affinity to MAO-B at a level comparable to THK-5351 and several other MAO-B inhibitors, such as rasagiline, lazabemide, and safinamide. Our SAR study of 18F-SMBT-1 derivatives demonstrated that the 2-methylpyridine derivative was the most ideal for generating high binding affinity to MAO-B (Supplemental Table 2). The binding of 18F-SMBT-1 to recombinant MAO-A was further investigated by an in vitro competitive binding assay using the reversible MAO-A binder 18F-fluoroethyl harmine. In contrast with its high affinity to MAO-B, 18F-SMBT-1 showed low binding affinity to MAO-A (half-maximal inhibitory concentration, 713 nM). Furthermore, 18F-SMBT-1 showed low binding affinity to amyloid-β and tau protein aggregates (half-maximal inhibitory concentration, >1,000 nM) (Supplemental Table 3). Receptor binding screen assays demonstrated no remarkable interaction with various receptors, ion channels, and transporters (Supplemental Table 4).
In Vitro Binding Studies of 18F-SMBT-1 to MAO-B
Next, we radiolabeled SMBT-1 with 18F and investigated the binding properties of 18F-SMBT-1 to MAO-B in detail. Saturation binding assays demonstrated a high binding affinity of 18F-SMBT-1 for recombinant MAO-B (dissociation constant [KD]= 3.7 nM; number of binding sites [Bmax] = 110.4 pmol/mg protein) (Fig. 2). As shown in Supplemental Figure 1, 18F-SMBT-1 also showed high binding affinity for MAO-B–rich AD brain homogenates (KD = 3.5 nM) and mouse brain homogenates (KD = 4.3 nM). However, the Bmax in AD brain homogenates (606 pmol/g of tissue) was approximately 3 times higher than that in mouse brain homogenates (153 pmol/g of tissue). The specific binding of 18F-SMBT-1 to human AD brain homogenates and recombinant MAO-B was reversible with similar kinetics (Fig. 3B). The off-rates were 0.0087 and 0.0072 min−1 for human AD brain homogenate and recombinant MAO-B, respectively.

In vitro saturation binding of 18F-SMBT-1 against recombinant MAO-B (A). In vitro binding kinetics of 18F-SMBT-1 against recombinant MAO-B and AD brain homogenate (B).
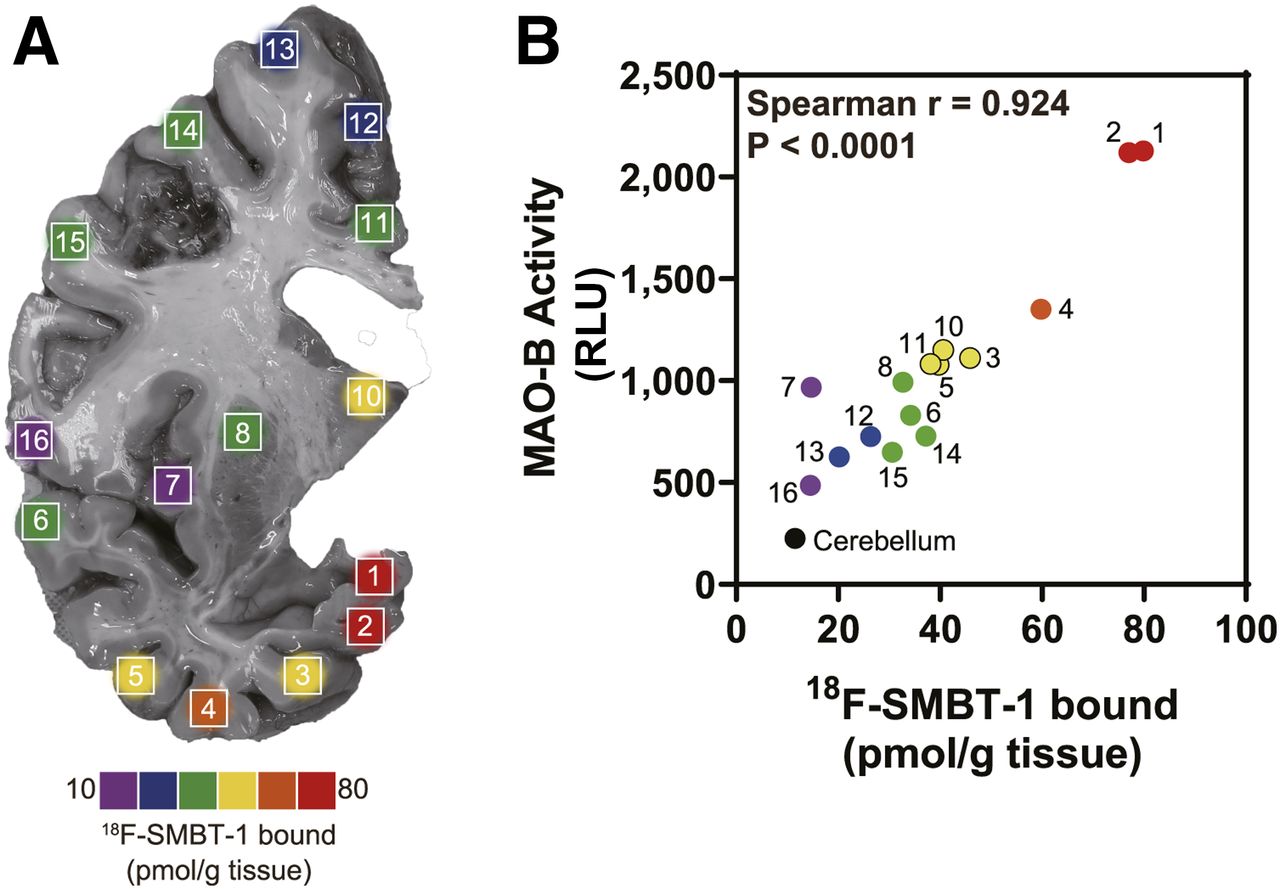
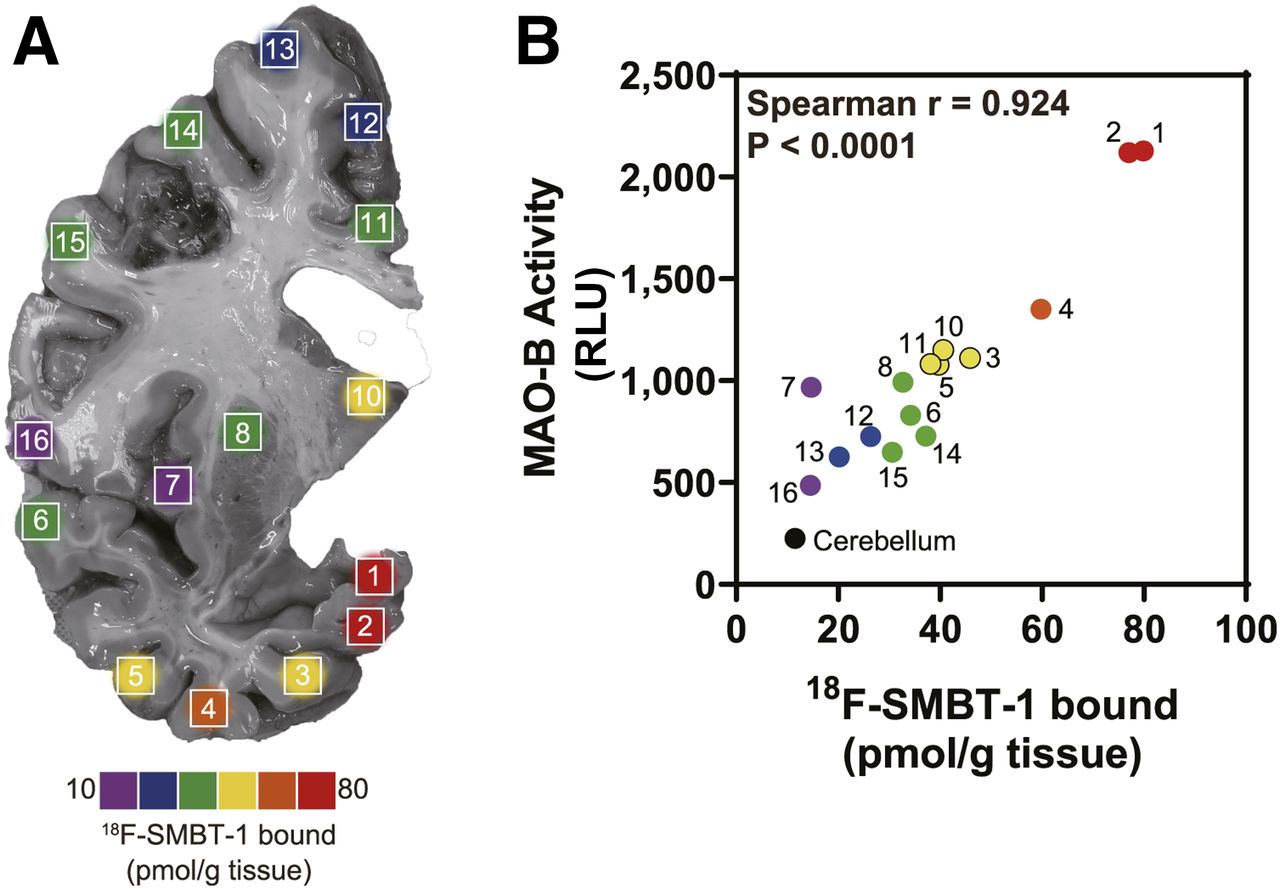
18F-SMBT-1 binding density map in 16 regions of coronal brain section of autopsy tissue from right hemisphere from patient with AD (81-y-old man) (A). Correlation of regional in vitro 18F-SMBT-1 binding and MAO-B activity (B). RLU = relative luminescence units.
A strong correlation was observed between regional in vitro 18F-SMBT-1 binding and MAO-B activity in a patient with AD (Spearman r = 0.923, P < 0.0001; Fig. 3). We also observed a significant correlation with sarkosyl insoluble tau (Spearman r = 0.678, P = 0.0049); however, this association was weaker than that observed with MAO-B activity. In contrast, no significant correlation was observed with insoluble total Aβ (Spearman r = −0.391, P = 0.135).
In Vitro Autoradiography of Postmortem Human Brain Sections
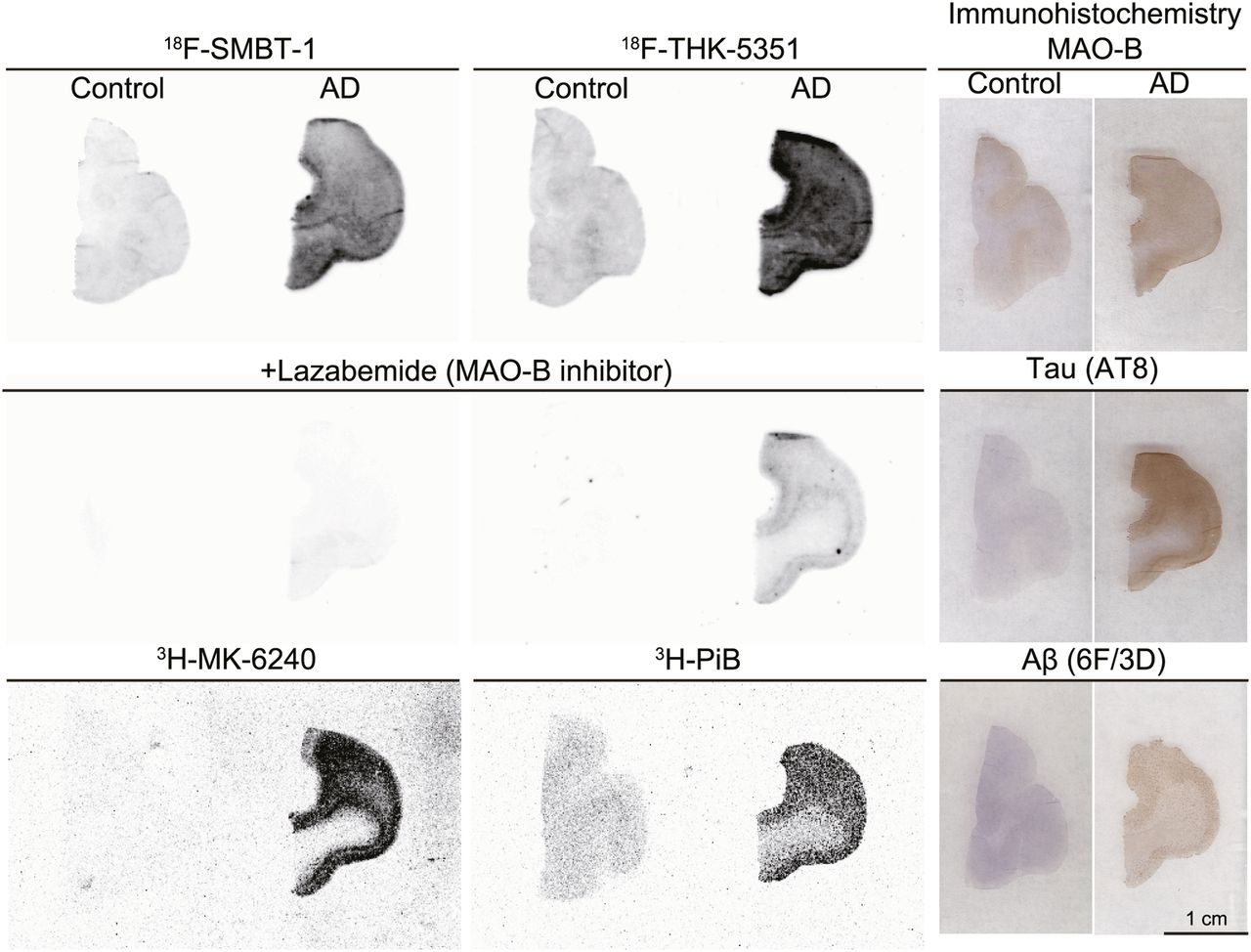
In vitro autoradiography of 18F-SMBT-1 was conducted to further evaluate the binding selectivity of 18F-SMBT-1 in human brain sections. Specific binding of 18F-SMBT-1 was greater in human AD brain sections than in control brain sections, as was consistent with the results of MAO-B immunohistochemistry (Fig. 4). This specific binding of 18F-SMBT-1 was completely displaced in the presence of the selective MAO-B inhibitor lazabemide, although 18F-THK-5351 binding remained detectable in human AD brain sections after lazabemide treatment. The distribution of 18F-SMBT-1 was also consistent with that of the reversible MAO-B tracer, 125I-Ro 43-0463 (Supplemental Fig. 2). Considerable amounts of 3H-Pittsburgh compound B and 3H-MK-6240 binding were detected in these sections. These results indicate that 18F-SMBT-1 binds to MAO-B with high selectivity.
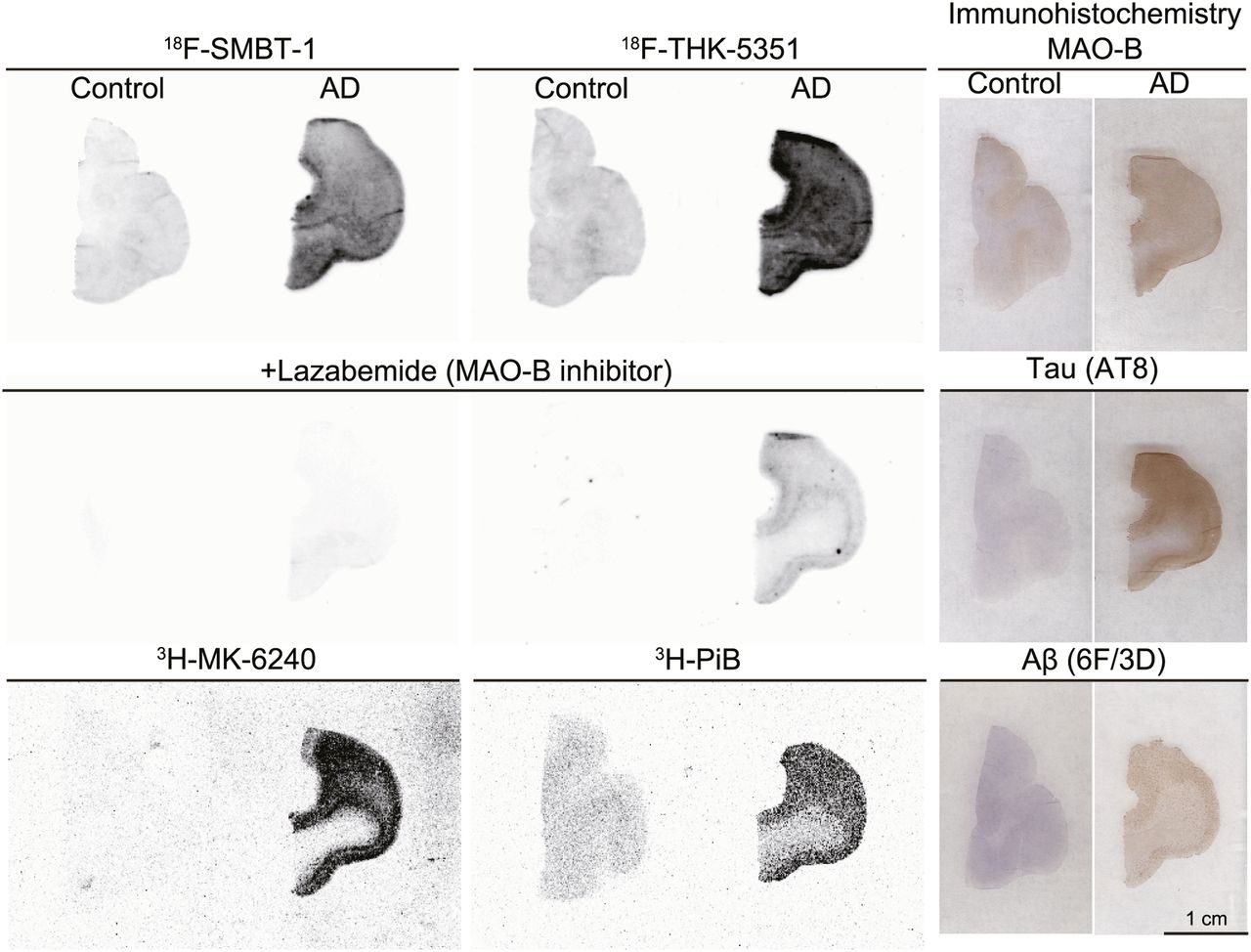
Comparative in vitro autoradiography of 18F-SMBT-1, 18F-THK-5351, 3H-MK-6240, and 3H-Pittsburgh compound B (PiB) in frontal cortex of control subject (55-y man) and patient with AD (68-y-old woman). Adjusted sections were stained by anti-MAO-B, anti-pTau (AT8), and antiamyloid-β (6F/3D) antibodies.
Biodistribution of 18F-SMBT-1 in Mice
18F-SMBT-1 showed excellent blood–brain barrier permeability in mice. The peak uptake of 18F-SMBT-1 (7.85% ± 0.76% injected dose/g at 2 min after injection) by the brain was greater than that of 18F-THK-5351. Furthermore, 18F-SMBT-1 showed rapid washout from normal brain tissue and no significant defluorination in mice (Table 1). 18F-SMBT-1 radiation exposure was estimated from the biodistribution data from mice (Supplemental Table 5). The resultant whole-body effective dose equivalents were 12.2 μSv/MBq (male) and 21.3 μSv/MBq (female), which were comparable to those of 18F-THK-5351 (15).
Biodistribution After Intravenous Injection of 18F-SMBT-1 in ICR Mice
In Vivo Metabolism in Mice
The extraction efficiencies from blood and brain were 89% and 87%, respectively. Figure 5 shows the time–activity kinetics of radiolabeled metabolites and 18F-SMBT-1 in the brain and plasma. 18F-SMBT-1 was dominantly metabolized to 1 polar metabolite. In plasma, 92%, 65%, and 19% of the parent compound remained at 2, 10, and 30 min after injection, respectively. On the other hand, most of the radioactivity in the brain was derived from the parent compound during the 30-min period after intravenous administration. As previously observed in 18F-THK-5351 (28), the major metabolite M2 exhibited the same Rf value as enzymatically produced O-sulfated 18F-SMBT-1.

(A and B) Time–activity curves of parent and radiolabeled metabolites in brain (A) and plasma (B). (C) Reverse-phase thin-layer chromatograms. (D) Chemical structure of O-sulfated 18F-SMBT-1 by human sulfotransferase 1E1 and reverse-phase thin-layer chromatograms. Lane 1: mouse plasma at 10 min after injection of 18F-SMBT-1; lane 2: 18F-SMBT-1 with human sulfotransferase 1E1.
Animal Toxicity Studies
A single intravenous administration of 18F-SMBT-1 at 1 mg/kg, equivalent to 100,000-fold the intended clinical dose for humans, caused no systemic toxicity in rats or mice.
DISCUSSION
18F-SMBT-1 is a single S-enantiomer and 2-methylpyridine derivative of 18F-THK-5351. 18F-THK-5351 was originally designed to detect tau aggregates in the form of paired helical filaments. However, recent studies showed high amounts of 18F-THK-5351 binding to MAO-B. Recent clinical studies have demonstrated the unique pattern of 18F-THK-5351 binding in various neurodegenerative diseases other than AD. However, the lack of binding selectivity of this tracer makes it difficult to interpret PET images. Therefore, we conducted lead optimization testing on THK-5351 derivatives to determine a selective MAO-B binding profile and a favorable pharmacokinetics profile. Previous structure–activity relationship analysis of 2-arylquinoline derivatives showed that the 2-amino group on the pyridine ring of 2-arylquinoline derivatives was essential for binding to tau aggregates. In the present study, structure–activity relationship analysis of MAO-B revealed that the hydroxy group in the (3-fluoro-2-hydroxy)propoxyl group of 2-arylquinoline plays an important role in achieving high binding affinity for MAO-B; (3-fluoro-2-hydroxy)propoxyl group has a chiral center. Previous studies demonstrated that the S-enantiomers of THK compounds showed better pharmacokinetics than the R-enantiomers (30,32). In addition, the present study demonstrated that the affinity of the S-enantiomer for MAO-B was higher than that of the R-enantiomer. Therefore, we selected the S-enantiomer as the novel MAO-B tracer candidate. We further explored the functional group at position 2 on the pyridine ring to reduce the binding affinity for tau aggregates. Among several derivatives, 18F-SMBT-1 showed the highest affinity for MAO-B and an excellent binding selectivity for MAO-B over MAO-A, amyloid, and tau aggregates. These findings were supported by in vitro autoradiography of 18F-SMBT-1 using human brain tissues.
The binding of the radiotracer can be predicted by the ratio of the Bmax to the binding affinity (KD) for the target. 18F-SMBT-1 showed low KD and high Bmax values (KD = 3.7 nM, Bmax = 110.4 pmol/mg protein) against recombinant MAO-B and human brain homogenates (KD = 3.5 nM, Bmax = 606 pmol/g tissue). The binding potential (Bmax/KD) was greater than that of currently available tau PET tracers, suggesting the ability of 18F-SMBT-1 to detect MAO-B in vivo. MAO-B levels in the human brain cortex were 2.5- to 4.7-fold higher than those in rodents (33). This finding was consistent with the Bmax determined via in vitro 18F-SMBT-1 binding assays. Since MAO-B is expressed predominantly in humans and not in rodents, the differences in MAO expression between these species should be considered when using 18F-SMBT-1 in preclinical studies.
Classic MAO-B PET tracers based on MAO-B inhibitors such as 11C-pargyline, 11C-l-deprenyl, and 18F-rasagiline show irreversible binding to MAO-B. This irreversibility is due to the formation of a covalent adduct with flavin adenine dinucleotide, which is a redox-active coenzyme associated with MAO-B (34–36). The irreversibility of tracer binding led to an underestimation of MAO-B in the high-concentration regions, as the rate of tracer binding may exceed the rate of tracer delivery (37). Deuterium-substituted radiotracers such as 11C-l-deprenyl-D2, 18F-fluorodeprenyl-D2, and 18F-rasagiline-D2 reduce the trapping rate of the tracers and partly overcome these limitations; however, they still possessed irreversible binding properties for MAO-B. Reversible radiotracers would be preferable for sensitive and quantitative MAO-B detection in vivo. 11C limits the use of the radiotracer to centers with an on-site cyclotron and 11C radiochemistry expertise. 18F-labeling of SMBT-1 permits centralized production and regional distribution of the radiotracer. The in vitro binding kinetics of 18F-SMBT-1 demonstrated its reversible binding to MAO-B in human brains. The advantages of 18F-SMBT-1 in terms of its reversibility would allow us to perform repeated PET scans on the same subject. This ability will help in longitudinal quantification of astrogliosis and measurement of MAO-B occupancy via therapeutics.
18F-SMBT-1 showed excellent pharmacokinetics profiles, such as high uptake by brain after intravenous administration and rapid washout from normal brain tissues without significant defluorination in mice. These characteristics were comparable to those of 18F-THK-5351. One of the drawbacks of 11C-l-deprenyl-D2 is the existence of radiolabeled metabolites, such as R(−)-methamphetamine and R(−)-amphetamine, that can penetrate the blood–brain barrier and bind to monoamine transporters. Conversely, the reversible MAO-B PET tracer 11C-SL.25.1188 showed no radiolabeled metabolites capable of permeating the blood–brain barrier. However, this tracer showed slow pharmacokinetics due to slow metabolism (10,38). 18F-SMBT-1 was completely metabolized in mice without leaving harmful radiolabeled metabolite remnants. The dominant radiolabeled metabolite (M2) in mouse plasma was O-sulfated 18F-SMBT-1, which was not detectable in the mouse brain. A previous study demonstrated that O-sulfated THK-5351 did not penetrate the blood–brain barrier in mice and showed little binding to human brain homogenates (28). This evidence suggests that the in vivo metabolic profile of 18F-SMBT-1 could be suitable for MAO-B imaging in the brain.
CONCLUSION
18F-SMBT-1 is a promising candidate as a selective and reversible imaging tracer for MAO-B. Ongoing clinical study will allow us to classify the clinical utility of this tracer in vivo.
DISCLOSURE
This study was supported by Cino Ltd., a Grant-in-Aid for Young Scientists (18K15538), a Grant-in-Aid for Scientific Research (B) (18H02771), a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) (26117003), a Grant-in-Aid for Fostering Joint International Research (B) (19KK0212) from MEXT, the Strategic Research Program for Brain Science (JP19dm0107157) from the Japan Agency for Medical Research and Development, and a Grant-in-Aid for Joint Research by Young Researchers from Tohoku University Graduate School of Medicine and Shimadzu Science Foundation. Yukitsuka Kudo and Nobuyuki Okamura own stock in Clino Ltd. Ryuichi Harada, Shozo Furumoto, Yukitsuka Kudo, and Nobuyuki Okamura have a patent pending for the technology described in this article. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: How do the preclinical properties of a newly generated MAO-B tracer, 18F-SMBT-1, compare with those of 18F-THK-5351?
PERTINENT FINDINGS: 18F-SMBT-1 possess high affinity and high selectivity for MAO-B in a reversible binding fashion, with preferable pharmacokinetic and metabolic profiles.
IMPLICATIONS FOR PATIENT CARE: 18F-SMBT-1 has potential for detecting MAO-B–expressing astrocytosis in humans.
Acknowledgments
We thank the staff at the Cyclotron and Radioisotope Center of Tohoku University for the HM-12 cyclotron operation. We acknowledge the support of the Biomedical Research Core of Tohoku University Graduate School of Medicine.
Footnotes
Published online Jul. 9, 2020.
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication March 12, 2020.
- Accepted for publication June 15, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Preclinical Characterization of the Tau PET Tracer [18F]SNFT-1: Comparison of Tau PET Tracers
- Relationship between reactive astrocytes, by [18F]SMBT-1 imaging, with amyloid-beta, tau, glucose metabolism, and microgliosis in mouse models of Alzheimers disease
- Preclinical Characterization of the Tau PET Tracer [18F]SNFT-1: Comparison of Tau PET Tracers
- First-in-Humans Evaluation of 18F-SMBT-1, a Novel 18F-Labeled Monoamine Oxidase-B PET Tracer for Imaging Reactive Astrogliosis
- Assessing Reactive Astrogliosis with 18F-SMBT-1 Across the Alzheimer Disease Spectrum
- Imaging Neuroinflammation in Neurodegenerative Disorders
- Aquaporin 4 is differentially increased and depolarized in association with tau and amyloid-beta
- Astrocyte Biomarkers in Alzheimer Disease: A Systematic Review and Meta-analysis