


Abstract
Abnormal deposition of amyloid-β and hyperphosphorylated tau protein in the brain are the pathologic hallmark of Alzheimer disease (AD). Noninvasive detection of the lesions is considered an effective tool for early diagnosis and staging of AD. In the past decade, we developed 2-arylquinoline (2-AQ) derivatives as PET tau tracers. In this study, we synthesized new derivatives and evaluated their properties. Methods: Fifteen 2-AQ derivatives were labeled with 18F, and their binding to tau lesions was evaluated by autoradiography using AD brain sections. The binding affinity for the AD brain homogenates was assessed by an in vitro competitive binding assay with 18F-THK-5105. 18F-labeled derivatives were injected into mice via the tail vein, and their pharmacokinetics over the first 120 min after injection were evaluated by an ex vivo biodistribution study. Tracer metabolism analysis was also assessed in mice. Results: The average log P value was 2.80. This study revealed that 2-AQ derivatives having 18F-labeled side chains on benzene or position 7 of the quinoline showed significantly lower binding affinity for tau than 6-substituted quinoline derivatives. The 2-AQ derivatives labeled with 18F-fluoroethoxy, 18F-fluoropropoxy, and 18F-fluoro-polyethyleneglycol groups displayed slow clearance from blood or a high accumulation in bone, whereas derivatives labeled with the 18F-(3-fluoro-2-hydroxy)propoxyl group did not. 18F-THK-5151 had outstanding tau binding properties and pharmacokinetics. Furthermore, the properties of its optically pure (S)-enantiomer (THK-5351) were superior to those of the (R)-enantiomer (THK-5451), particularly in terms of its clearance from the brain and its resistance to defluorination in mice. Conclusion: The structure–activity relationship study of 2-AQ derivatives revealed the optimal structural features for tau imaging agents. On the basis of these results, 18F-THK-5351 ((S)-18F-THK-5151) was selected as a potential agent for tau imaging.
Alzheimer disease (AD) is the most common form of dementia, clinically characterized by memory impairment and affecting the activities of daily living. The typical pathologic hallmarks of AD are senile plaques and neurofibrillary tangles consisting of amyloid-β (Aβ) and hyperphosphorylated tau protein aggregates, respectively (1). In sporadic AD, Aβ aggregates are thought to accumulate in the brain because of impaired clearance of Aβ peptides. This process precedes the clinical manifestation of AD by more than a decade (2). Tau protein is a microtubule-associated protein and can be divided into 6 isoforms according to the number of microtubule-binding domains and the presence or absence of 2 N-terminal inserts in the adult human brain (3). The behavior of tau is regulated by posttranslational modifications including phosphorylation and acetylation, and the number of phosphorylations per molecule is significantly greater in the brains of AD and other tauopathies than in healthy brains (4,5). It is believed that these pathologic abnormalities precede brain atrophy and cognitive impairment (6); hence, they are regarded as targets for therapeutic intervention and diagnostic imaging of AD.
During the last decade, several radiolabeled Aβ binding probes were developed to visualize Aβ aggregates in AD brains by PET (7). The probes mainly consist of lipophilic heterocyclic rings, which allow them to cross the blood–brain barrier, and are labeled with 11C or 18F. Although 11C-Pittsburgh compound B is the most prevalent Aβ probe, 3 18F-labeled probes have already been approved by the Food and Drug Administration and the European Medicines Agency (8). The development of tau PET probes has been of increasing interest over the past few years (9). Tau PET imaging is expected to provide a better indication of the severity and prognosis of AD given the stronger association between neurodegeneration and tau pathology (10). Similar to Aβ probes, most tau probes are small molecules radiolabeled with 11C or 18F and have planar aromatic rings that enable them to bind to the β-sheet structure of tau aggregates. First-in-human PET studies in AD or other tauopathies of some tau probes have already been performed using novel tau tracers (11–13).
After screening more than 2,000 small molecules, we identified quinoline derivatives that could preferentially bind tau over Aβ (14). Since then, we have been developing 2-arylquinoline (2-AQ) derivatives for tau PET imaging (15–17) and 3 18F-labeled derivatives, 18F-THK-523, 18F-THK-5105, and 18F-THK-5117, have already been evaluated in clinical studies (13,18,19). In recent basic and clinical studies, 18F-THK-5117 demonstrated high binding selectivity and affinity for tau over Aβ pathology and better pharmacokinetics in the living brain than other THK probes (18). However, 18F-THK-5117 showed marked white matter retention in both healthy controls and AD patients (18). Additionally, the white matter radioactivity might spill over and result in an overestimation of the radioactivity in the adjacent gray matter in which neurofibrillary tangles are located (20,21). These observations prompted us to develop an optimized PET probe with lower background radioactivity in the brain than the existing probes. In this study, we performed a structure–activity relationship study of 18F-labeled 2-AQ derivatives and evaluated their preclinical characteristics to determine their suitability as tau PET probes.
MATERIALS AND METHODS
Synthesis of 2-AQ Derivatives
Methods for the synthesis and characterization data of 2-AQ derivatives are described in the supplemental data (available at http://jnm.snmjournals.org).
Radiosynthesis of 18F-2-AQ Derivatives
A mixture of the corresponding precursor (3 mg) and an activated 18F-KF (1.7 GBq) and Kryptofix222 (16 mg) in dimethyl sulfoxide was heated at 110°C for 10 min. As necessary, 2 M HCl were then added and stirred for an additional 3 min to remove the tetrahydropyranyl group, followed by neutralization with 4 M AcOK. The products were extracted with a Sep-Pak tC18 cartridge (Waters) and then purified by high-performance liquid chromatography (Inertsil ODS-4, 10 × 250 mm, 5 μm [GL Sciences, Inc.]; acetonitrile/20 mM NaH2PO4). The radiolabeled product was isolated using a Sep-Pak tC18 cartridge from the high-performance liquid chromatography fraction. For the injectable, the product was solubilized in saline with polysorbate 80 (<0.1%). The radiochemical purities of all 18F-2-AQ derivatives were greater than 99%. The radiochemical yields ranged from 11% to 72% (decay-corrected). The specific activities ranged from 16.1 to 118 GBq/μmol.
Log P Determination
Log P values were estimated by a reversed-phase high-performance liquid chromatography method as described in the supplemental data.
In Vitro Autoradiography
Experiments with human samples were performed under the regulations of the ethics committee of Tohoku University School of Medicine. Brain sections (8 μm, medial temporal lobe) from an AD patient (82-y-old woman) were incubated with each 18F-2-AQ derivative (0.22 MBq/mL) for 10 min at room temperature and then washed with water and 50% ethanol. The sections were exposed to a BAS-III imaging plate (Fuji Film) overnight. Autoradiograms were obtained using a Typhoon FLA 9500 (GE Healthcare Bio-Sciences). Gallyas–Braak silver staining and anti-Aβ immunostaining were performed with the adjacent sections (supplemental data).
In Vitro Binding Assays
According to the previous methods (16), the inhibitory constant (Ki) and dissociation constant (Kd) were determined using the AD brain homogenates of temporal gray matter and hippocampus, respectively. Details are described in the supplemental data.
Biodistribution Assay in Control Mice
Study protocols using animals were approved by the Institutional Animal Care and Use Committee of the Tohoku University Environmental and Safety Committee. Biodistribution assays in control mice were conducted by the method described in the supplemental data. In this study, a 2 min–to–10 min ratio of brain uptake was calculated and used as an index of the clearance rate of each 18F-2-AQ derivative.
Metabolite Analysis
The 18F-labeled tracer (18.5 MBq) was injected into male ICR mice (6 wk old; n = 3 per group) via the tail vein. Mice were sacrificed by decapitation under anesthesia at 2, 10, and 30 min after injection, and the hemibrain and cardiac blood were collected. The plasma fractions were deproteinized with acetonitrile (1:1.5 v/v) followed by centrifugation at 14,000g for 5 min (extraction efficiency, >94%). Brain tissue homogenized with phosphate-buffered saline (0.4 mL) was also deproteinized with acetonitrile (1 mL) as described above (extraction efficiency, >83%). The supernatants were applied to a high-performance thin-layer chromatography silica gel 60 RP-18 W plate (Merck KGaA), which was then developed with acetonitrile/20 mM NaH2PO4 (pH 6.5) (=1/1), dried, and exposed to a BAS-III imaging plate overnight. Autoradiograms were obtained and analyzed using a Typhoon FLA 9500 and Multi Gauge software (version 3.0; Fujifilm).
RESULTS
Preparation of 2-AQ Derivatives
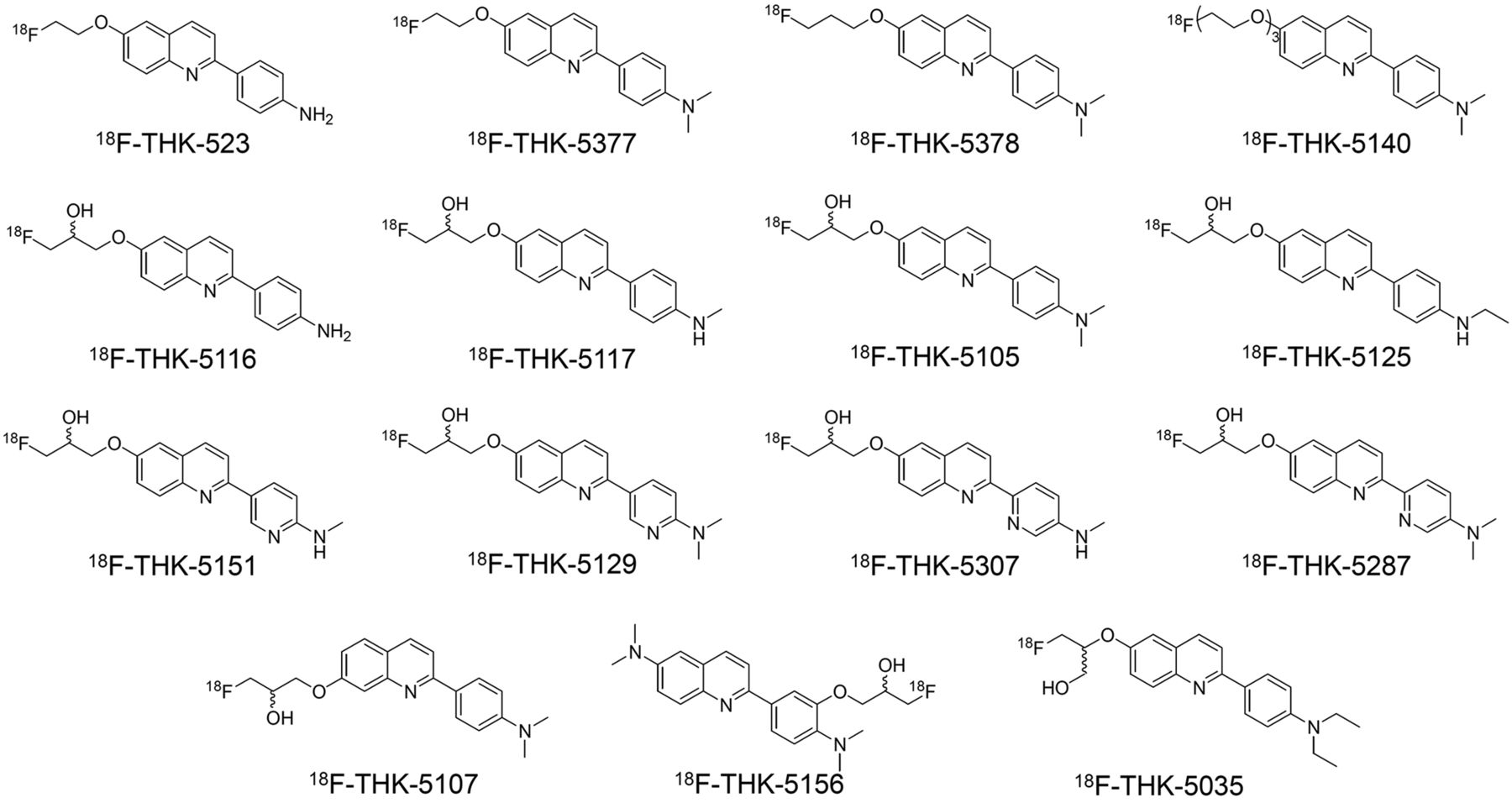
We prepared 15 2-AQ derivatives for this structure–activity relationship study (Fig. 1). The fluoroalkoxylated and fluoropegylated derivatives and their tosylate precursors were synthesized via reactions of the corresponding side chains and 2-AQ frameworks (Supplemental Schemes 1 and 2). The 2-AQ framework was basically constructed by coupling quinoline triflate/chloride and phenylboronic/pyridinylboronic acid pinacol ester followed by a Mitsunobu reaction with a fluoro side chain (Supplemental Schemes 3–7). The frameworks of 2-(5-aminopyridin-2-yl)quinoline derivatives were constructed by coupling 2-chloroquinoline and 2-(tributylstannyl)pyridine (Supplemental Schemes 8 and 9). The average molecular weight of the 2-AQ derivatives was 337.7 (282.31–398.47), and the average log P value was 2.80 (1.47–4.39) (Supplemental Table 1).

Chemical structures of 18F-2-AQ derivatives.
In Vitro Autoradiography
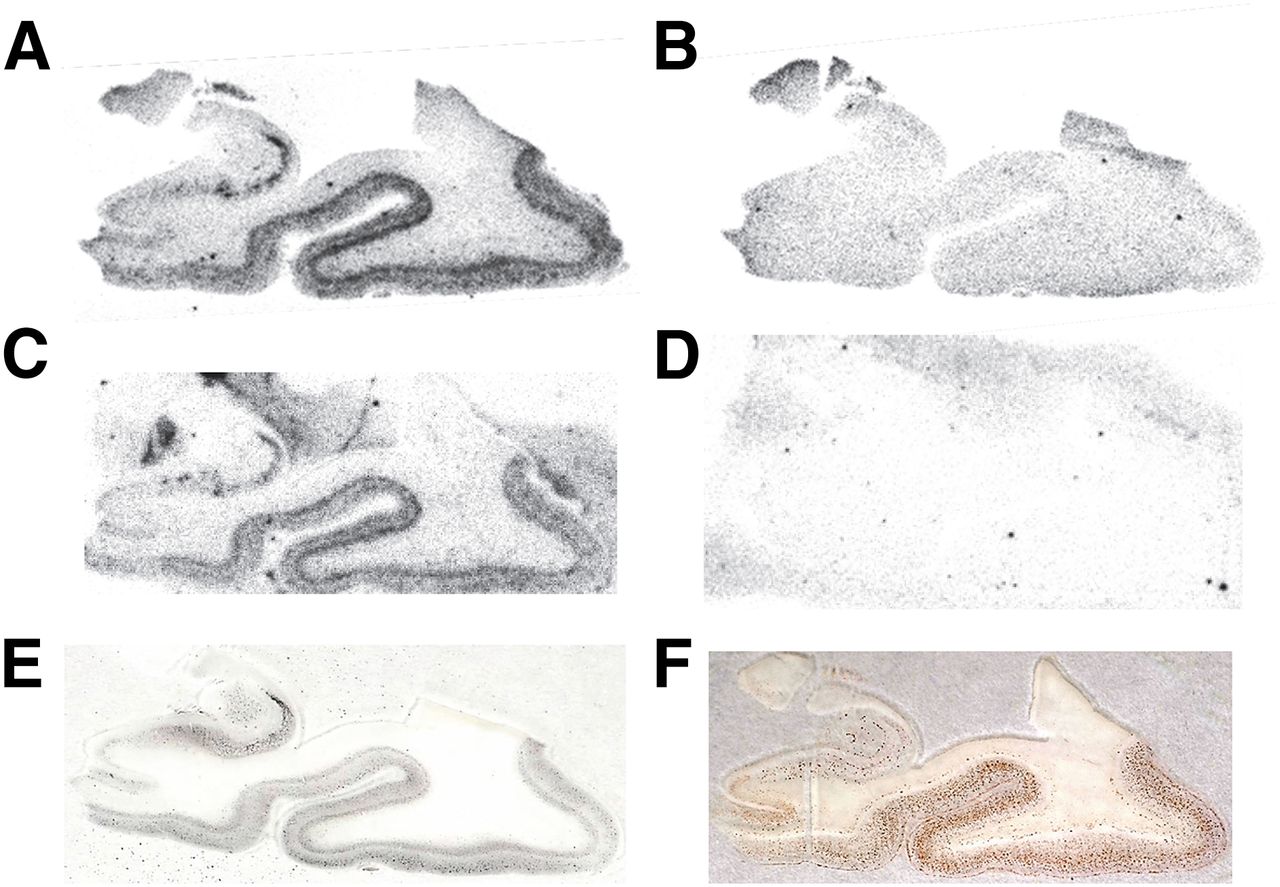
In the autoradiography assay, we evaluated the binding characteristics of the 18F-2-AQ derivatives for medial temporal lesions of the AD brain. The semiquantitative results are summarized in Table 1. Most derivatives with a side chain on position 6 of the quinoline showed tau-characteristic laminar distribution close to the boundary between the gray matter and white matter. Among them, 18F-THK-5117 and 18F-THK-5151 showed strong and highly selective binding for tau (Figs. 2A and 2C, respectively) and smaller amounts of nonspecific binding in white matter (Figs. 2B and 2D, respectively). A (3-fluoro-1-hydoxy)-2-propoxyl derivative (18F-THK-5035) showed a weak binding to tau in the AD brain. Conversely, other derivatives with a side chain on position 7 of the quinoline (18F-THK-5107) or benzene (18F-THK-5156) did not show any binding to both tau and Aβ lesions observed by Gallyas–Braak staining (Fig. 2E) and Aβ immunostaining (Fig. 2F), respectively.
Autoradiographic Characteristics and Ki and Kd Values of THK Compounds

Autoradiograms of representative 2-arylquinoline derivatives and images of Gallyas–Braak staining and Aβ immunostaining. (A) 18F-THK-5117. (B) 18F-THK-5117 + 25 µM THK-5117. (C) 18F-THK-5151. (D) 18F-THK-5151 + 25 µM THK-5151. (E) Gallyas–Braak staining. (F) Aβ immunostaining.
In Vitro Binding to AD Brain Homogenate
The binding affinity of 2-AQ derivatives was evaluated by saturation binding assays to determine their Kd values or competitive binding assay with 18F-THK-5105 using AD brain homogenate (Table 1). With the exception of THK-5116, the 6-subsituted quinoline (3-fluoro-2-hydroxy)-1-propoxy derivatives had lower Ki values than other derivatives. All the derivatives that showed no binding to tau lesions in the autoradiography assay had Ki values of more than approximately 100 nM. The Kd values of the tested derivatives ranged from 2.60 to 11.5 nM. N,N-dimethylamino compounds tended to have lower Kd values than their corresponding N-methylamino derivatives, similar to the trend observed for the Aβ probe (22).
Biodistribution in Control Mice
The pharmacokinetics of the 18F-2-AQ derivatives were evaluated by ex vivo biodistribution assay, and the results are summarized in Table 2. All derivatives showed high initial brain uptake ranging from 2.72 percentage injected dose per gram (%ID/g) to 9.20 %ID/g. Highly lipophilic 18F-THK-5378 and 18F-THK-5035 exhibited slower brain uptake with a peak at 10 min after injection, whereas the radioactivity in the brain of other derivatives decreased time-dependently after 2 min after injection. The highest three 2 min–to–10 min ratios of brain uptake were 3.6 for 18F-THK-5151, 2.8 for 18F-THK-5307, and 2.2 for 18F-THK-5117 (16). Comparison of the radioactivity in blood and bone at 120 min after injection revealed that the structure of the side chain largely affects the pharmacokinetic characteristics of the radiotracer in mice. High blood radioactivity retention both decreases the signal-to-background ratio and increases bone accumulation due to defluorination of the 18F-labeled radiotracer. The 2-AQ derivatives with either fluoropropoxyl or 3-fluorohydroxypropoxyl side chains showed low blood radioactivity (0.22–0.77 %ID/g). By comparison, fluoroethoxyl and fluoropegylated derivatives demonstrated higher blood radioactivity of more than 1.5 %ID/g at 120 min after injection. In the bone, the radioactivity accumulation of the fluoropropoxyl derivative (THK-5378) was the highest (15.5 %ID/g) of all tested compounds and marked with THK-5377 and THK-5140 (5.39 and 6.37 %ID/g, respectively). No noticeable accumulation in the bone was observed with the other derivatives.
Pharmacokinetic Properties of Compounds in Mice
Evaluation of THK-5151 Enantiomers
From the results of autoradiography and pharmacokinetic studies, we selected THK-5151 as the most promising candidate for tau PET imaging. THK-5151 has a chiral center in its side chain and thus has 2 optical isomers (Fig. 3A), and it was anticipated that the biologic activities would be different between the enantiomers (23). Therefore, we synthesized the THK-5151 enantiomers and evaluated their properties as tau imaging probes (Supplemental Schemes 10 and 11; Supplemental Fig. 1). The autoradiograms of 18F-THK-5151 enantiomers used to evaluate their binding properties were apparently indistinguishable from each other. The Kd value for binding of the (S)-enantiomer (18F-THK-5351, 2.9 nM) to AD brain homogenate was lower than that of the (R)-enantiomer (18F-THK-5451, 28 nM). Obvious differences were demonstrated in a pharmacokinetic study in mice (Figs. 3B and 3C). The (S)-enantiomer had a faster clearance from the brain and blood than 18F-THK-5451, and the brain uptake ratios after 10 min after injection of 18F-THK-5351 were much higher than those of 18F-THK-5451 although the brain-to-blood ratios of both enantiomers were around 1.0 after 10 min after injection (Table 3). Furthermore, the bone radioactivity of 18F-THK-5451 was about 5 times higher than that of 18F-THK-5351 at 120 min after injection (Fig. 3D).

(A) Chemical structures of THK-5351 and THK-5451. (B–D) Time–activity curves of 18F-THK-5351 (blue) and 18F-THK-5451 (red) in mice brain (B), blood (C), and bone (D).
Pharmacokinetic Properties of 18F-THK-5351 and 18F-THK-5451 in Mice
Metabolite Analysis
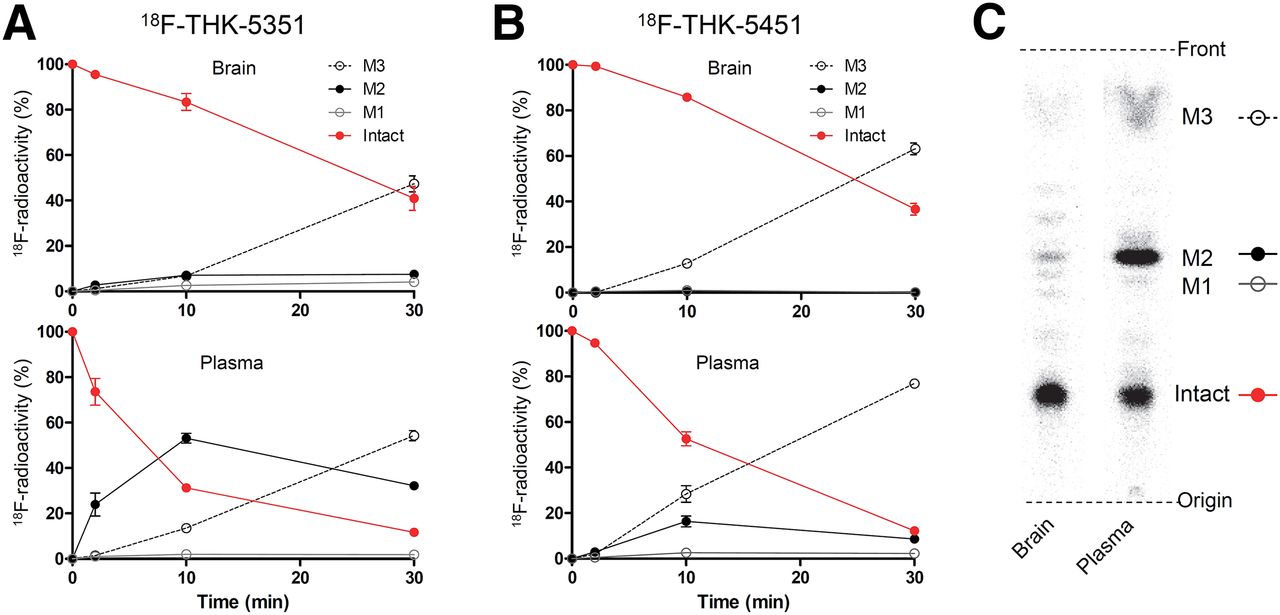
We analyzed the metabolism of 18F-THK-5351 and 18F-THK-5451 in control mouse brain and plasma. Time–activity curves of radiometabolites in brain and plasma are indicated in Figure 4. Both tracers gradually degraded in plasma after injection, and 2 prominent polar radiometabolites (M2 and M3 in Fig. 4) were observed. In the brain, degradation of both tracers was relatively slow compared with that in plasma, and the unchanged radiotracer decreased nearly 40% at 30 min after injection.
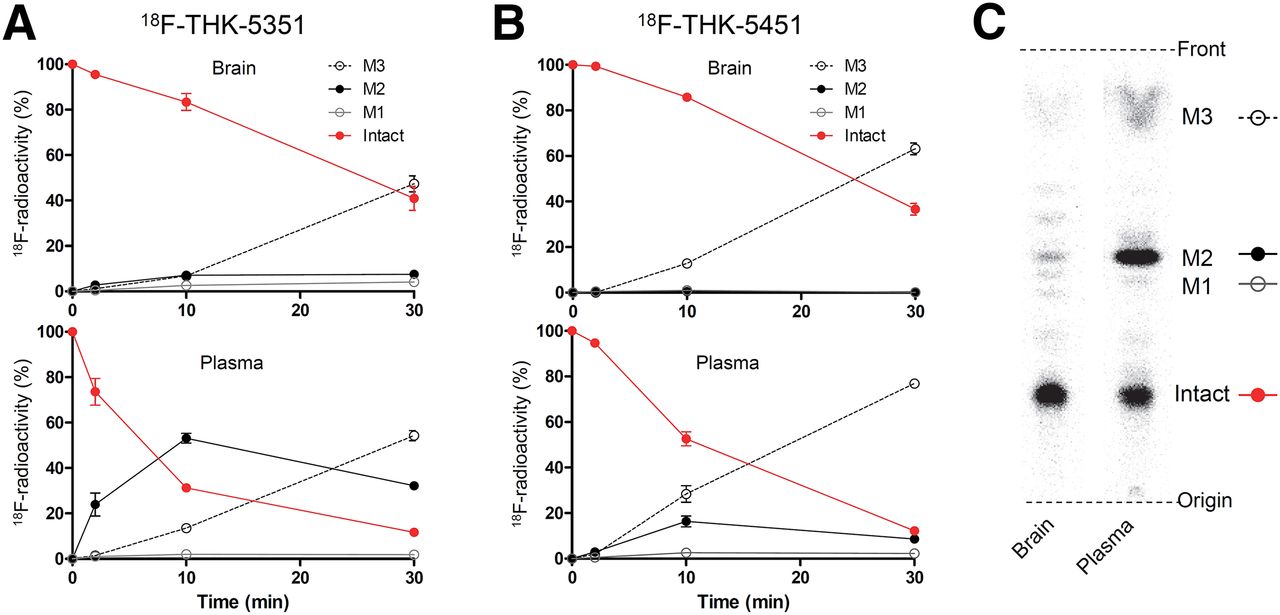
In vivo metabolism of 18F-THK-5351 and 18F-THK-5451 in mice. Time–activity curves of unchanged radiotracer and radiometabolites in plasma and brain for 18F-THK-5351 (A) and 18F-THK-5451 (B). (C) Representative radio–thin-layer chromatography images in plasma and brain at 10 min after injection.
DISCUSSION
Previously, we demonstrated the potential of THK compounds to image AD tau pathology in a PET study, although visual PET image inspection was complicated as a result of the relatively high nonspecific binding of the compounds in white matter (13,19). For further structural optimization of THK compounds, we synthesized 2-AQ derivatives including pyridine derivatives and evaluated the relationship between their structures and performance as a tau PET probe.
With respect to binding affinity, we assessed Kd or Ki values of the derivatives. Generally, the lower value meant higher binding affinity. The Kd values of the (3-fluoro-2-hydroxy)-1-propoxy derivatives were below 20 nM, suggesting that the derivatives have sufficient binding affinity to be used as tau imaging probes (10). Compared with the Kd values, the Ki values were higher except that for THK-5105, and their variation may have been caused by differences in the binding sites between the inhibitors and 18F-THK-5105. Difference of Kd values between enantiomers of 18F-THK-5151 was assumed to be caused by differences in binding kinetics as well as 18F-THK-5105 (24).
The lipophilicity of the radiotracer is one of the primary factors that contributes to its pharmacokinetics in the brain. Ideally, a tau PET tracer should be cleared quickly from the brain to avoid prolongation of the time required to reach equilibrium in a PET study and to increase the signal-to-background ratio (10). Radiotracers need sufficient lipophilicity with log P values of 1–3 to cross the blood–brain barrier; however, too high of a lipophilicity results in a high level of nonspecific binding and slow clearance from the brain (22,25). Except for THK-523, the 2-AQ derivatives showed a common parabolic relationship between their log P values and brain uptake at 2 min after administration similar to what was observed in other studies (Fig. 5A) (22,25). Interestingly, 2 min–to–10 min ratios of brain uptake showed a linear negative correlation with log P values (r = −0.72, P = 0.0023, Fig. 3B), whereas 2 min–to–30 min and 2 min–to–60 min ratios did not (r = −0.60, P = 0.017, and r = −0.39, P = 0.15, respectively). These results suggest that a lower lipophilic derivative tends to exhibit faster washout from the brain in the early phase after administration, and log P value could be useful for estimating the brain kinetics of the early phase. In previous studies, 2 min–to–30 min or 2 min–to–60 min ratios of brain uptake were used as an index of the tracer clearance (16,22). In this study, however, the clearance rates of some highly lipophilic compounds with high brain uptake were overestimated when they were evaluated by a 2 min–to–60 min ratio. Additionally, the brain uptake of other test compounds with fast brain clearance, such as 18F-THK-5129, bottomed out by 30 min after injection; thus, their clearance rates were underestimated by a 2 min–to–30 min ratio. These results also support the use of a 2 min–to–10 min ratio as an index of the tracer clearance. Judging from the 2 min–to–10 min ratio, 2-/3-methylaminopyridine is a favorable partial structure of 2-AQ scaffold for rapid tracer clearance from the brain.

Relationship between lipophilicity (log P) and brain uptake of 2-arylquinoline derivatives in early phase after administration. (A) Scatterplot between log P and brain uptake 2 min after injection. (B) Scatterplot between log P and 2 min–to–10 min ratios of brain uptake, which was evaluated by two-tailed Pearson product–moment correlation analysis.
The effects of varying the 18F-labeled side chains were prominently demonstrated in the biodistribution assay (Table 2). Previously, Pan et al. demonstrated that 18F-2-fluoroethanol had slow clearance from major organs including the brain, and this phenomenon was believed to be caused by intracellular trapping of radiometabolites generated by oxidative metabolism (26,27). By contrast, the radioactive clearance of 18F-3-fluoropropanol was rapid, but remarkable bone accumulation was observed (26). Exactly the same trends as their study were observed in this study, suggesting the need for further optimization of these 18F-labeling groups. Considering the results that (3-fluoro-2-hydroxy)-1-propoxy derivatives were commonly cleared from blood smoothly and only negligibly accumulated in bone, addition of the hydroxyl group to the fluoropropoxyl group could be an effective way to optimize the side chain. However, the modification raised a new issue concerning a chirality at the hydroxyl carbon.
(S)-enantiomer, 18F-THK-5351, showed faster kinetics in the brain and blood and lower radioactivity uptake in the bone than the (R)-enantiomer. Similar differences between enantiomers were also observed in the metabolism of 18F-THK-5105 and revealed to be caused by stereospecific hepatic metabolism (24). There was a large difference in the generation of M2 at 10 min after injection between 18F-THK-5351 and 18F-THK-5451, suggesting that it is possible that the rapid metabolism to M2 stimulates the elimination of 18F-THK-5351 from the mouse body.
In contrast to the observations in plasma, the amount of M2 was less obvious in the brain, indicating that M2 was less likely to cross the blood–brain barrier (Fig. 4). Therefore, M2 would have little effect on the quantitative analysis of the brain kinetics. However, M3 was observed in the brain to a similar extent as in plasma. This result suggests that M3 is likely to cross the blood–brain barrier and may potentially affect the quantitative analysis. Thus, it is important to clarify whether M3 is generated in human plasma or not in a human PET study.
The mechanism responsible for the defluorination of 18F-THK-5151 enantiomers was not clear, but the difference in bone accumulation seemed to be caused by the stereospecificity of some enzymatic defluorination pathways (28,29). There are several (3-18F-fluoro-2-hydroxy)-1-propoxy–containing PET tracers currently under development, and our study suggests that preparation of pure single enantiomers may produce significant improvement in their properties (30,31).
In summary, 18F-THK-5351 (18F-(S)-THK-5151), which has both methylaminopyridine and 3-fluorohydroxypropoxyl structures, showed the most favorable properties as a tau radiotracer candidate. However, it is generally difficult to predict and conclude the clinical utility of a PET tracer only from the preclinical data. In the case of 18F-THK-5117, which has a lower lipophilicity than 18F-THK-5105, the tracer showed faster brain kinetics in both mice and humans than 18F-THK-5105 (Table 4), leading to higher-contrast PET images than 18F-THK-5105. Because of the same reason, 18F-THK-5351 could exhibit faster brain kinetics and produce better-contrast PET images than 18F-THK-5117. Actually, preliminary PET study in humans demonstrated faster brain kinetics of 18F-THK-5351 than 18F-THK-5117 (Table 4) (32). However, such approach for optimization might not resolve an issue of off-target binding to the basal ganglia commonly observed in both 18F-THK-5105 and 18F-THK-5117. Further optimization study to address the issue of off-target binding through a different approach may be necessary in the future.
Summary of Representative THK Tau Probe Properties
CONCLUSION
This study revealed the influence of the 18F-labeled side chain on the characteristics of brain imaging radiotracers. 18F-THK-5351 was selected as the most promising candidate and is currently under clinical evaluation for its utility as an AD tau PET radiotracer.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported by GE Healthcare; CLINO Co. Ltd.; Sumitomo Electric Industries, Ltd.; Group CSR Foundation; a grant-in-aid for Scientific Research (B) (25293259); a grant-in-aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) (26117003) from MEXT; the Industrial Technology Research grant program of the NEDO in Japan (09E51025a); and Health and Labor Sciences Research grants from the Ministry of Health, Labor, and Welfare of Japan. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank the staff at Cyclotron and Radioisotope Center of Tohoku University for operation of the HM-12 cyclotron.
Footnotes
Published online Dec. 23, 2015.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication September 28, 2015.
- Accepted for publication November 29, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}