


Visual Abstract
Abstract
A neuroinflammatory reaction in Alzheimer disease (AD) brains involves reactive astrocytes that overexpress monoamine oxidase-B (MAO-B). 18F-(S)-(2-methylpyrid-5-yl)-6-[(3-fluoro-2-hydroxy)propoxy]quinoline (18F-SMBT-1) is a novel 18F PET tracer highly selective for MAO-B. We characterized the clinical performance of 18F-SMBT-1 PET across the AD continuum as a potential surrogate marker of reactive astrogliosis. Methods: We assessed 18F-SMBT-1 PET regional binding in 77 volunteers (76 ± 5.5 y old; 41 women, 36 men) across the AD continuum: 57 who were cognitively normal (CN) (44 amyloid-β [Aβ]–negative [Aβ−] and 13 Aβ-positive [Aβ+]), 12 who had mild cognitive impairment (9 Aβ− and 3 Aβ+), and 8 who had AD dementia (6 Aβ+ and 2 Aβ−). All participants also underwent Aβ and tau PET imaging, 3-T MRI, and neuropsychologic evaluation. Tau imaging results were expressed in SUV ratios using the cerebellar cortex as a reference region, whereas Aβ burden was expressed in centiloids. 18F-SMBT-1 outcomes were expressed as SUV ratio using the subcortical white matter as a reference region. Results: 18F-SMBT-1 yielded high-contrast images at steady state (60–80 min after injection). When compared with the Aβ− CN group, there were no significant differences in 18F-SMBT-1 binding in the group with Aβ− mild cognitive impairment. Conversely, 18F-SMBT-1 binding was significantly higher in several cortical regions in the Aβ+ AD group but also was significantly lower in the mesial temporal lobe and basal ganglia. Most importantly, 18F-SMBT-1 binding was significantly higher in the same regions in the Aβ+ CN group as in the Aβ− CN group. When all clinical groups were considered together, 18F-SMBT-1 correlated strongly with Aβ burden and much less with tau burden. Although in most cortical regions 18F-SMBT-1 did not correlate with brain volumetrics, regions known for high MAO-B concentrations presented a direct association with hippocampal and gray matter volumes, whereas the occipital lobe was directly associated with white matter hyperintensity. 18F-SMBT-1 binding was inversely correlated with Mini Mental State Examination and the Australian Imaging Biomarkers and Lifestyle’s Preclinical Alzheimer Cognitive Composite in some neocortical regions such as the frontal cortex, lateral temporal lobe, and supramarginal gyrus. Conclusion: Cross-sectional human PET studies with 18F-SMBT-1 showed that Aβ+ AD patients, but most importantly, Aβ+ CN individuals, had significantly higher regional 18F-SMBT-1 binding than Aβ− CN individuals. Moreover, in several regions in the brain, 18F-SMBT-1 retention was highly associated with Aβ load. These findings suggest that increased 18F-SMBT-1 binding is detectable at the preclinical stages of Aβ accumulation, providing strong support for its use as a surrogate marker of astrogliosis in the AD continuum.
The neuropathologic hallmarks of Alzheimer disease (AD)—neurofibrillary tangles (NFT) of tau protein and amyloid-β (Aβ) plaques—are accompanied by reactive gliosis, cellular degeneration, and diffuse synaptic and neuronal loss (1). Astrocytes are the most abundant glial cells in the brain and are involved in several functions critical for the normal functioning and preservation of brain homeostasis (2,3). Loss of these regulatory and compensatory mechanisms in astrogliosis likely translates into increased vasculature-related vulnerability (4), affecting the brain’s ability to compensate for the accumulating Aβ and tau burden and impaired cerebrovascular function in AD. The complex spectrum of toxic and protective pathways (5,6) of reactive astrogliosis plays a crucial role in the pathophysiology of AD (7–9) and other neurodegenerative conditions (10–16). Reactive astrogliosis and microgliosis have been observed around both dense-core Aβ plaques and neurofibrillary tangles, and they are believed to contribute greatly to neurodegeneration throughout the course of AD (9,17). In contrast to microgliosis (18), reactive astrogliosis occurs early in the disease (19), and is a particularly attractive target for understanding its contribution to the development of AD dementia, and, as such, is a potential therapeutic target for AD (20).
Reactive astrocytes overexpress monoamine oxidase B (MAO-B) (21), and molecular neuroimaging studies have used MAO-B tracers such as 11C-l-deprenyl-D2 and 11C-SL25.1188 (22,23) as surrogate markers of astrogliosis (15,19,24–26). Some of these studies have shown that reactive astrogliosis is observed at the prodromal stages in both sporadic and familial AD (19,24). 11C-BU99008—a tracer for the imidazoline 2 binding sites, with some partial binding to MAO-B (27)—has also been proposed as a surrogate marker of astrogliosis (28,29). Unfortunately, these tracers are labeled with 11C, which has a 20-min half-life, preventing widespread clinical or research applications.
A novel 18F MAO-B tracer, 18F-(S)-(2-methylpyrid-5-yl)-6-[(3-fluoro-2-hydroxy)propoxy]quinoline (18F-SMBT-1), with high in vitro binding affinity (dissociation constant, 3.5 nM) and selectivity to MAO-B has recently been developed (30). Comparison of in vitro 18F-SMBT-1 binding against MAO-B activity showed SMBT-1 binding to be highly correlated with the regional activity of MAO-B in AD brain tissue homogenates (30). Autoradiography analysis showed significantly higher specific binding in the frontal cortex tissue section from an AD patient than in a control (30), and this binding was completely displaced by the selective MAO-B inhibitor lazabemide (30).
In a first-in-humans study (31), 18F-SMBT-1 showed robust entry into the brain and reversible binding kinetics, with quantitative and semiquantitative measures of 18F-SMBT-1 binding being strongly associated. More than 85% of 18F-SMBT-1 signal was blocked by selegiline across the brain, indicating high selectivity for MAO-B and low nonspecific binding. 18F-SMBT-1 regional binding followed the known regional brain distribution of MAO-B (R2 = 0.84) while also capturing the known MAO-B increases with age (R2 > 0.94), suggesting that 18F-SMBT-1 can potentially be used as a surrogate marker of reactive astrogliosis in AD.
The aim of this study was to characterize 18F-SMBT-1 binding across the AD continuum, assessing its relation to Aβ and tau pathology burden, as well as brain volumetrics, white matter hyperintensity, and cognitive performance.
MATERIALS AND METHODS
Participants
In total, 77 nonsmoking elderly participants (76 ± 5.5 y old; age range, 58–89 y; 40 women, 35 men) were included in the study: 57 cognitively normal (CN) controls, 12 subjects meeting the criteria for mild cognitive impairment (MCI) (32), and 8 subjects meeting the NINDS-ADRDA and NIAA-AA criteria for AD (33). Subjects with AD and MCI were recruited from memory disorder clinics. CN subjects were recruited by advertisement in the community. Some participants (46 CN, 6 MCI, and 4 AD) were included in a previous study (31).
All participants were screened for unstable medical or psychiatric disease and concomitant medication. Participants with known use of MAO-B inhibitors or with a diagnosis of a psychiatric disorder were excluded. For participants with a recognized memory impairment, this information was collected from the next of kin or a caregiver. The study protocol was approved by the Austin Health Human Research Ethics Committee, and all participants gave written informed consent.
A detailed neuropsychologic evaluation is provided in the supplemental materials (available at http://jnm.snmjournals.org).
Image Acquisition
All SMBT-1 PET scans were acquired on a Philips TF64 PET/CT device. Aβ and tau PET scans were acquired on 1 of 2 scanners, a Philips TF64 PET/CT or a Siemens Biograph mCT. A low-dose CT scan was obtained for attenuation correction.
18F-SMBT-1 PET
All 77 participants underwent 18F-SMBT-1 PET. As previously reported (31), 10 nondemented participants (4 CN and 6 MCI) underwent a 90-min dynamic scan after receiving an intravenous bolus injection of 185 MBq (±10%) of 18F-SMBT-1. The remaining 67 participants received an intravenous bolus injection of 185 MBq (±10%) of 18F-SMBT-1 and underwent a 20-min emission scan (4 × 5 min) starting at 60 min after injection.
18F-SMBT-1 was synthesized in-house in the Department of Molecular Imaging and Therapy, Austin Health, as previously described (31). 18F-SMBT-1 yielded more than 95% radiochemical purity after high-performance liquid chromatography purification, with an average decay-corrected radiochemical yield of 40%. The molar activity at the end of 18F-SMBT-1 synthesis was more than 400 GBq/μmol.
Aβ PET
All 77 participants underwent Aβ PET imaging with 18F-flutemetamol (n = 3), 18F-florbetapir (n = 2), or 18F-NAV4694 (n = 72) to ascertain Aβ status. 18F-NAV4694 and 18F-florbetapir were synthesized in-house in the Department of Molecular Imaging and Therapy, Austin Health, as previously described (34,35). 18F-flutemetamol was manufactured by Cyclotek Pty Ltd. The 18F-NAV4694 and 18F-florbetapir PET acquisitions consisted of 20-min (4 × 5 min) dynamic scans obtained at 50 min after an intravenous bolus injection of 200 MBq (±10%) of the tracers. Similarly, the18F-flutemetamol PET acquisition also was a 20-min (4 × 5 min) scan starting at 90 min after injection of 185 MBq (±10%) of the tracer. All Aβ imaging results were expressed in centiloids (34–38). In a subset of participants (n = 31) who had available longitudinal Aβ imaging data, rates of Aβ accumulation—expressed as centiloids/y—were generated from the linear regression of the data as previously described (39).
Tau PET
Seventy-four participants underwent tau imaging with either 18F-MK6240 (n = 70) or 18F-PI2620 (n = 2). Both tau imaging tracers were synthesized in-house in the Department of Molecular Imaging and Therapy, Austin Health, as previously reported (40,41). The 18F-MK6240 PET acquisition consisted of a 20-min (4 × 5 min) dynamic scan obtained at 90 min after an intravenous bolus injection of 185 MBq (±10%) of the tracer. The 18F-PI2620 PET acquisition consisted of a 20-min (4 × 5 min) dynamic scan acquired at 80 min after an intravenous bolus injection of 200 MBq (±10%) of the tracer. All tau imaging results were expressed as SUV ratios using the cerebellar cortex as a reference region. A metatemporal region comprising the entorhinal cortex, amygdala, hippocampus, parahippocampal gyrus, fusiform gyrus, and inferior and middle temporal gyri—as adapted from a previous publication (42)—was used to determine tau status and for correlational analysis.
MRI
Seventy-three participants underwent structural MRI on a 3-T TIM Trio scanner (Siemens Medical Solutions) to obtain high-resolution T1-weighted anatomic magnetization-prepared rapid gradient-echo and fluid-attenuated inversion recovery sequences.
Image Analysis
Aβ and tau PET scans were spatially normalized using CapAIBL (38). The standard centiloid method was applied to determine Aβ burden (36). A threshold of 20 centiloids was used to categorize participants as high Aβ (Aβ-positive [Aβ+]) or low Aβ (Aβ-negative [Aβ−]) (43). Thresholds of 1.19 SUV ratio for 18F-MK6240 and 1.20 SUV ratio for 18F-PI2620 in the metatemporal composite region (42) were used to categorize participants as high tau (tau-positive) or low tau (tau-negative). As previously described (31), assessment of the stability of potential reference regions for 18F-SMBT-1 across age and across groups showed no associations with age, but the cerebellar cortex was significantly higher in Aβ+ CN subjects than in Aβ− CN subjects, precluding its use as a reference region. Therefore, 18F-SMBT-1 regional SUVs at 60–80 min after injection were normalized using the subcortical white matter as a reference region to generate SUV ratios. 18F-SMBT-1 PET images were spatially normalized using CapAIBL, and no correction for partial-volume effects was applied.
The T1-weighted magnetization-prepared rapid gradient-echo images for all participants were first segmented into gray matter, white matter, and cerebrospinal fluid using an expectation maximization algorithm (44) and subsequently used to measure hippocampal, gray matter, white matter, and ventricular volumes. The hippocampus region of interest was extracted using a multiatlas approach based on the Harmonized Hippocampus Protocol (45). Cortical volumes were normalized by total intracranial volume. The white matter hyperintensity volume was quantified from fluid-attenuated inversion recovery images using the HyperIntensity Segmentation Tool (46).
Statistical Analyses
All statistical analyses were performed with JMP Pro (version 16.0; SAS Institute Inc.) for Macintosh (Apple). Data are presented as mean ± SD unless otherwise stated. Groups were compared using the Tukey honest significance test followed by a Dunnett test against Aβ− CN. Effect size was measured with Cohen d. Correlations were assessed by Spearman and Pearson correlation coefficients. Vertexwise correlations between 18F-SMBT-1 and Aβ and tau imaging were performed with CapAIBL. The 2 participants who underwent tau imaging with 18F-PI2620 were excluded from the correlational analysis. Group comparisons of 18F-SMBT-1 and correlations between SMBT-1 and Aβ or tau burdens were adjusted for age and sex. Correlations between 18F-SMBT-1 and brain volumes were adjusted for age, sex, and Aβ and tau burdens. Correlations between 18F-SMBT-1 and cognitive parameters were also adjusted for age, sex, hippocampal volume, and Aβ and tau burdens. Significance was set at a P value of less than 0.05, uncorrected for multiple comparisons.
RESULTS
Table 1 shows the demographics of the clinical groups. There were no significant differences in age between groups. The MCI group had twice as many men as women, in contrast to the CN and AD groups, which had more women than men. The MCI and AD groups had significantly fewer years of education. As expected, the MCI and AD groups had cognitive performance significantly worse than the CN group (Table 1). The AD group had significantly more atrophic hippocampi and cortical gray matter, more extensive white matter hyperintensity, higher Aβ and tau burdens, and a higher prevalence of APOE4 (Table 1). The demographics of participants classified by Aβ status is provided in Supplemental Table 1.
Demographics
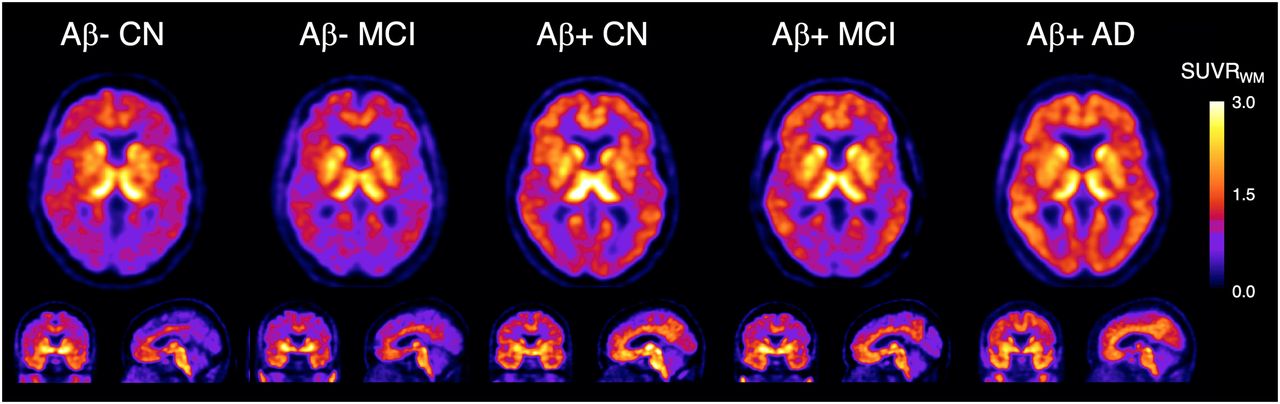
18F-SMBT-1 yielded high-contrast SUV images at steady state (60–80 min after injection). Figure 1 shows representative 18F-SMBT-1 images from Aβ− CN, Aβ+ CN, Aβ− MCI, Aβ+ MCI, and Aβ+ AD subjects. When the clinical groups were compared with the CN group, 18F-SMBT-1 binding was significantly higher in several regions in the AD group (including the 2 Aβ− “AD” subjects), namely the posterior cingulate gyrus, supramarginal gyrus, and lateral occipital lobe, but also was significantly lower in the globus pallidus, hippocampus, and parahippocampal gyrus.

Representative transaxial, coronal, and sagittal 18F-SMBT-1 PET images in Aβ− CN subject (84-y-old man; MMSE, 28; CDR, 0; 7 centiloids); Aβ− MCI subject (69-y-old woman; MMSE, 27; CDR, 0.5; 9 centiloids); Aβ+ CN subject (72-y-old man; MMSE, 29; CDR, 0; 24 centiloids); Aβ+ MCI subject (72-y-old man; MMSE, 27; CDR, 0.5; 144 centiloids); and Aβ+ AD subject (78-y-old woman; MMSE, 25; CDR, 1; 173 centiloids). Aβ− CN and MCI participants show normal distribution of 18F-SMBT-1 in brain, highlighting cortical areas with high concentration of MAO-B such as basal ganglia, thalamus, mesial temporal lobe, and anterior cingulate gyrus, as well as different nuclei in brain stem. Higher cortical 18F-SMBT-1 binding is observed in Aβ+ CN, MCI, and AD participants, with binding extending to frontal cortex, temporal lobe, occipital lobe, and posterior cingulate gyrus. SUVRWM = SUV ratio using white matter as reference region.
Then, the clinical groups were classified on the basis of their Aβ status. Of the 57 CN subjects, 44 were Aβ− and 13 Aβ+. Of the 12 MCI subjects, 9 were Aβ− and 3 Aβ+, whereas of the 8 patients with probable AD, 6 were Aβ+ and 2 Aβ−. When compared with the Aβ− CN group, there were no significant differences in 18F-SMBT-1 binding in either the Aβ− MCI group or the Aβ− AD group (Supplemental Table 2). In contrast, when compared with the Aβ− CN group, 18F-SMBT-1 binding was significantly higher in several cortical regions in the Aβ+ AD group (Fig. 2), namely the posterior cingulate gyrus, supramarginal gyrus, lateral occipital lobe, gyrus angularis, and primary visual cortex, but also was significantly lower in the globus pallidus and hippocampus, with Cohen effect sizes (d) ranging from 2.65 and 2.36 in the lateral occipital lobe and supramarginal gyrus, respectively, to −1.66 and −1.88 in the hippocampus and globus pallidus, respectively (Supplemental Table 2). Several regions were also significantly higher in the Aβ+ CN group than in the Aβ− CN group (Fig. 2). In addition to the posterior cingulate gyrus, supramarginal gyrus, and lateral occipital lobe—the same regions as those with high binding in the Aβ+ AD group—18F-SMBT-1 binding was also significantly higher in the orbitofrontal, lateral, and inferior temporal gyri. 18F-SMBT-1 binding was also significantly lower in the globus pallidus (Supplemental Table 2). The effect sizes (d) ranged from 1.21 and 1.00 in the supramarginal gyrus and lateral temporal lobe, respectively, to −1.29 in the globus pallidus (Supplemental Table 2). Similar regions, such as the supramarginal gyrus, showed higher 18F-SMBT-1 binding in the Aβ+ MCI group, but none reached significance (Fig. 2).

Bar graphs showing that regional 18F-SMBT-1 SUV ratios were significantly higher in posterior cingulate gyrus, supramarginal gyrus, lateral occipital lobe, gyrus angularis, and primary visual cortex in Aβ+ AD group but also were significantly lower in globus pallidus and hippocampus. In addition, 18F-SMBT-1 binding was significantly higher in orbitofrontal, lateral, and inferior temporal gyri and significantly lower in globus pallidus. Similar regions, such as supramarginal gyrus, showed higher 18F-SMBT-1 binding in Aβ+ MCI group, but none reached significance. *Significantly higher than in Aβ− CN group (P < 0.05). †Significantly lower than in Aβ− CN group (P < 0.05). SUVRWM = SUV ratio using white matter as reference region.
When all clinical groups were considered together, 18F-SMBT-1 PET correlated strongly with Aβ burden and much less with tau burden (Table 2). Figure 3A shows that this close regional relationship between Aβ deposition and astrogliosis was present in only some regions of the brain, such as the supramarginal gyrus, posterior cingulate gyrus, lateral occipital lobe, and inferior and middle temporal gyri, but was much lower in other regions also characterized by high Aβ deposition, such as the frontal cortex, and relatively absent in the superior temporal gyrus. When further exploring the relationship between 18F-SMBT-1 and Aβ, we observed that a nonlinear fit seemed to better describe the relationship between 18F-SMBT-1 and Aβ in, for example, the supramarginal gyrus, where the 18F-SMBT-1 signal seems to be increasing before Aβ becomes abnormal (Supplemental Fig. 1). This relationship was not affected by adjustment for age, white matter hyperintensity, and hippocampal volume. There were no correlations between 18F-SMBT-1 and Aβ in brain regions with a high density of MAO-B, such as the anterior cingulate gyrus and the mesial temporal lobe. Further, in the basal ganglia, the region with the highest density of MAO-B in the brain, 18F-SMBT-1 correlated inversely with Aβ (Table 2). A similar picture was observed for the correlation between 18F-SMBT-1 and tau in the metatemporal region, although the associations were less extensive (Fig. 3B) and were less significant than for Aβ (Table 2). When examining the relationship between 18F-SMBT-1 and Aβ accumulation in a subset of participants (n = 31) who had available longitudinal Aβ imaging data, we observed a significant association in the temporal lobe (lateral and inferior temporal) (Table 2). Trend levels (P < 0.09) were also observed in the parahippocampus, temporooccipital region, and supramarginal gyrus region (Table 2).
Association Between Regional 18F-SMBT-1 Binding and Global Aβ Burden, Metatemporal Tau, and Aβ Accumulation

(A) There were high regional vertexwise associations between 18F-SMBT-1 binding and Aβ across brain areas of early Aβ deposition such as temporoparietal junction, supramarginal gyrus, and posterior cingulate gyrus, but also much lower associations were observed in areas also characterized by high Aβ, such as frontal cortex and superior temporal gyrus. Some areas, particularly those characterized by high MAO-B concentrations in Aβ− CN subjects, such as basal ganglia, mesial temporal lobe, and anterior cingulate gyrus, presented negative correlations (not shown). (B) Much less extensive regional associations were observed with tau. Color scale represents regression coefficients (r).
When all clinical groups were considered together, Aβ and tau burdens were highly associated with hippocampal volume and gray matter volume (Supplemental Table 3). Overall, sex was the major contributor to the variation in gray matter volume, whereas age was the major contributor to the variation in white matter hyperintensity. A few associations survived after covariate adjustment. 18F-SMBT-1 binding in the supramarginal gyrus and lateral occipital lobe was inversely associated with gray matter (r = −0.28, P = 0.017) and hippocampal volume (r = −0.28, P = 0.016), respectively (Supplemental Table 3). Conversely, 18F-SMBT-1 binding in the hippocampus was significantly associated with hippocampal volume (r = 0.34, P = 0.003), cortical gray matter volume (r = 0.34, P = 0.004), and white matter volume (r = 0.23, P = 0.048), but theirs was a direct relationship—in other words, the lower the 18F-SMBT-1 binding, the lower the respective volume (Supplemental Table 2). Similar findings were observed in the caudate nuclei, pallidus, thalamus, and pons, where 18F-SMBT-1 binding was also directly associated with gray matter volume (Supplemental Table 3). In contrast to the findings in the mesial temporal lobe, no atrophy was detected in these regions. 18F-SMBT-1 binding in the occipital lobe was significantly associated with white matter hyperintensity (Supplemental Table 3).
When all groups were considered together, and after adjusting for age, sex, Aβ, tau, and hippocampal volume, 18F-SMBT-1 binding correlated with Mini Mental State Examination (MMSE) and the Australian Imaging Biomarkers and Lifestyle’s Preclinical Alzheimer Cognitive Composite (AIBL PACC) in some neocortical regions such as the frontal cortex, lateral temporal lobe, supramarginal gyrus, and angular gyrus, contributing 24%–35% of the variance in MMSE and 18%–28% of the variance in AIBL PACC (Table 3). Overall, all cognitive domains were driven by tau, with hippocampal volume contributing to CDR SoB, nonmemory, and AIBL PACC and sex contributing to episodic memory and AIBL PACC.
Association Between Regional 18F-SMBT-1 Binding and Cognitive Performance
DISCUSSION
To the best of our knowledge, 18F-SMBT-1 represents the first available 18F MAO-B radiotracer to be used in a clinical study to assess reactive astrogliosis. In the present clinical study, we evaluated the performance of 18F-SMBT-1 PET across the AD spectrum.
When comparing clinical groups, we found that the AD group had both significantly higher (posterior cingulate gyrus, supramarginal gyrus) and lower (hippocampus, globus pallidus) SMBT-1 binding than the CN elderly controls. This difference was better defined when the clinical groups were separated according to high or low Aβ PET burden. It became clear that in regions such as the posterior cingulate gyrus, supramarginal gyrus, and lateral occipital lobe, the Aβ+ CN and Aβ+ AD groups became better separated from the Aβ− CN group but also that the globus pallidus and hippocampus remained significantly lower. Interestingly, the same regions tended to be higher in the Aβ+ MCI group, but it was a group with only 3 participants and none of the regions achieved significance. Most importantly, the Aβ+ CN group had significantly higher 18F-SMBT-1 binding than the Aβ− CN group in the same regions as those that were significantly higher in the AD group (posterior cingulate gyrus, supramarginal gyrus, and lateral occipital lobe), but 18F-SMBT-1 binding was also significantly higher in the orbitofrontal, lateral, and inferior temporal gyri. These findings match recent reports from fluid biomarker studies that found plasma levels of glial fibrillary acidic protein, an astrocytic marker, to be higher in Aβ+ CN subjects than in Aβ− CN subjects (47), as well as predicting future conversion to AD in MCI subjects (48).
As in the AD group, when compared with the Aβ− CN group the Aβ+ CN group had significantly lower 18F-SMBT-1 binding in the globus pallidus. The first thing to consider when looking at the mesial temporal structures is that the PET results are not corrected for partial-volume effects, and as expected, the hippocampi were significantly more atrophic in the Aβ+ AD group than in the Aβ− CN group (5.94 ± 0.5 vs. 4.50 ± 0.5, P < 0.0001)—likely the best explanation for lower 18F-SMBT-1 binding in a region that otherwise has a high density of MAO-B. On the other hand, partial-volume effects are not likely to explain the lower 18F-SMBT-1 binding in the region with the highest density of MAO-B in the brain. Although only the globus pallidus reached significance, all basal ganglia regions showed lower 18F-SMBT-1 binding (Fig. 2). An alternative explanation might be that, as in vitro studies using autoradiography and 11C-l-deprenyl-D2 showed, MAO-B presented an inverse correlation with Braak and Braak stages in AD brains (49), suggesting that as the neurodegeneration progresses, the expression of MAO-B decreases, most noticeably in those areas with a high density of MAO-B such as the mesial temporal lobe and basal ganglia, while also reflecting the regional loss of astrocytes similar to what is observed in neurons (50) associated with progressive Aβ deposition.
Early Aβ deposition is known to occur in brain regions with significantly higher SMBT-1 binding in Aβ+ CN and Aβ+ AD subjects, such as the supramarginal gyrus (identified as the “temporoparietal junction” in 2012 (51) and recently rebranded as the “banks of the superior temporal sulcus” (52)) along with the orbitofrontal and the posterior cingulate gyri (51) . This fact suggests that reactive astrogliosis, as detected with 18F-SMBT-1, is associated with early Aβ deposition at the preclinical stages of AD and likely plays a moderating or modulating role over neurodegeneration, cognitive trajectories, and clinical progression. This possibility was further confirmed by the correlational and vertexwise analysis in which 18F-SMBT-1 binding in the same regions correlated strongly with Aβ. But the association between 18F-SMBT-1 binding and Aβ merits closer examination. The highest correlations were in areas of early Aβ deposition, whereas other areas, characterized by high Aβ such as the frontal cortex, had much lower correlations. In contrast, brain areas known for a high density of MAO-B either lacked correlation or had even an inverse correlation, as observed in the mesial temporal lobe, anterior cingulate gyrus, and basal ganglia. And although the association between 18F-SMBT-1 binding and Aβ accumulation in areas such as the inferior temporal lobe followed a linear relationship (Supplemental Fig. 2), the association between 18F-SMBT-1 binding and Aβ was better described by a nonlinear fit. This finding suggests that the increase in 18F-SMBT-1 signal likely precedes the time that Aβ becomes abnormal (Supplemental Fig. 1), in agreement with the hypothesis that reactive astrogliosis precedes the significant build-up of Aβ plaques in the brain (24,53). The consequent suggestion is that astrocytes could be reactive and increase MAO-B expression or activity in response to Aβ changes that precede plaque deposition (e.g., an increased brain concentration of Aβ oligomers and protofibrils). The high association of reactive astrogliosis with insoluble Aβ, coupled with its early manifestation, has prompted the postulation that the neuroinflammatory reaction is driven by soluble Aβ oligomers (24,25). Astrocytes do not constitute a homogeneous population and have—and adopt—different morphologic, biochemical, and functional properties reflecting a complex mix of toxic and protective pathways (6). Thus, the relationship with Aβ and tau, given the diverse morphologic and biochemical diversity of astrocytes, is not likely to be straightforward or the same across different brain regions, indicating a complex regional relationship rather than a global response.
The associations between 18F-SMBT-1 binding and brain volumetrics or white matter hyperintensity were not strong. Hippocampal and parahippocampal 18F-SMBT-1 binding was associated with hippocampal volume, as 18F-SMBT-1 binding in the caudate nuclei, pallidus, thalamus, and pons was also associated with gray matter volume, but these were all direct associations, with lower 18F-SMBT-1 binding being associated with smaller volumes. The expedient explanation would be that these regions—regions with normally high concentrations of MAO-B—are atrophic. As explained in the section dealing with the correlations with Aβ, the hippocampi in the Aβ+ AD group were significantly more atrophic. Therefore, the lower 18F-SMBT-1 binding in the hippocampus and parahippocampal gyrus can be explained by partial-volume effects. But the basal ganglia were not atrophic, and there were no significant differences across the groups; partial-volume effects thus cannot explain the lower 18F-SMBT-1 binding. We believe that the results in the basal ganglia truly reflect a reduction in 18F-SMBT-1 binding in these areas, especially in AD, a condition in which, as neurodegeneration progresses, the expression of MAO-B likely decreases, most noticeably in those areas with a high density of MAO-B such as the basal ganglia.
In regard to the associations between 18F-SMBT-1 binding and cognitive parameters, the main driver of cognitive impairment across all domains was tau burden, contributing more than 50% of the variance, followed by hippocampal volume. Sex was also a contributor to episodic memory and AIBL PACC, with women performing worse than men. 18F-SMBT-1 binding correlated mainly with MMSE and AIBL PACC in some neocortical regions such as the frontal cortex, with the lateral temporal lobe, supramarginal gyrus, and angular gyrus contributing 24%–35% of the variance of MMSE and 18%–28% of the variance of AIBL PACC. These findings suggest that reactive astrogliosis makes a detrimental contribution to general cognition and some memory tasks that are independent of tau, Aβ, or hippocampal atrophy and that early amelioration of neuroinflammation might be a complementary therapeutic avenue for AD (20).
There are several limitations and caveats to this study. Similarly to microglial activation, only surrogate markers are available to assess reactive astrogliosis. In contrast to neuroreceptor PET studies using tracers that bind directly to the receptor, the study of neuroinflammation is based on using surrogate markers to assess their change of state from resting to activated. Thus, microglial activation has been studied for decades by assessing overexpression of mitochondrial translocator protein 18 kDa (18) and, more recently, by using tracers for macrophage colony-stimulating factor 1 receptor (54) or purinergic receptors (55), whereas reactive astrogliosis has been assessed with markers of MAO-B (19) and imidazoline 2 binding sites (29). It needs to be clearly understood that 18F-SMBT-1 is a MAO-B tracer, and as such, several factors and conditions that affect MAO-B can affect 18F-SMBT-1 binding. Besides the obvious effect of MAO-B inhibitors, MAO-B also increases with age (56), is affected by smoking (57), and has been found to be altered in psychiatric conditions such as major depression (22). Therefore, careful clinical anamnesis and an itemized listing of exclusionary criteria are required before participants undergo 18F-SMBT-1 PET. Although MAO-B is present mainly in astrocytes, it is also found, in a much smaller concentration, in neurons (58). 18F-SMBT-1 can detect increases and decreases in MAO-B in the brain but cannot discriminate between the fluid change or transition of reactive astrocytes from a protective to a toxic state, and not all reactive astrocytes overexpress MAO-B (58). The small number of Aβ+ MCI subjects precludes drawing any conclusion on group differences in the SMBT-1 signal. A larger sample size, especially Aβ+ MCI and Aβ+ AD patients, will be required to further validate the finding that 18F-SMBT-1 captures the reported increases in MAO-B across the AD continuum. There is also a chance of spurious correlations from analyses of subgroups. These analyses were undertaken to dissect the relation between the different aspects of 18F-SMBT-1 binding in the presence or absence of Aβ and tau and how 18F-SMBT-1 binding relates to other variables. Finally, the participants were volunteers who were not randomly selected from the community and were generally well educated and had high scores on cognitive tests; thus, these findings might not apply to the general population.
The introduction of biomarker-based approaches for the identification of brain pathology has informed new strategies for the design of clinical trials aimed at preventing the onset of cognitive impairment and dementia. Markers of Aβ and tau pathology and markers of neurodegeneration have been incorporated into a recently proposed biomarker-based framework (59). The advantage of the modular design of the framework, and considering that reactive gliosis is a critical aspect of the neuropathology of AD, is that the biomarker framework might be expanded to include reactive gliosis.
Our studies showed that 18F-SMBT-1 can be used as a surrogate marker of reactive astrogliosis. Despite the limited sample size in some of the groups, there was a distinctive degree and pattern of tracer binding across the AD continuum that was associated mainly with the presence of Aβ burden in the brain. 18F-SMBT-1 will allow a better understanding of the pathophysiology of AD, enabling more accurate staging and determination of prognosis at earlier stages of the disease. It will also be necessary to examine the relationship between 18F-SMBT-1 binding in the brain and plasma glial fibrillary acidic protein. Longitudinal studies will be required to assess the effects of reactive astrogliosis over the clinical expression of AD and also, given the intimate relationship between astrocytes and blood vessels, of cerebrovascular disease. Longitudinal studies will also be required to fully elucidate the complex interaction between reactive astrogliosis, AD pathology, and cerebrovascular disease and their moderating or modulating impact over neurodegeneration, cognitive decline, and clinical progression.
CONCLUSION
Cross-sectional human PET studies with 18F-SMBT-1, a highly selective 18F-labeled MAO-B tracer, showed that Aβ+ AD subjects but, most importantly, Aβ+ CN subjects have significantly higher regional 18F-SMBT-1 binding than Aβ− CN subjects. Moreover, in several regions of the brain, 18F-SMBT-1 retention was highly associated with Aβ burden. These findings suggest that increased 18F-SMBT-1 binding occurs at the preclinical stages of Aβ accumulation, providing strong support for its use as a surrogate marker of astrogliosis and a biomarker of early stages in the AD continuum.
DISCLOSURE
The study was supported in part by National Health Medical Research Council (NHMRC) of Australia grants G1005121 and 19KK0212 from Japan. Yukitsuka Kudo and Nobuyuki Okamura own stock in Clino Ltd., licensing SMBT-1. Ryuichi Harada, Shozo Furumoto, Yukitsuka Kudo, and Nobuyuki Okamura have a patent pending for the technology described in this article. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can 18F-SMBT-1 be used to assess reactive astrogliosis in vivo?
PERTINENT FINDINGS: A clinical study on 77 elderly participants showed that 18F-SMBT-1, a novel 18F MAO-B tracer used as a surrogate marker of reactive astrogliosis, was significantly higher in AD patients and, most importantly, in elderly CN controls with high Aβ in the brain.
IMPLICATIONS FOR PATIENT CARE: 18F-SMBT-1 can be used as a surrogate and early marker of reactive astrogliosis across the AD continuum.
ACKNOWLEDGMENTS
We thank the Brain Research Institute for support in acquiring the MRI data. We thank Drs. Chester Mathis, William E. Klunk, Oscar Lopez, Ann Cohen, Brian Lopresti, Howard Aizenstein, Scott Mason, Beth Snitz, and Beth Shaaban at the University of Pittsburgh for extremely fruitful discussions about SMBT-1, MAO-B, and reactive astrogliosis. We thank the participants in this study and their families. This paper is dedicated to the first author’s father, Victor E. Villemagne (January 8, 1935–May 7, 2021).
Footnotes
Published online Jan. 27, 2022.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication September 22, 2021.
- Accepted for publication January 14, 2022.





{kind=link}
{kind=link}
{kind=link}
{kind=link}