


Abstract
Several methods have been developed to label peptides with 18F. However, in general these are laborious and require a multistep synthesis. We present a facile method based on the chelation of 18F-aluminum fluoride (Al18F) by 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA). The method is characterized by the labeling of NOTA-octreotide (NOTA-d-Phe-cyclo[Cys-Phe-d-Trp-Lys-Thr-Cys]-Throl (MH+ 1305) [IMP466]) with 18F. Methods: Octreotide was conjugated with the NOTA chelate and labeled with 18F in a 2-step, 1-pot method. The labeling procedure was optimized with regard to the labeling buffer, peptide, and aluminum concentration. Radiochemical yield, specific activity, in vitro stability, and receptor affinity were determined. Biodistribution of 18F-IMP466 was studied in AR42J tumor–bearing mice and compared with that of 68Ga-labeled IMP466. In addition, small-animal PET/CT images were acquired. Results: IMP466 was labeled with Al18F in a single step with 50% yield. The labeled product was purified by high-performance liquid chromatography to remove unbound Al18F and unlabeled peptide. The radiolabeling, including purification, was performed in 45 min. The specific activity was 45,000 GBq/mmol, and the peptide was stable in serum for 4 h at 37°C. Labeling was performed at pH 4.1 in sodium citrate, sodium acetate, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 2-(N-morpholino)ethanesulfonic acid buffer and was optimal in sodium acetate buffer. The apparent 50% inhibitory concentration of the 19F-labeled IMP466 determined on AR42J cells was 3.6 nM. Biodistribution studies at 2 h after injection showed a high tumor uptake of 18F-IMP466 (28.3 ± 5.2 percentage injected dose per gram [%ID/g]; tumor-to-blood ratio, 300 ± 90), which could be blocked by an excess of unlabeled peptide (8.6 ± 0.7 %ID/g), indicating that the accumulation in the tumor was receptor-mediated. Biodistribution of 68Ga-IMP466 was similar to that of 18F-IMP466. 18F-IMP466 was stable in vivo, because bone uptake was only 0.4 ± 0.2 %ID/g, whereas free Al18F accumulated rapidly in the bone (36.9 ± 5.0 %ID/g at 2 h after injection). Small-animal PET/CT scans showed excellent tumor delineation and high preferential accumulation in the tumor. Conclusion: NOTA-octreotide could be labeled rapidly and efficiently with 18F using a 2-step, 1-pot method. The compound was stable in vivo and showed rapid accretion in somatostatin receptor subtype 2–expressing AR42J tumors in nude mice. This method can be used to label other NOTA-conjugated compounds with 18F.
During the past decade, radiolabeled receptor-binding peptides have emerged as an important class of radiopharmaceuticals that have changed radionuclide imaging. Peptides have been labeled with 111In and 99mTc for SPECT and, more recently, with positron emitters such as 68Ga, 64Cu, 86Y, and 18F for PET. 18F is the most widely used radionuclide in PET and has excellent characteristics for peptide-based imaging: the half-life (110 min) matches the pharmacokinetics of most peptides, and the low positron energy of 635 keV results in short ranges in tissue and excellent preclinical imaging resolution (<2 mm) (1). A wide range of methods to label peptides with 18F have been investigated. In general, an 18F-labeled synthon is produced by nucleophilic substitution, which is subsequently reacted with the functionalized peptide. One of the first generally applicable methods is based on conjugation of the synthon N-succinimidyl-4-18F-fluorobenzoate to a primary amino group of the peptide (2). Although widely used, the method requires a multistep synthesis and is, therefore, time-consuming and laborious. Poethko et al. have developed an improved 18F-labeling method by reacting 18F-fluorobenzaldehyde with an aminooxy-derivatized peptide, forming an oxime bond (3). Alternatively, 18F-fluorobenzaldehyde was reacted with hydrazine-nicotinamide–conjugated peptides. However, this method resulted in increased lipophilicity of the peptide that may lead to unfavorable characteristics in vivo (4). Carbohydration of the peptide may counteract the enhanced lipophilicity (5). More recently, 18F-FDG was explored for the labeling of aminooxy-derivatized peptides (6,7). This approach requires the use of carrier-free 18F-FDG, necessitating high-performance liquid chromatography (HPLC) purification of 18F-FDG before conjugation with the functionalized peptide. Furthermore, methods based on the Huisgen cycloaddition of alkynes and azides were explored for the fluorination of peptides (8–11). Recently, silicon-based building blocks were used to fluorinate bombesin peptides functionalized with 2 tertiary butyl groups. However, this method also resulted in a lipophilic 18F peptide and loss of tumor targeting (12,13).
We reported recently that a 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA)–conjugated pretargeting peptide could be labeled directly with 18F (14). To demonstrate that this 2-step, 1-pot method can be applied to other peptides, we report a new approach to label the somatostatin analog NOTA-octreotide with 18F. Radiolabeled somatostatin analogs, such as diethylenetriaminepentaacetic acid (DTPA)–octreotide, DOTA-Tyr3-octreotide, NOTA-octreotide, and DOTA-Tyr3-octreotate, can be used to image somatostatin receptor subtype 2–expressing tumors. With this 2-step, 1-pot fluorination method, the peptide could be stably labeled with a 50% radiochemical yield at a high specific activity within 45 min.
MATERIALS AND METHODS
General
The octreotide peptide analog NOTA-d-Phe-cyclo[Cys-Phe-d-Trp-Lys-Thr-Cys]-Throl (MH+ 1305), designated IMP466, was synthesized using standard Fmoc-based solid-phase peptide synthesis. After cleavage from the resin, the peptide was cyclicized by incubation with dimethyl sulfoxide overnight. The Throl resin and protected amino acids were purchased from CreoSalus Inc. The bis-t-butyl NOTA ligand was provided by Immunomedics, Inc. All other chemicals were purchased from Sigma-Aldrich or Fisher Scientific. All buffers used for the radiolabeling procedures were metal-free.
Radiolabeling
18F Labeling
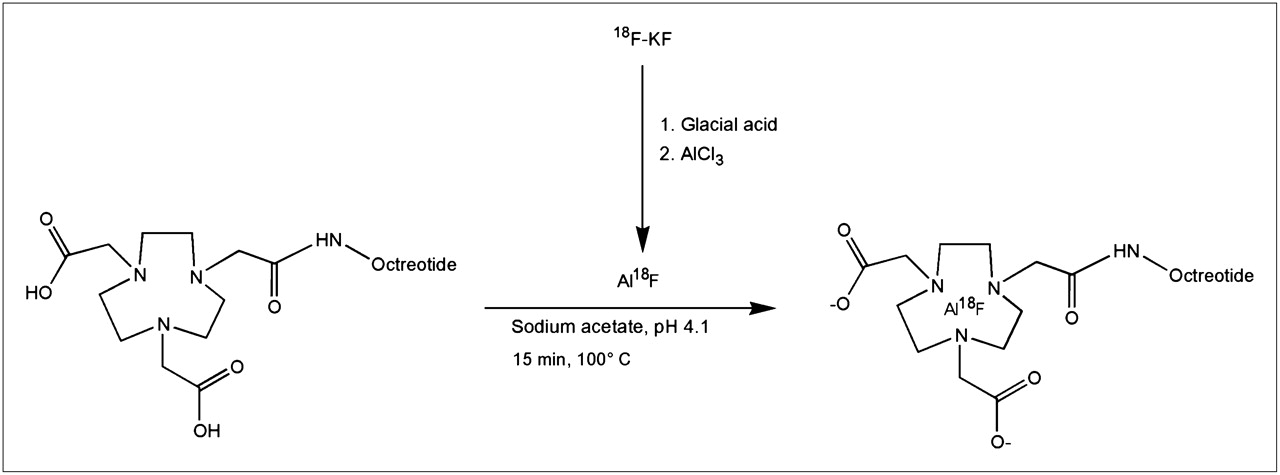
The 18F-labeling reaction is summarized in Figure 1. A QMA SepPak Light cartridge (Waters) with 2–6 GBq of 18F (BV Cyclotron VU) was washed with 3 mL of metal-free water. 18F was eluted from the cartridge with 0.4 M KHCO3, and fractions of 200 μL were collected. The pH of the fractions was adjusted to 4 with 10 μL of metal-free glacial acetic acid. Three microliters of 2 mM AlCl3 in 0.1 M sodium acetate buffer, pH 4, were added. Then, 10–50 μL of IMP466 (10 mg/mL) were added in 0.5 M sodium acetate, pH 4.1. The reaction mixture was incubated at 100°C for 15 min unless stated otherwise. The radiolabeled peptide was purified on reversed-phase (RP) HPLC. The 18F-IMP466–containing fractions were collected and diluted 2-fold with H2O and purified on a 1-mL Oasis HLB cartridge (Waters) to remove acetonitrile and trifluoroacetic acid. In brief, the fraction was applied on the cartridge, and the cartridge was washed with 3 mL of H2O. The radiolabeled peptide was then eluted with 2 × 200 μL of 50% ethanol. On injection in mice, the peptide was diluted with 0.9% NaCl.

Preparation of Al18F and chelation with NOTA-octreotide.
Effect of Buffer
The effect of the buffer on the labeling efficiency of IMP466 with 18F− was investigated (n = 3 for each buffer). IMP466 was dissolved in sodium citrate buffer, sodium acetate buffer, 2-(N-morpholino)ethanesulfonic acid (MES) or 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer at 10 mg/mL (7.7 mM). The molarity of all buffers was 1 M, and the pH was 4.1. To 200 μg (153 nmol) of IMP466, 100 μL of 18F-aluminum fluoride (Al18F) (pH 4) were added and incubated at 100°C for 15 min. Radiolabeling yield and specific activity were determined with RP HPLC as described below.
68Ga Labeling
IMP466 was labeled with 68GaCl3 eluted from a TiO2-based 1,110-MBq 68Ge/68Ga generator (Cyclotron Co. Ltd.) using 0.1 M HCl (Ultrapure; J.T. Baker). Five 1-mL fractions were collected, and an aliquot of the second fraction was used for labeling the peptide. IMP466 (4 μg) was dissolved in 2.5 M HEPES buffer, pH 7.0. 68Ga eluate (120–240 MBq, 4 times the volume of the peptide) was added, and the mixture was heated at 95°C for 20 min. Then 50 mM ethylenediaminetetraacetic acid was added to a final concentration of 5 mM to complex the nonincorporated 68Ga3+. The 68Ga-labeled IMP466 was purified on a 1-cm3 Oasis HLB cartridge and eluted with 200 μL of 50% ethanol. The specific activity of 68Ga-IMP466 was 20,000 GBq/mmol at the time of injection.
HPLC Analysis
The radiolabeled preparations were analyzed by RP HPLC on an Agilent 1200 system (Agilent Technologies). A C18 column (Onyx monolithic, 4.6 × 100 mm; Phenomenex) was used at a flow rate of 2 mL/min with the following buffer system: buffer A, 0.1% v/v trifluoroacetic acid in water; buffer B, 0.1% v/v trifluoroacetic acid in acetonitrile; and a gradient of 97% buffer A at 0–5 min and 80% buffer A to 75% buffer A at 5–35 min. The radioactivity of the eluate was monitored using an in-line NaI radiodetector (Raytest GmbH). Elution profiles were analyzed using Gina-star software (version 2.18; Raytest GmbH). The specific activity was determined by HPLC using calibration curves based on the ultraviolet signal.
Octanol–Water Partition Coefficient (log Poctanol/water)
To determine the lipophilicity of the radiolabeled peptides, approximately 50,000 counts per minute of the radiolabeled peptide were diluted in 0.5 mL of phosphate-buffered saline. An equal volume of 1-octanol was added to obtain a binary phase system. After stirring in a vortex mixer for 2 min, we separated the 2 layers by centrifugation (100g, 5 min). Three 100-μL samples were taken from each layer, and radioactivity was measured in a well-type γ-counter (Wallac Wizard 3"; Perkin-Elmer).
Stability
Ten microliters of the 18F-labeled IMP466 were incubated in 500 μL of freshly collected human serum and incubated for 4 h at 37°C. An equal volume of acetonitrile was added and stirred in a vortex mixer, then followed by centrifugation at 1,000g for 5 min to pellet the precipitated serum proteins. The supernatant was analyzed on RP HPLC as described above.
The in vivo stability of 18F-IMP466 was examined by injecting 18.5 MBq of 18F-IMP466 in a BALB/c nude mouse. After 30 min, the mouse was euthanized, and blood and urine were collected and analyzed by HPLC.
Cell Culture
The AR42J rat pancreatic tumor cell line was cultured in Dulbecco's modified Eagle's medium (Gibco Life Technologies) supplemented with 4,500 mg of d-glucose per milliliter, 10% (v/v) fetal calf serum, 2 mmol of glutamine per liter, 100 U of penicillin per milliliter, and 100 μg of streptomycin per milliliter. Cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Apparent 50% Inhibitory Concentration (IC50) Determination
The apparent IC50 for binding the somatostatin receptors on AR42J cells was determined in a competitive binding assay using 19F-IMP466, 69Ga-IMP466, or 115In-DTPA-octreotide to compete for the binding of 111In-DTPA-octreotide (OctreoScan; Covidien).
19F-IMP466 was formed by mixing an AlF solution (0.02 M AlCl3 in 0.5 M NaAc, pH 4, with 0.1 M NaF in 0.5 M NaAc, pH 4) with IMP466 and heating at 100°C for 15 min. The reaction mixture was purified by RP HPLC on a C-18 column (30 × 150 mm, Sunfire; Waters), as described above.
69Ga-IMP466 was prepared by dissolving gallium nitrate (2.3 × 10−8 mol) in 30 μL mixed with 20 μL of IMP466 (1 mg/mL) in 10 mM NaAc, pH 5.5, and heated at 90°C for 15 min. Samples of the mixture were used without further purification.
115In-DTPA-octreotide was made by mixing indium chloride (1 × 10−5 mol) with 10 μL of DTPA-octreotide (1 mg/mL) in 50 mM NaAc, pH 5.5, and incubated at room temperature for 15 min. This sample was used without further purification. 111In-DTPA-octreotide was radiolabeled according to the manufacturer's protocol.
AR42J cells were grown to confluency in 12-well plates and washed twice with binding buffer (Dulbecco's modified Eagle's medium with 0.5% bovine serum albumin). After 10 min of incubation at room temperature in binding buffer, 19F-IMP466, 69Ga-IMP466, or 115In-DTPA-octreotide was added at a final concentration ranging from 0.1 to 1,000 nM, together with a trace amount (10,000 counts per minute) of 111In-DTPA-octreotide (radiochemical purity > 95%). After incubation at room temperature for 3 h, the cells were washed twice with ice-cold phosphate-buffered saline. Cells were scraped, and cell-associated radioactivity was determined. Under these conditions, a limited extent of internalization may occur. Therefore, we describe the results of this competitive binding assay as apparent IC50 values rather than IC50. The apparent IC50 was defined as the peptide concentration at which 50% of binding without competitor was reached. Apparent IC50 values were calculated using GraphPad Prism software (version 4.00 for Windows; GraphPad Software).
Biodistribution Studies
Male nude BALB/c mice (6–8 wk old) were injected subcutaneously in the right flank with 0.2 mL of AR42J cell suspension of 1 × 107 cells/mL. Approximately 2 wk after inoculation, when tumors were 5–8 mm in diameter, 370 kBq of 18F-labeled or 68Ga-labeled IMP466 (both 0.2 nmol) were administered intravenously (n = 5). Separate groups of mice (n = 5) were coinjected with a 1,000-fold molar excess of unlabeled IMP466. One group of 3 mice was injected with unchelated Al18F. All mice were killed by CO2/O2 asphyxiation 2 h after injection. Tissues of interest were dissected, weighed, and counted in a γ-counter. The percentage injected dose per gram of tissue (%ID/g) was calculated for each tissue on the basis of a dilution of the product for injection. The animal experiments were approved by the local animal welfare committee and performed according to national regulations.
PET/CT
Mice with subcutaneous AR42J tumors were injected intravenously with 10 MBq of 18F-IMP466 or 68Ga-IMP466 (both 0.7 nmol) per mouse. One and 2 h after the injection of peptide, mice were scanned on an animal PET/CT scanner (Inveon; Siemens Preclinical Solutions) with an intrinsic spatial resolution of 1.5 mm (1). The animals were placed supine in the scanner. PET emission scans were acquired over 15 min, followed by a CT scan for anatomic reference (spatial resolution, 113 μm; 80 kV, 500 μA). Scans were reconstructed using Inveon Acquisition Workplace software (version 1.2; Siemens Preclinical Solutions), using an ordered-set expectation maximization 3-dimensional maximum a posteriori algorithm with the following parameters: matrix, 256 × 256 × 159; pixel size, 0.43 × 0.43 × 0.8 mm3; and β-value, 0.1.
Statistical Analysis
All mean values are given as ±SD. Statistical analysis was performed using a Welch's corrected unpaired Student t test or 1-way ANOVA using GraphPad InStat software (version 3.06; GraphPad Software). The level of significance was set at P less than 0.05.
RESULTS
18F-Labeling Procedure
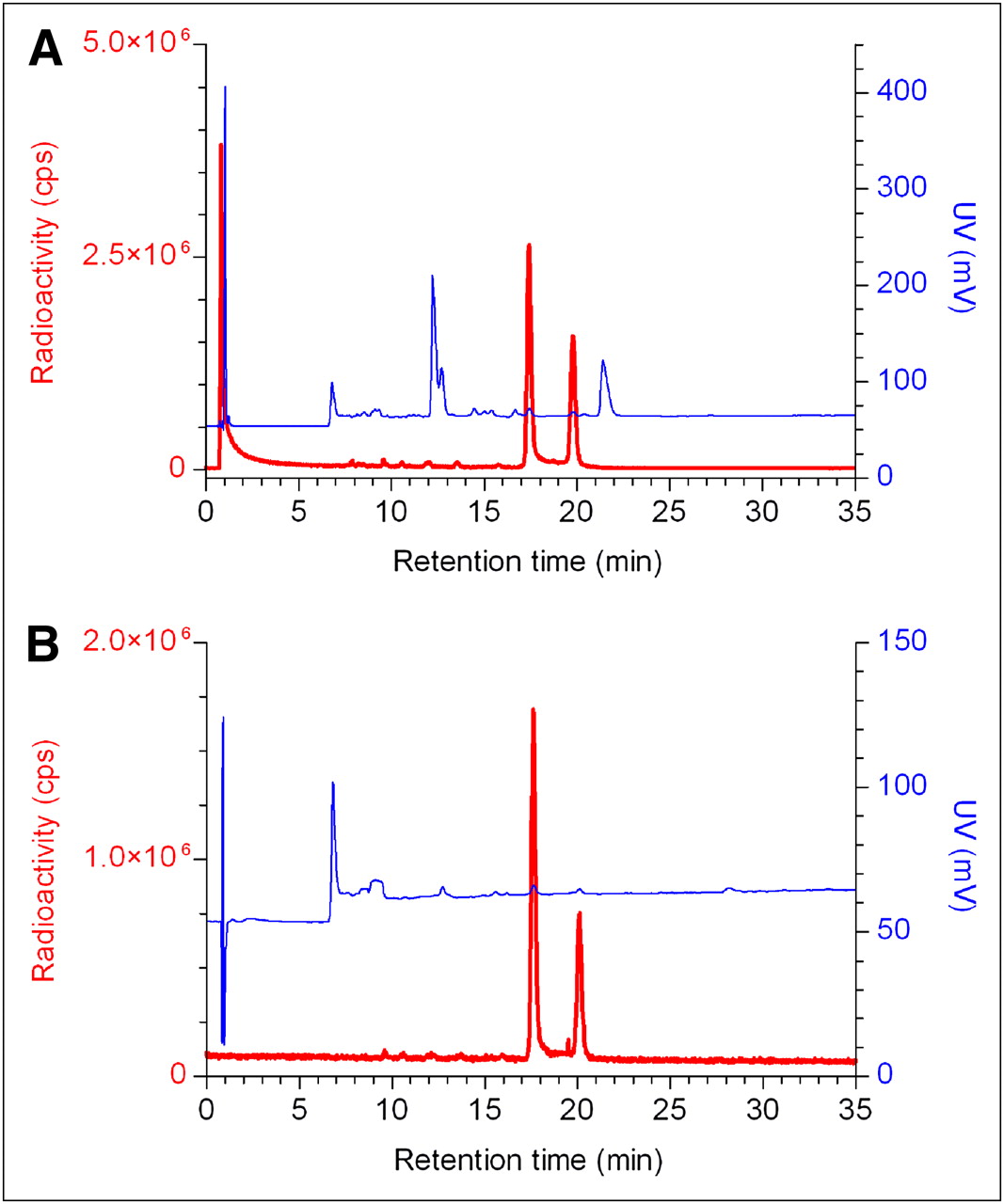
HPLC analysis of the IMP466 labeling mixture (Fig. 2) showed the presence of unbound Al18F (retention time [Rt] = 0.8 min) and 2 radioactive peptide peaks (Rt = 17.4 and 19.8 min), indicating the formation of two 18F-IMP466 stereoisomers. Moreover, an ultraviolet peak of unlabeled IMP466 is present (Rt = 21.4 min). After HPLC and HLB purification, both the unbound Al18F and the unlabeled IMP466 ultraviolet peaks disappeared (Fig. 2).

RP HPLC chromatograms of IMP466 18F-labeling mix (A) and purified 18F-IMP466 (B). Red traces represent radioactivity (left y-axis), and blue traces represent ultraviolet signal (right y-axis). In HPLC chromatogram of crude mixture, unbound Al18F eluted with void volume (Rt = 0.8 min). Two radioactive peaks correspond to times of stereoisomers of radiolabeled peptide (Rt = 17.4 and 19.8 min). Finally, unlabeled IMP466 was present in ultraviolet channel (Rt = 21.4 min). After purification, only 2 radioactive peptide peaks are observed, indicating formation of 2 stereoisomers. cps = counts per second; UV = ultraviolet.
Effect of Buffer
For sodium acetate, MES, or HEPES, the radiolabeling yield was 49% ± 2%, 46% ± 2%, and 48% ± 3%, respectively (n = 3 for each buffer). In sodium citrate, no labeling was observed. When the labeling reaction was performed in sodium acetate buffer, the specific activity was 32,000 ± 17,000 GBq/mmol, whereas in MES and HEPES buffer, specific activities of 29,000 ± 14,000 and 31,000 ± 23,000 GBq/mmol, respectively, were obtained.
Effect of Peptide Concentration
The effect of peptide concentration on the labeling efficiency also was investigated. IMP466 was dissolved in sodium acetate buffer, pH 4.1, at a concentration of 7.7 mM (10 mg/mL). Either 38, 153, or 363 nmol of IMP466 were added to 200 μL of Al18F (581–603 MBq) to yield a final IMP466 concentrations of 190, 765, and 1,815 μM, respectively. The radiolabeling yield increased with increasing amounts of peptide. At a concentration of 190 μM, the radiolabeling yield was 8% ± 1%; at 765 μM, the yield increased to 42% ± 3%; and at the highest concentration, the radiolabeling yield was 50% ± 2%. The specific activity of the products obtained at each concentration was 48,000 GBq/mmol.
Effect of AlCl3 Concentration
Because AlCl3 is used to form Al18F, the added amount of AlCl3 is critical in the labeling procedure. Five stock solutions with various AlCl3 concentrations were prepared: 0.2, 0.5, 1.0, 2.0, and 20 mM. From these solutions in sodium acetate, 3 μL were added to 200 μL of 18F-fluoride, pH 4, to form Al18F, resulting in final amounts of AlCl3 added of 0.6, 1.5, 3.0, 6.0, and 60 nmol, respectively. To these samples, 153 nmol of IMP466 (final concentration, 765 μM) were added and incubated for 15 min at 100°C. Radiolabeling yield was optimal (50% ± 2%, n = 5) after incubation with 6 nmol of AlCl3. Lowering the AlCl3 concentration resulted in reduced yields, ranging from 42% at 3 nmol to 10% at 0.6 nmol of AlCl3. Increasing the amount led to a similar effect. Incubation with 60 nmol of AlCl3 resulted in a radiolabeling yield of only 6%.
Octanol–Water Partition Coefficient
To determine the lipophilicity of the 18F- and 68Ga-labeled IMP466, the octanol–water partition coefficients were determined. The log Poctanol/water value for the 18F-IMP466 was −2.44 ± 0.12, and that of 68Ga-IMP466 was −3.79 ± 0.07, indicating that the 18F-IMP466 was slightly less hydrophilic than 68Ga-IMP466.
IC50 Determination
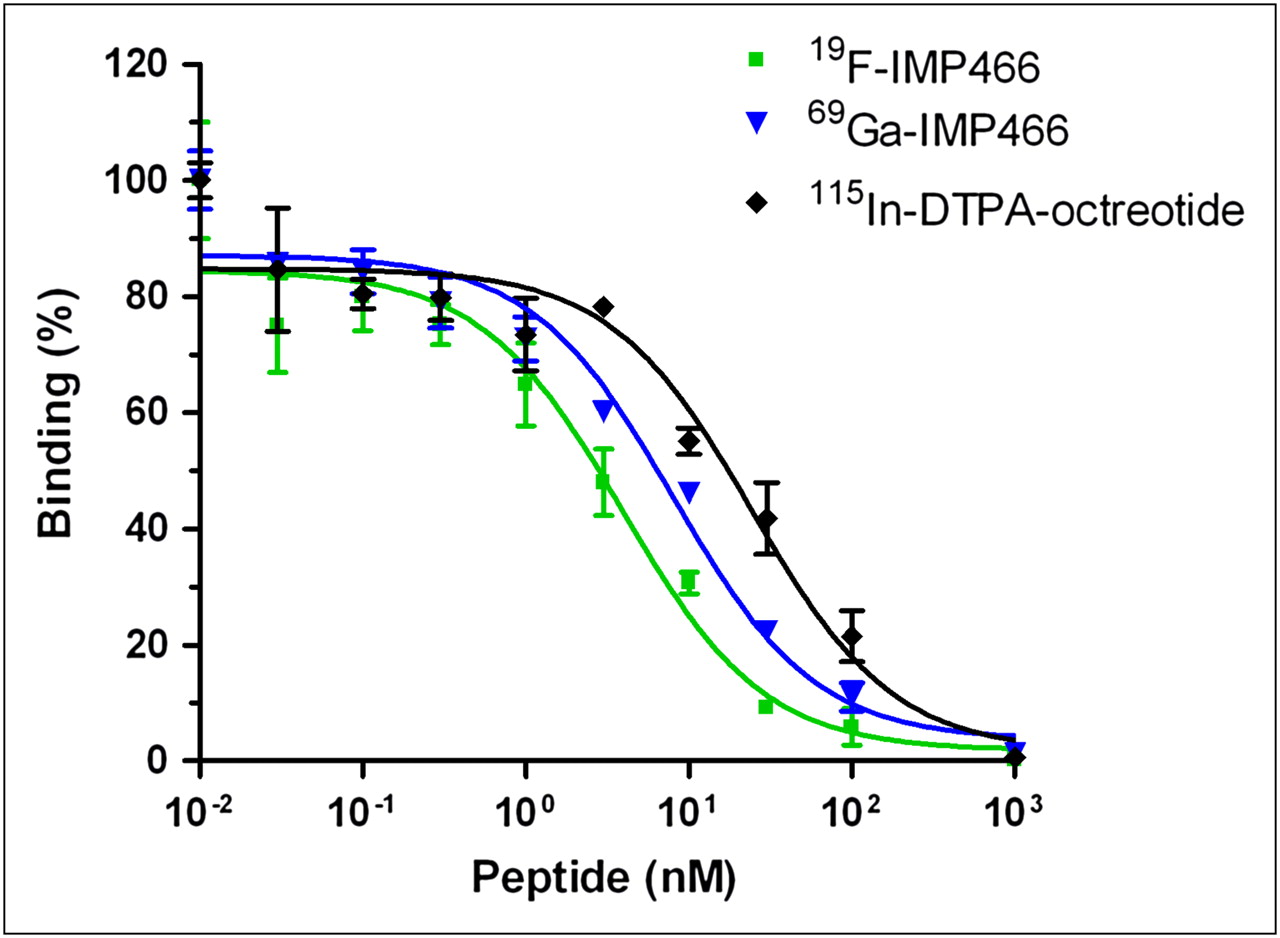
The affinity profiles are shown in Figure 3. The apparent IC50 of Al19F-labeled IMP466 was 3.6 ± 0.6 nM, whereas that for 69Ga-labeled IMP466 was 13 ± 3 nM. The apparent IC50 of the reference peptide, 115In-DTPA-octreotide (OctreoScan), was 6.3 ± 0.9 nM.
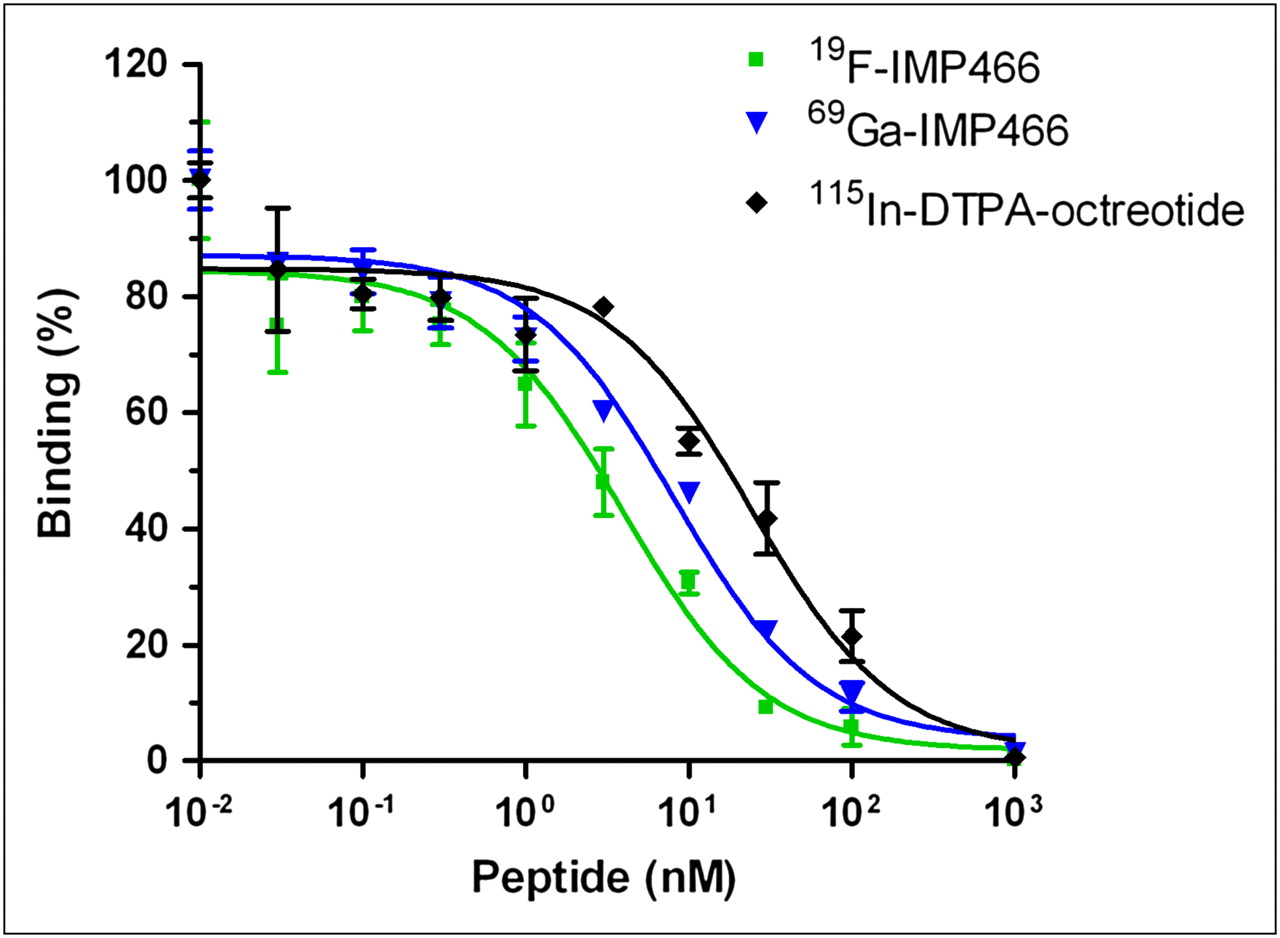
Competitive binding assay (apparent IC50) of 19F-IMP466, 69Ga-IMP466, and 115In-DTPA-octreotide determined on AR42J tumor cells. Values on y-axis represent binding expressed as percentage of binding without competitor.
Stability
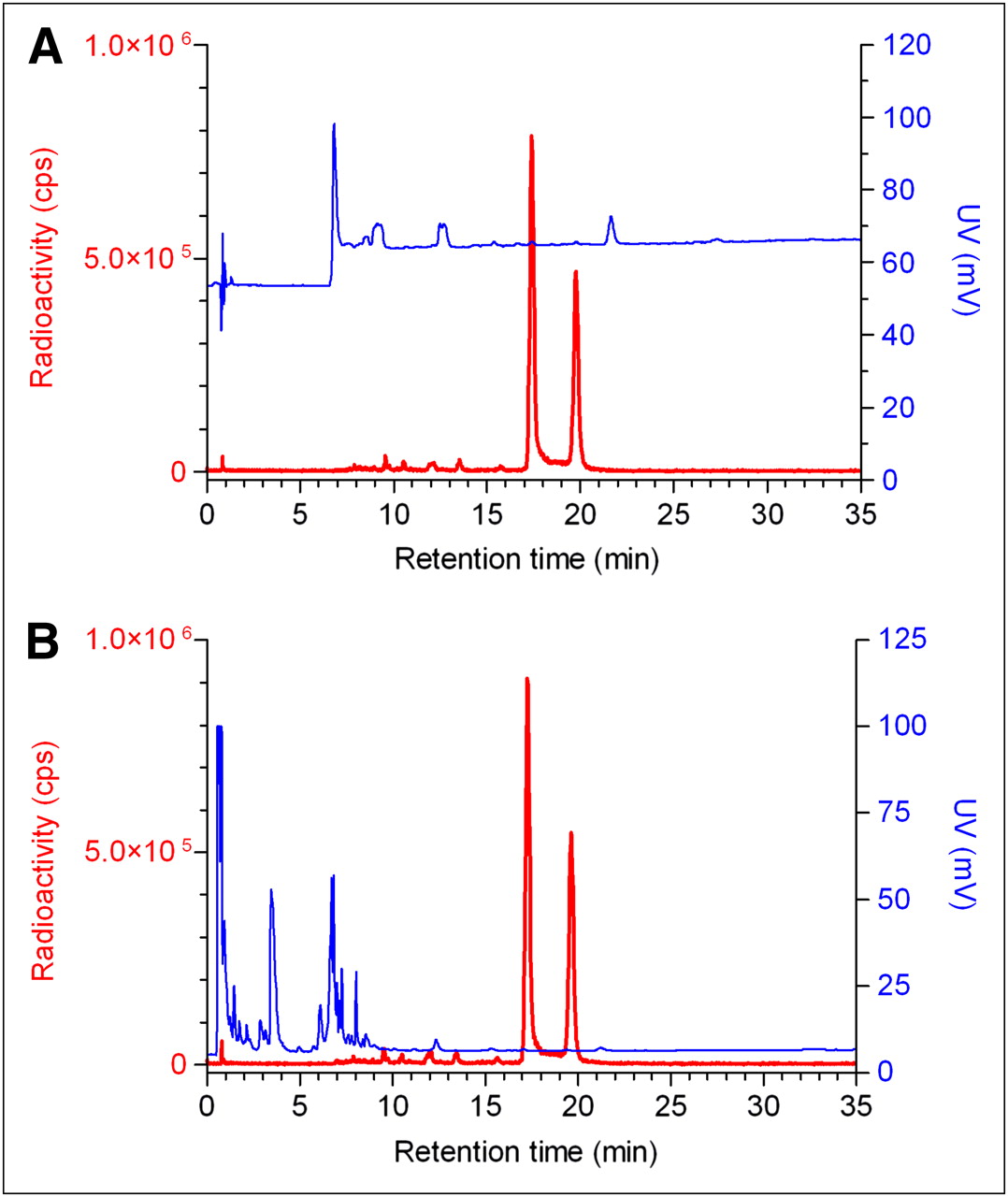
18F-labeled IMP466 did not release Al18F after incubation in human serum at 37°C for 4 h, indicating excellent stability of the Al18F-NOTA-octreotide. These findings were confirmed in vivo. After 30 min, only intact radiolabeled peptide was found in urine (Fig. 4) and no free 18F. In addition, PET/CT scans did not reveal any bone uptake, indicating the absence of free 18F-fluoride or Al18F.
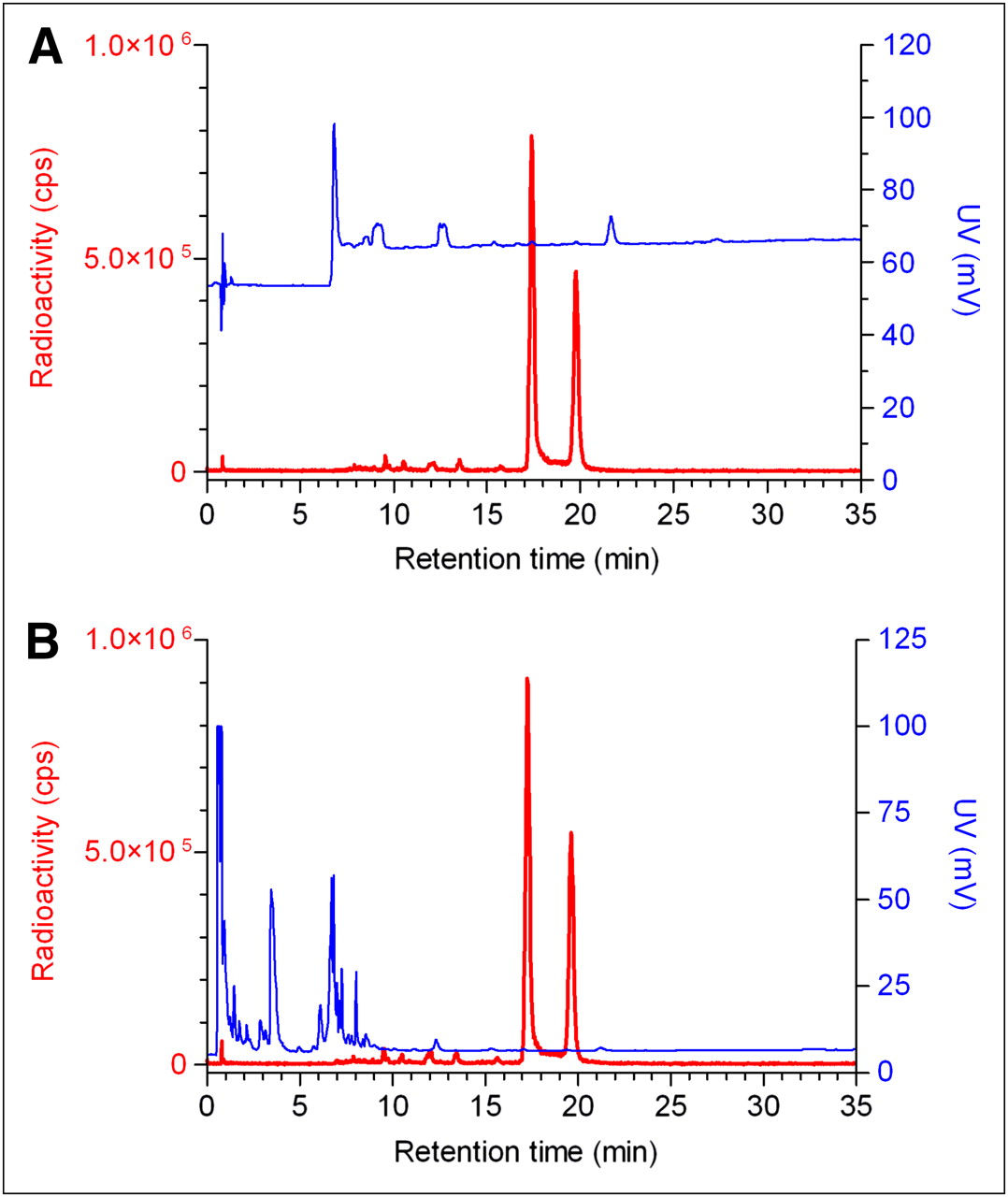
HPLC chromatograms of purified 18F-IMP466 before injection (A) and urine sample 30 min after injection (B). Red traces represent radioactivity (left y-axis), and blue traces represent ultraviolet signal (right y-axis). HPLC traces of 2 samples are similar, indicating that excreted product is intact 18F-IMP466. cps = counts per second; UV = ultraviolet.
Biodistribution Studies
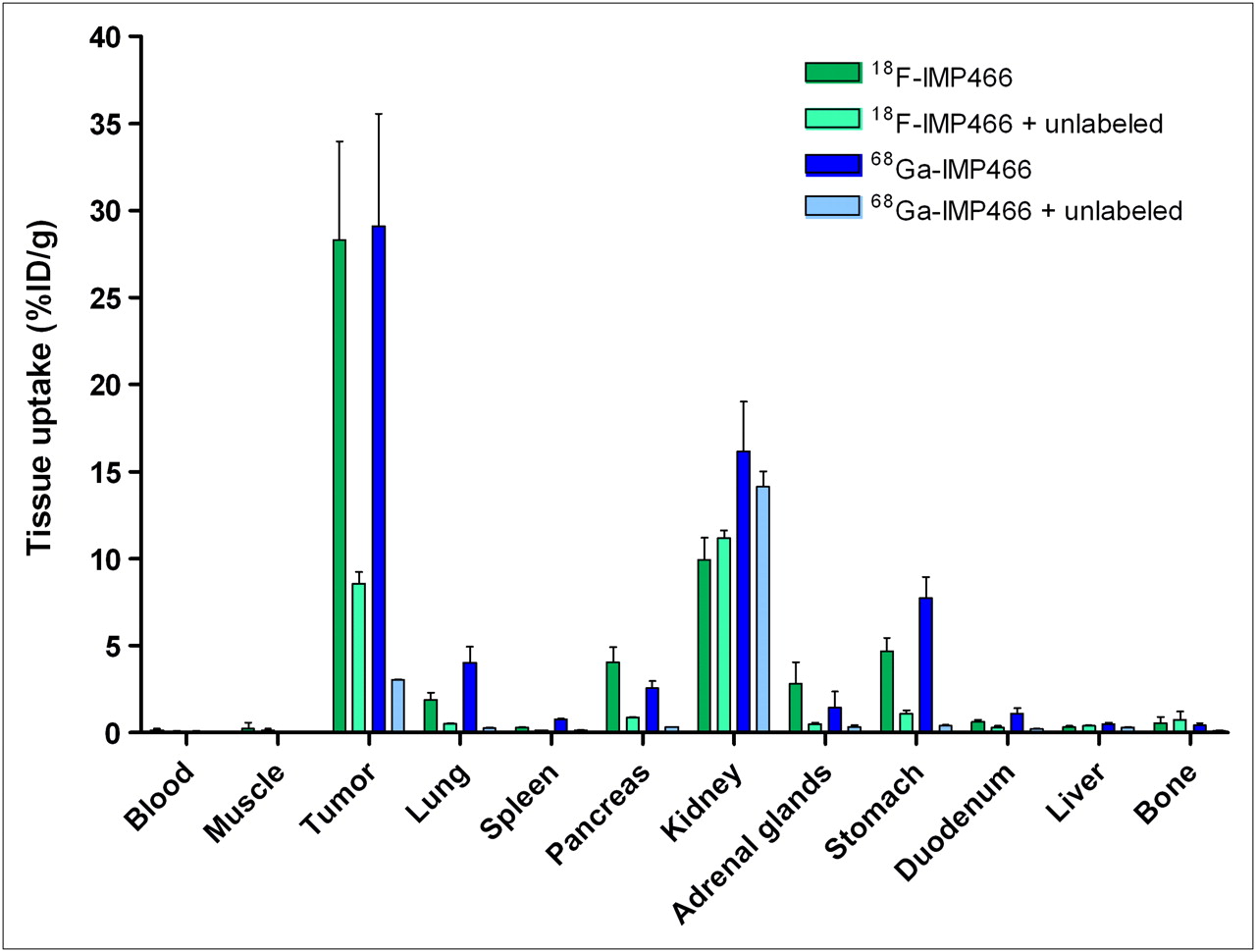
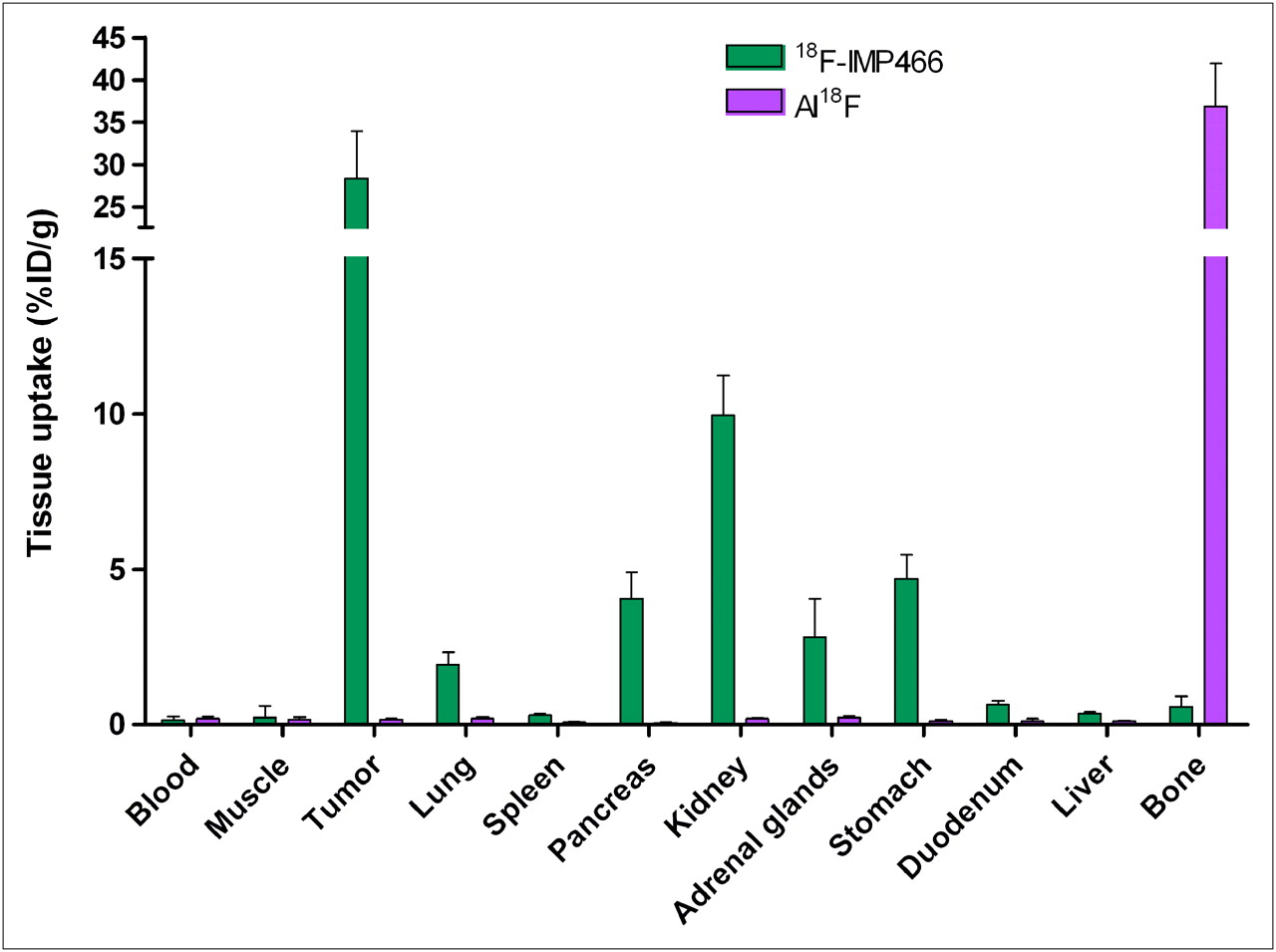
The biodistribution of both 18F-IMP466 and 68Ga-IMP466 in BALB/c nude mice with subcutaneous AR42J tumors at 2 h after injection is summarized in Figures 5 and 6. Unchelated Al18F was included as a control. Tumor uptake of 18F-IMP466 was 28.3 ± 5.7 %ID/g at 2 h after injection and reduced in the presence of an excess of unlabeled IMP466 to 8.6 ± 0.7 %ID/g, indicating that uptake was receptor-mediated. Nonspecific uptake of 18F-IMP466 was somewhat higher than that of 68Ga-IMP466, as is illustrated by the less efficient blocking of the tumor uptake of 18F-IMP466. Blood levels were low (0.10 ± 0.07 %ID/g, 2 h after injection), resulting in a tumor-to-blood ratio of 300 ± 90. Uptake in normal tissues, except in the kidneys, was low, with specific uptake in somatostatin receptor subtype 2–expressing tissues, such as adrenal glands, pancreas, and stomach. Bone uptake of 18F-IMP466 was negligible, as compared with uptake after injection of nonchelated Al18F (0.33 ± 0.07 %ID/g vs. 36.9 ± 5.0 %ID/g at 2 h after injection, respectively; P < 0.001), indicating good in vivo stability of the 18F-IMP466.
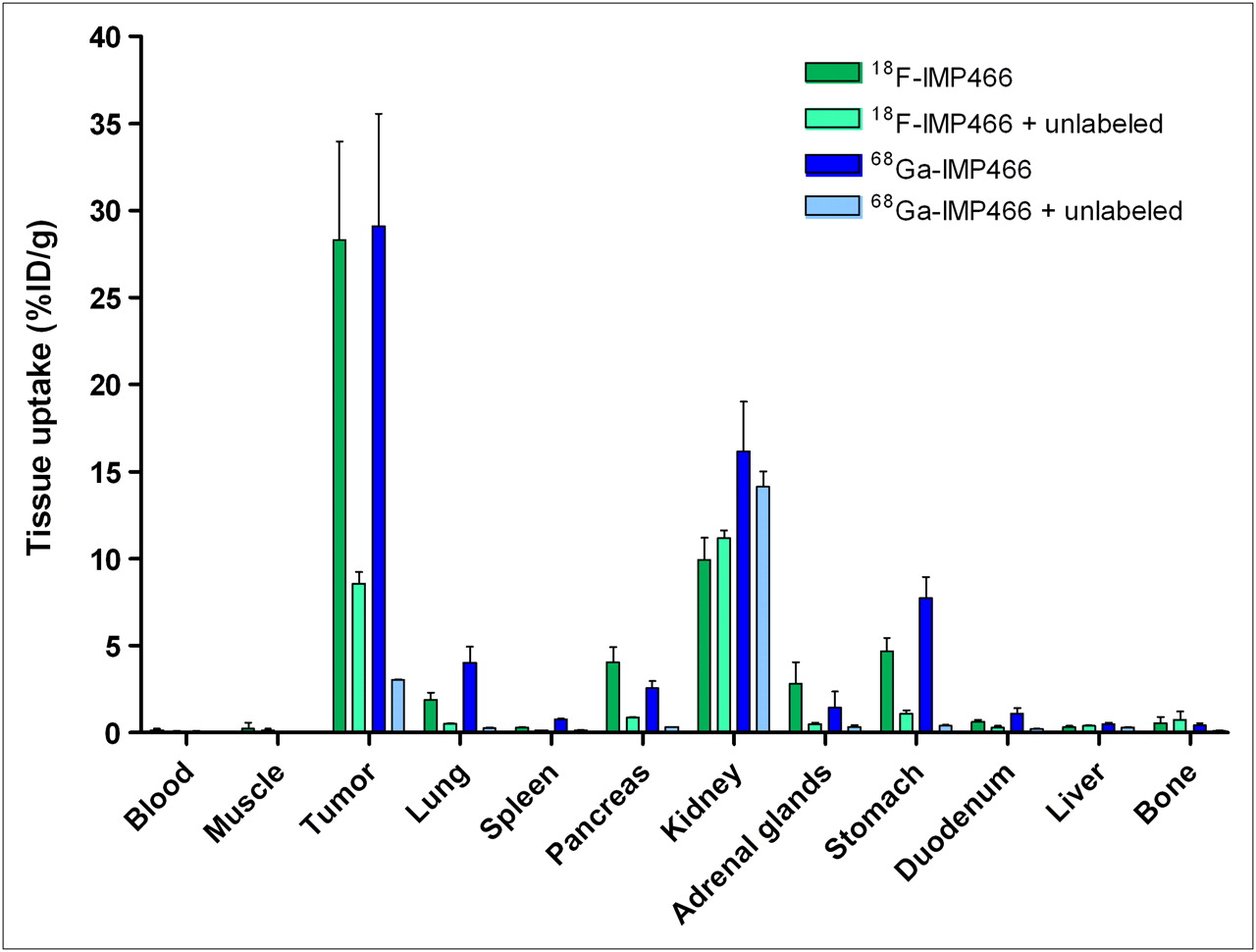
Biodistribution of 18F-IMP466 and 68Ga-IMP466 at 2 h after injection in AR42J tumor–bearing mice (n = 5/group). As control, mice in separate groups (n = 3/group) received excess of unlabeled octreotide to demonstrate receptor specificity. Tumors weighed 0.04–0.33 g.
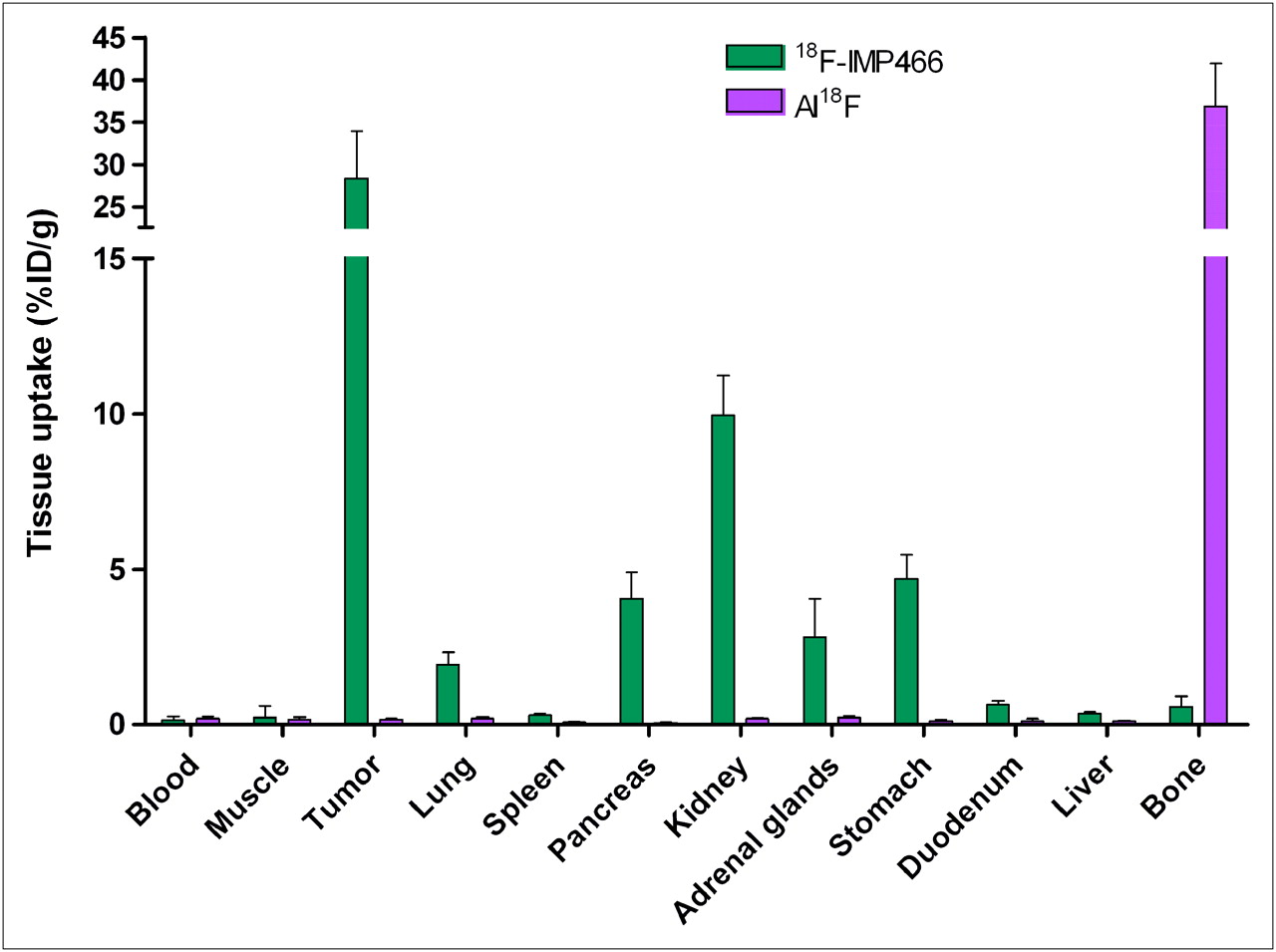
Biodistribution of 18F-IMP466 and unbound Al18F at 2 h after injection in AR42J tumor–bearing mice (n = 5/group). Tumors weighed 0.07–0.36 g.
Tumor uptake of 68Ga-IMP466 (29.2 ± 0.5 %ID/g, 2 h after injection) was similar to that of 18F-IMP466 (P = 0.7). Lung uptake of 68Ga-IMP466 was 2-fold higher than that of 18F-IMP466 (4.0 ± 0.9 %ID/g vs. 1.9 ± 0.4 %ID/g, respectively). In addition, kidney retention of 68Ga-IMP466 was significantly higher than that of 18F-IMP466 (16.2 ± 2.86 %ID/g vs. 10.0 ± 1.3 %ID/g, respectively; P < 0.01).
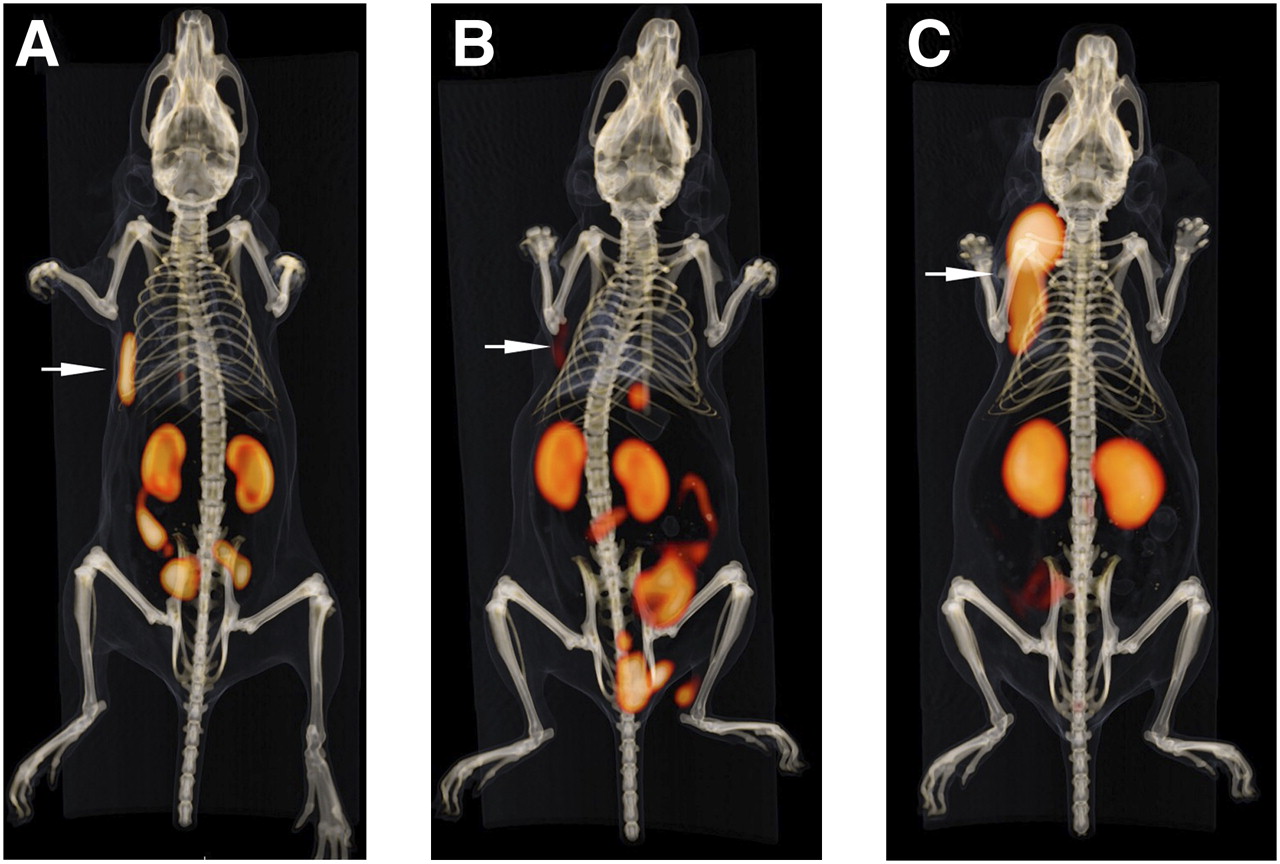
Fused PET and CT scans are shown in Figure 7. PET scans largely corroborated the biodistribution data. Both 18F-IMP466 and 68Ga-IMP466 showed high uptake in the tumor and high retention in the kidneys. The activity in the kidneys was localized mainly in the renal cortex. Some intestinal uptake was observed on the scans of 18F-IMP466. The PET/CT scans also demonstrated that the Al18F was stably chelated by the NOTA chelator, because no bone uptake was observed.

Anterior 3-dimensional volume-rendered projections of fused PET and CT scans of mice with subcutaneous AR42J tumor on right flank injected with 18F-IMP466 (A), 18F-IMP466 in presence of excess of unlabeled IMP466 (B), and 68Ga-IMP466 (C). Arrows indicate tumors. Scans were recorded at 2 h after injection.
DISCUSSION
The radiolabeling of peptides with 18F involves laborious and time-consuming procedures and first requires the synthesis of an 18F-labeled synthon. We showed recently that a NOTA-conjugated pretargeting peptide (IMP449) could be labeled with 18F in a 2-step, 1-pot reaction by creating Al18F (14). Here, we describe the optimized 18F-labeling of NOTA-conjugated octreotide (IMP466) using the same approach. The biodistribution of the 18F-NOTA-octreotide was compared with that of 68Ga-NOTA-octreotide. We demonstrated that the affinity of 18F-NOTA-octreotide was at least as good as that of 111In-DTPA-octreotide and was comparable with values reported in the literature for DOTA-octreotate and DOTA-d-Phe1,Tyr3-octreotide (15).
In the present study, we used the directly coupled NOTA derivative rather than the isothiocyanatobenzyl-derivatized NOTA to diminish the effect of the phenyl group on the lipophilicity of the peptide. For 68Ga labeling, it has been demonstrated that this NOTA variant is as good as the isothiocyanatobenzyl-activated NOTA (16). After preparing the Al18F complex, the chelation of this complex by NOTA was investigated in various buffers. We showed that chelation of Al18F could be performed in sodium acetate, MES, and HEPES buffer, but, remarkably, the chelation failed in sodium citrate buffer. The labeling reaction requires fairly high NOTA-peptide concentrations. At the concentrations studied, the reaction was most efficient at an IMP466 concentration of 1.8 mM. This optimal concentration is in the same range as reported for other peptide fluorination methods (3,4) but lower than the optimal concentration required in the classic N-succinimidyl-4-18F-fluorobenzoate method (2).
Subsequently, we investigated the effect of the amount of Al3+ on the radiolabeling yield. The Al3+ concentration greatly affects the labeling yield. First, the 18F− complexes with the Al3+, then the Al18F complex is chelated by NOTA. After incubation at elevated temperature, the reaction mixture is purified using HPLC to separate the Al18F-labeled IMP466 from unchelated Al18F, Al3+-labeled IMP466, and unlabeled IMP466. We showed that the optimal concentration of AlCl3 was 2 mM, whereas both at lower and at higher concentrations, the radiolabeling yield decreased. These data indicate that there is a delicate balance between 18F− and Al3+. Lowering the amount of Al3+ will lead to less 18F associated with Al3+. In contrast, a higher amount will yield more undesired NOTA-peptide labeled with Al3+ rather than labeled with Al18F. Because the formation of Al18F is performed at pH 4.1, we believe that the form of the aluminum in sodium acetate buffer is probably Al3+(AcO−)3F−. In any case, the molar ratio of aluminum to 18F is such that it is unlikely that any AlF2 or AlF3 is formed (17). We attempted to isolate the 2 isomers but observed that there was a rapid equilibrium between them. Therefore, no further studies of separate isomers were possible.
The lipophilicity of both the 18F-labeled and the 68Ga-labeled IMP466 was in the same range as reported for other 111In-labeled octreotide analogs (18). Other 18F-labeled octreotide analogs, such as 18F-fluoropropionyl octreotide (log P, −0.07 ± 0.01) and Nα-(1-deoxy-d-fructosyl)-Nε-(2-18F-fluoropropionyl)-Lys0-Tyr3octreotate (18F-FP-Gluc-TOCA) (log P, −1.70 ± 0.02), displayed a higher lipophilicity (19). The biodistribution of 18F-FP-Gluc-TOCA was similar to the biodistribution of 18F-IMP466, but tumor uptake of the latter compound was 2-fold higher than that of the carbohydrate analog.
The biodistribution of 18F-IMP466 was similar to that of 68Ga-IMP466, indicating that the Al18F complex did not affect the in vivo characteristics of octreotide. Both peptides showed an equally high tumor uptake at 2 h after injection, with lower uptake in all other organs. The in vivo studies also showed the excellent stability of the Al18F-NOTA complex, because no significant bone uptake could be measured, and the intact product was isolated in the urine.
The current method can be performed in 1 pot, is fast (45 min), yields carrier-free fluorinated peptide, and does not affect the pharmacokinetics of octreotide. In most 18F-labeling strategies for peptides and proteins, a fluorinated synthon needs to be synthesized first. In general, this fluorination is based on a nucleophilic substitution that requires laborious azeotropic drying of the 18F-fluoride–kryptofix complex. Examples of such a synthon are succinimidyl-18F-fluorobenzoate (2), 4-18F-fluorobenzaldehyde (3), and 2-18F-fluoropropionic acid 4-nitrophenyl ester (20). Subsequently, the synthon is reacted with the (functionalized) peptide, leading to longer synthesis times and lower overall yields. In addition, these techniques also lead to increased lipophilicity, because most synthons contain aromatic groups and this might affect the biodistribution profile. It has been reported that this effect can be counteracted by carbohydration (5). However, this requires considerable peptide modification.
More recently, a method based on silicon–fluorine has been published, in which the 18F is bound to a silicon-containing building block in a single step (12,13). Although somewhat similar to our approach, the silicon–18F initially proved to be unstable but could be stabilized by the addition of tertiary butyl groups. This, however, led to a strong increase in lipophilicity (log P, 1.3 ± 0.1).
Finally, click chemistry has been explored for the radiofluorination of peptides (8–10). Although the yield of these click chemistry–based labeling procedures based on the alkyne–azide cycloaddition is excellent (>80%), the method starts with the fluorination of an azide or alkyne, such as fluoro(ethyl)azide or a fluoroalkyne. This requires azeotropic drying of the fluoride, resulting in a time-consuming multistep procedure.
Compared with 68Ga labeling, the Al18F method is easy and versatile, mainly because both methods are based on a chelator-derivatized peptide. One of the advantages of the Al18F method is the longer half-life of 18F, allowing PET at later time-points after injection of the tracer.
CONCLUSION
Our new approach combines the ease of chelator-based radiolabeling methods with the advantages of 18F (i.e., half-life, availability, and positron energy). The F-labeled NOTA-octreotide could be synthesized in less than 45 min without the need to synthesize an 18F synthon. Moreover, the fluorinated peptide was stable in vitro and in vivo and has excellent tumor-targeting properties. Therefore, this fluorination method is a promising facile and versatile fluorination procedure.
Acknowledgments
We thank Maarten Brom, Jonathan Disselhorst, and Bianca Lemmers-de Weem for technical assistance. This work was funded in part by NIH grant 1R43 EB003751-01A1 from the National Institute of Biomedical Imaging and Bioengineering, Bethesda, Maryland. William J. McBride and David M. Goldenberg are employed or have financial interest in Immunomedics, Inc.
Footnotes
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication June 3, 2009.
- Accepted for publication November 16, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- 18F-AlF-NOTA-Octreotide Outperforms 68Ga-DOTATATE/NOC PET in Neuroendocrine Tumor Patients: Results from a Prospective, Multicenter Study
- Radiohybrid Ligands: A Novel Tracer Concept Exemplified by 18F- or 68Ga-Labeled rhPSMA Inhibitors
- 18F-AlF-Labeled Biomolecule Conjugates as Imaging Pharmaceuticals
- Molecular Imaging of Gastroenteropancreatic Neuroendocrine Tumors: Current Status and Future Directions
- Preclinical Evaluation of a High-Affinity 18F-Trifluoroborate Octreotate Derivative for Somatostatin Receptor Imaging
- A Comparative Study of Radiolabeled Bombesin Analogs for the PET Imaging of Prostate Cancer
- Three Methods for 18F Labeling of the HER2-Binding Affibody Molecule ZHER2:2891 Including Preclinical Assessment
- PET of Tumors Expressing Gastrin-Releasing Peptide Receptor with an 18F-Labeled Bombesin Analog
- Imaging of Human Epidermal Growth Factor Receptor Type 2 Expression with 18F-Labeled Affibody Molecule ZHER2:2395 in a Mouse Model for Ovarian Cancer
- Targeting Somatostatin Receptors: Preclinical Evaluation of Novel 18F-Fluoroethyltriazole-Tyr3-Octreotate Analogs for PET