


Abstract
Radiolabeled bombesin (BBN) analogs that bind to the gastrin-releasing peptide receptor (GRPR) represent a topic of active investigation for the development of molecular probes for PET or SPECT of prostate cancer (PCa). RM1 and AMBA have been identified as the 2 most promising BBN peptides for GRPR-targeted cancer imaging and therapy. In this study, to develop a clinically translatable BBN-based PET probe, we synthesized and evaluated 18F-AlF- (aluminum-fluoride) and 64Cu-radiolabeled RM1 and AMBA analogs for their potential application in PET imaging of PCa. Methods: 1,4,7-triazacyclononane, 1-glutaric acid-4,7 acetic acid (NODAGA)–conjugated RM1 and AMBA were synthesized and tested for their GRPR-binding affinities. The NODAGA-RM1 and NODAGA-AMBA probes were further radiolabeled with 64Cu or 18F-AlF and then evaluated in a subcutaneous PCa xenograft model (PC3) by small-animal PET imaging and biodistribution studies. Results: NODAGA-RM1 and NODAGA-AMBA can be successfully synthesized and radiolabeled with 64Cu and 18F-AlF. 64Cu- and 18F-AlF-labeled NODAGA-RM1 demonstrated excellent serum stability and tumor-imaging properties in the in vitro stability assays and in vivo imaging studies. 64Cu-NODAGA-RM1 exhibited tumor uptake values of 3.3 ± 0.38, 3.0 ± 0.76, and 3.5 ± 1.0 percentage injected dose per gram of tissue (%ID/g) at 0.5, 1.5, and 4 h after injection, respectively. 18F-AlF-NODAGA-RM1 exhibited tumor uptake values of 4.6 ± 1.5, 4.0 ± 0.87, and 3.9 ± 0.48 %ID/g at 0.5, 1, and 2 h, respectively. Conclusion: The high-stability, efficient tumor uptake and optimal pharmacokinetic properties highlight 18F-AlF-NODAGA-RM1 as a probe with great potential and clinical application for the PET imaging of prostate cancer.
Prostate cancer (PCa) is the second leading cause of cancer death in men in the United States (1). Conventional imaging techniques (e.g., CT) have played a relatively minor role in the management of organ-confined PCa, and novel imaging techniques including PET could play an important role in the detection of localized and locally recurrent PCa (2). It is crucial to develop novel imaging probes and techniques for the primary diagnosis, follow-up, and monitoring of the recurrence of PCa. The clinical evaluation of certain PET probes, such as 18F-FDG and radiolabeled acetate and choline, has highlighted the pivotal role that PET might play in the imaging and characterization of PCa patients. The discovery of novel PET molecular probes is expected to dramatically facilitate the diagnosis, prognosis, and stratification of PCa patients for effective therapeutic regimens (3–8).
Serum prostate-specific antigen testing has been a valuable tool in the detection, staging, and monitoring of PCa, but lack of specificity results in a high negative biopsy rate. Some biomarkers with high specificity for the disease in tissue have been identified, such as the gastrin-releasing peptide receptor (GRPR). Because it takes advantage of highly sensitive molecular imaging methods such as PET, GRPR-targeted PET imaging can be used for the detection, staging, and monitoring of PCa in a noninvasive and specific manner. GRPR is overexpressed in many human epithelial malignancies, including PCa (9,10), breast cancer (11), and lung cancer (12). GRPR-binding ligands represent potential diagnostic and therapeutic agents for the targeting of GRPR-positive tumors. Examples of such GRPR-binding ligands include mammalian gastrin-releasing peptide (GRP) and its amphibian homolog bombesin (BBN) (13). These 2 peptides share homology in a C-terminal region, BBN(7-14), which is composed of the following 8 amino acid residues: Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2. BBN(7-14) has been extensively used for the development of molecular probes for the imaging of PCa (14–19). Moreover, a variety of BBN analogs, including both agonists and antagonists, has been radiolabeled with different radionuclides for GRPR-targeted PCa imaging and therapy (20–28).
Among those GRPR-targeted peptides, RM1 (DOTA-CH2CO-G-4-aminobenzoyl-f-W-A-V-G-H-Sta-L-NH2, antagonist) (24) and AMBA (DOTA-CH2CO-G-4-aminobenzoyl-Q-W-A-V-G-H-L-M-NH2, agonist) (23) have been shown to be 2 promising GRPR-targeted peptide platforms for developing imaging and therapeutic agents for PCa. These 2 peptides have been radiolabeled with radiometals (111In, 68Ga, and 64Cu) for SPECT and PET imaging of PCa (24–27). AMBA has also been radiolabeled with 177Lu for radionuclide therapy of PCa (23,28). All of these radiocomplexes exhibit efficient tumor accumulation and favorable in vivo properties, which support their potential clinical application. More interestingly, 68Ga-AMBA–based PET imaging has recently been found to be superior to metabolism-based imaging using 18F-methylcholine for scintigraphy of PCa (25). These studies clearly suggest that the increased likelihood of successfully translating a BBN probe into the clinic can be achieved using AMBA and RM1 scaffolds.
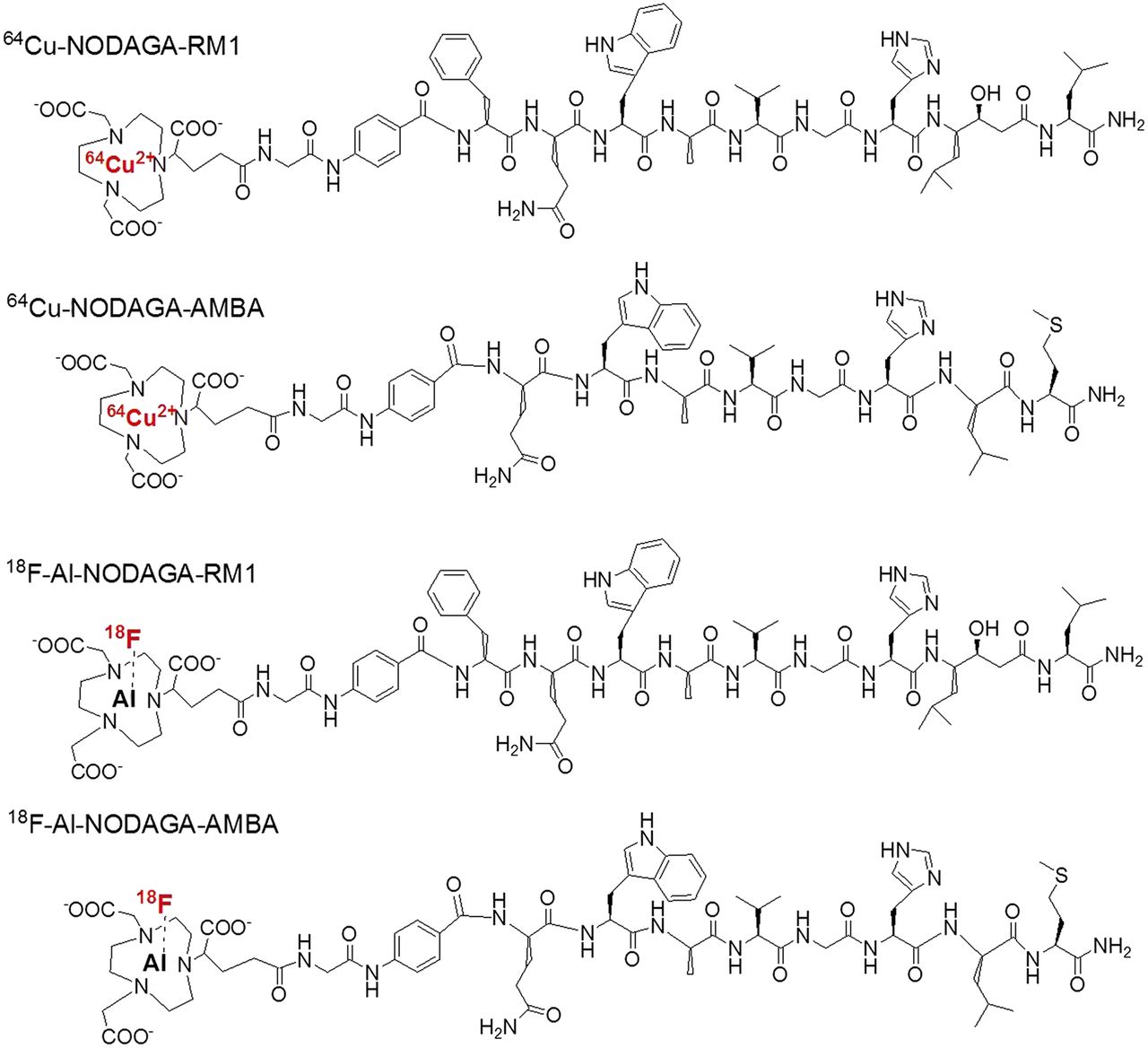
However, 18F-labeled RM1 and AMBA have not been explored, and currently there are no published data providing a head-to-head comparison of the performance of 18F-labeled RM1 and AMBA analogs. 18F is the most widely used radionuclide for clinical PET because of its ideal physical properties (∼100% positron efficiency, low β+ energy [0.64 MeV], and physical half-life of 109.7 min). Moreover, 64Cu has also gained extensive interest in the context of PET probe development and has been actively explored to label peptides for clinical imaging because of its many advantages including simple radiolabeling chemistry. Compared with 18F, however, 64Cu suffers from lower availability, longer half-life, and potential higher radiation dose (29). In this study, we synthesized 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA)–conjugated RM1 and AMBA (NODAGA-RM1, NODAGA-AMBA) peptides and then radiolabeled them via a simple 1-step chelation reaction (18F-AlF [aluminum-fluoride] or 64Cu) in aqueous phase (30–33) to generate 18F-AlF-NODAGA-RM1, 18F-AlF-NODAGA-AMBA, 64Cu-NODAGA-RM1, and 64Cu-NODAGA-AMBA (Fig. 1). We further evaluated these PET probes and compared them head-to-head in PC3 tumor–bearing mice.
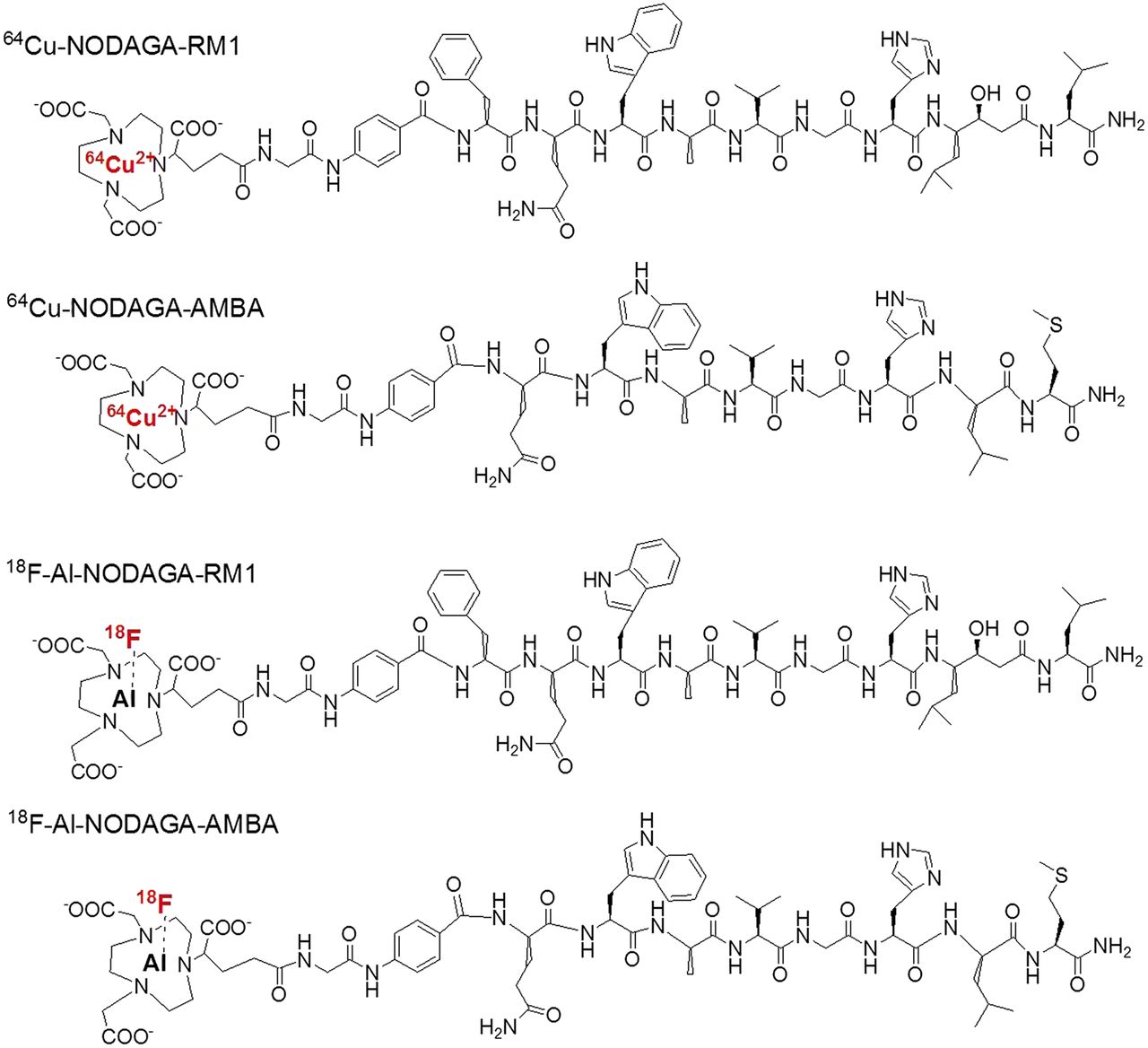
Schematic structures of 64Cu-NODAGA-RM1, 64Cu-NODAGA-AMBA, 18F-AlF-NODAGA-RM1, and 18F-AlF-NODAGA-AMBA.
MATERIALS AND METHODS
All of the chemicals obtained commercially were of analytic grade and used without further purification. The PC3 human prostate carcinoma cell line was obtained from the American Type Culture Collection. Male nude mice were purchased from the Charles River Laboratory. 64Cu was provided by the Department of Medical Physics, University of Wisconsin at Madison. No-carrier-added 18F was obtained from an in-house PETtrace cyclotron (GE Healthcare). The Sep-Pak C18 cartridges were obtained from Waters. The syringe filters and polyethersulfone membranes (pore size, 0.22 μm; diameter, 13 mm) were obtained from Nalgene Nunc International. 125I-[Tyr4]BBN was purchased from PerkinElmer. 2,2′-(7-(1-carboxy-4-((2,5-dioxopyrrolidin-1-yl)oxy)-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (NOTAGA-NHS) was purchased from CheMatech.
Reversed-phase high-performance liquid chromatography (HPLC) was performed using a Dionex 680 chromatography system with a UVD 170U absorbance detector and 105S single-channel model radiation detector (Carroll & Ramsey Associates). A Vydac protein and peptide column (218TP510; C18, 5 μm, 250 × 10 mm) was used for semipreparative HPLC at a flow rate of 4 mL/min. The mobile phase was maintained at a constant 95% for solvent A (0.1% trifluoroacetic acid [TFA] in water) and 5% for solvent B (0.1% TFA in acetonitrile [MeCN]) at 0–5 min and was subsequently changed to 35% for solvent A and 65% for solvent B at 35 min. A Vydac protein and peptide column (218TP510; C18, 5 μm, 250 × 4.6 mm) was used for the analytic HPLC at a flow rate of 1 mL/min. The mobile phase was changed from an initial 95% for solvent A and 5% for B (from 0 to 2 min) to 35% for solvent A and 65% for solvent B at 32 min. The recorded data were processed using Chromeleon software (version 6.50; Dionex). The ultraviolet absorbance was monitored at 218 nm, and the identification of the peptides was confirmed based on the ultraviolet spectrum using a PDA detector (Dionex).
Chemistry and Radiochemistry
Preparation of NODAGA-RM1 and NODAGA-AMBA
The G-4-aminobenzoyl-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (abbreviated as AMBA) and G-4-aminobenzoyl-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 (abbreviated as RM1) peptides (Fig. 1) were synthesized. Then the 2 peptides were conjugated with NODAGA-NHS to obtain NODAGA-AMBA and NODAGA-RM1, respectively. The detailed procedures are described in the supplemental file (supplemental materials are available at http://jnm.snmjournals.org).
64Cu and 18F Labeling
The 64Cu labeling was performed as previously described (34). Briefly, NODAGA-RM1 or NODAGA-AMBA (2 nmol each) was dissolved in NaOAc buffer (0.1 M, pH 5.0) and labeled with 64Cu (74 MBq [2 mCi]) for 60 min at 37°C. The labeled peptides were then purified by analytic HPLC. The radioactive peaks containing the desired products were collected and rotary-evaporated to remove the solvent. The products were then formulated in phosphate-buffered saline (1×, pH 7.4) and passed through a 0.22-μm Millipore filter into a sterile vial for the subsequent in vitro and in vivo experiments. The labeling yield was above 90% for all of the probes.
The 18F-AlF labeling was performed as previously described (33). A QMA SepPak Light cartridge (Waters) fixed with 18F (1.1 GBq [30 mCi]) was washed with 2.5 mL of metal-free water. The 18F was then eluted from the cartridge with 400 μL of 0.4 M KHCO3, from which 200-μL fractions were taken. The pH of the solution was adjusted to 4 with metal-free glacial acetic acid. AlCl3 (2 mM, 3 μL) in 0.1 M sodium acetate buffer (pH 4) and 10 μL of peptide (1 mg/mL in dimethyl sulfoxide) were then added to the reaction solution sequentially. The reaction mixtures were incubated at 100°C for 15 min. The labeled peptides were then purified by semipreparative HPLC. The fractions containing the desired products were collected and rotary-evaporated to remove the solvent. The products were reconstituted in phosphate-buffered saline and passed through a 0.22-μm Millipore filter into sterile vials for the subsequent in vitro and in vivo experiments.
Cell-Binding Assays
The cell-binding assay was performed similarly as previously reported (19,35). The detailed procedures are included in the supplemental file.
Mouse Serum Stability
The in vitro stability of the PET probes was evaluated by incubation with mouse serum (1 mL) at 37°C. The solutions were filtered using a NanoSep 10 K centrifuge (Pall Corp.) to isolate low-molecular-weight radiocomplexes. The samples were analyzed by the radio-HPLC, and the percentages of intact PET probe were determined by quantifying the peaks corresponding with the intact probe and degradation products.
Small-Animal PET Imaging
The animal procedures were performed according to a protocol approved by the Stanford University Institutional Animal Care and Use Committee. The establishment of the PC3 tumor mice model and the procedure for small-animal PET are shown in the supplemental file.
The PC3 xenograft–bearing mice were injected with approximately 1.85 MBq (50 μCi) of either 64Cu-NODAGA-RM1 or 64Cu-NODAGA-AMBA via the tail vein (n = 4 for each group). At the indicated times after injection (0.5, 1.5, and 4 h), the mice were anesthetized with isoflurane (5% for induction and 2% for maintenance in 100% O2) using a knock-down box. Five-minute static scans were then obtained. The PC3 xenograft–bearing mice were injected with 0.37 MBq (10 μCi) of either 18F-AlF-NODAGA-RM1 or 18F-AlF-NODAGA-AMBA probe via the tail vein (n = 5 for each group). Blocking studies were performed via tail vein injection of the 18F probe with cold AMBA (10 mg/kg of body weight) (n = 5). At 0.5, 1, and 2 h after injection, the small-animal PET images were obtained.
The small-animal PET images were reconstructed using the 2-dimensional ordered-subsets expectation maximization algorithm. No background correction was performed. The regions of interest (ROIs; 5 pixels for coronal and transaxial slices) were designated over the tumor on decay-corrected whole-body coronal images. The maximum counts per pixel per minute were obtained from the ROIs and converted to counts per milliliter per minute using a calibration constant. On the basis of an assumed tissue density of 1 g/mL, the ROIs were converted to counts per gram per min. The image ROI–derived percentage of the injected radioactive doses per gram of tissue (%ID/g) values were determined by dividing counts per gram per minute by injected dose. No attenuation correction was performed.
Animal Biodistribution Studies
The PC3 xenograft–bearing nude mice (n = 4 for each group) were injected with approximately 1.85 MBq (50 μCi) of 64Cu-AlF-NODAGA-RM1 via the tail vein and sacrificed at 2 and 24 h after injection. The tumor and normal tissues of interest were removed and weighed, and their levels of radioactivity were measured using a γ-counter. The uptake of radioactivity in the tumor and normal tissues was expressed as %ID/g.
Similarly, PC3 xenograft–bearing nude mice (n = 5 for each group) were injected with approximately 0.37 MBq (10 μCi) of 18F-AlF-NODAGA-RM1 via the tail vein and sacrificed at 2 h after injection. The 18F-AlF-NODAGA-RM1 blocking study was performed by coinjection of the probe with the AMBA peptide (10 mg/kg of body weight) via the tail vein.
Statistical Analysis
The quantitative data are expressed as the mean values ± SD. The mean values were compared using a 1-way ANOVA and Student t test. P values of less than 0.05 were considered statistically significant.
RESULTS
Chemistry and Radiochemistry
The G-4-aminobenzoyl-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (chemical formula for [M+H]+: C64H88N16O13+; calculated molecular weight [MW], 1,289.7 Da; measured MW, 1,289.6 Da) and G-4-aminobenzoyl-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 (chemical formula for [M+H]+: C53H73N15O11+; calculated MW, 1,116.5 Da; measured MW, 1,116.5 Da) peptides were synthesized successfully using a solid-phase peptide synthesizer. The NODAGA peptides were then prepared by direct conjugation of NODAGA-NHS with AMBA or RM1, resulting in 80% yield and greater than 90% purity. Both HPLC and mass spectroscopy were used to confirm the identity of the product. The retention time of the purified NODAGA-RM1 was 25.6 min, and the molecular mass was measured as 1,646.8 Da for [M+H]+ (chemical formula: C79H112N19O20; calculated MW, 1,646.8 Da). The retention time of NODAGA-AMBA was 21.2 min, and the m/z was measured as 1,473.7 Da for [M+H]+ (chemical formula: C67H97N18O18S; calculated MW, 1,473.7 Da).
64Cu-NODAGA-RM1 and 64Cu-NODAGA-AMBA were synthesized in high yield (>85%), and their specific activities were calculated as greater than 29.6 GBq/μmol (800 mCi/μmol). The radiosynthesis of the 18F-AlF-NODAGA-RM1 and 18F-AlF-NODAGA-AMBA was completed within 40 min, with decay-corrected yields of 5.6% ± 1.1% and 4.9% ± 1.3%, respectively, and the radiochemical purity of the probes was more than 95%. The specific activities of the purified 18F-AlF-NODAGA peptides were calculated as greater than 1.85 GBq/μmol (50 mCi/μmol).
Cell-Binding Affinity Assay
The receptor-binding affinity assay results for the RM1, NODAGA-RM1, AMBA, and NODAGA-AMBA probes are shown in Figure 2. All of these peptides inhibited the GRPR binding of 125I-[Tyr4]BBN on the PC3 cells in a concentration-dependent manner. The half maximal inhibitory concentration values for RM1, NODAGA-RM1, AMBA, and NODAGA-AMBA were 0.25 ± 0.04, 4.6 ± 1.0, 0.17 ± 0.07, and 1.9 ± 0.50 nM (n = 4), respectively. These results suggest that the incorporation of the NODAGA motif reduced the GRPR-binding affinity of the original peptides but that the resulting bioconjugates still possessed significantly high affinities, warranting further biologic evaluation.

Inhibition of GRPR-binding by 125I-[Tyr4]BBN on PC3 cells by RM1, NODAGA-RM1, AMBA, and NODAGA-AMBA (n = 4, mean ± SD).
Serum Stability of Radiolabeled NODAGA Peptides
Both 64Cu-NODAGA-RM1 and 18F-AlF-NODAGA-RM1 exhibited favorable stability in mouse serum. More than 90% of the probes remained intact after 1 h of incubation in mouse serum at 37°C. However, 64Cu-NODAGA-AMBA and 18F-AlF-NODAGA-AMBA exhibited degradation under the same conditions (supplemental data).
Small-Animal PET Imaging Studies
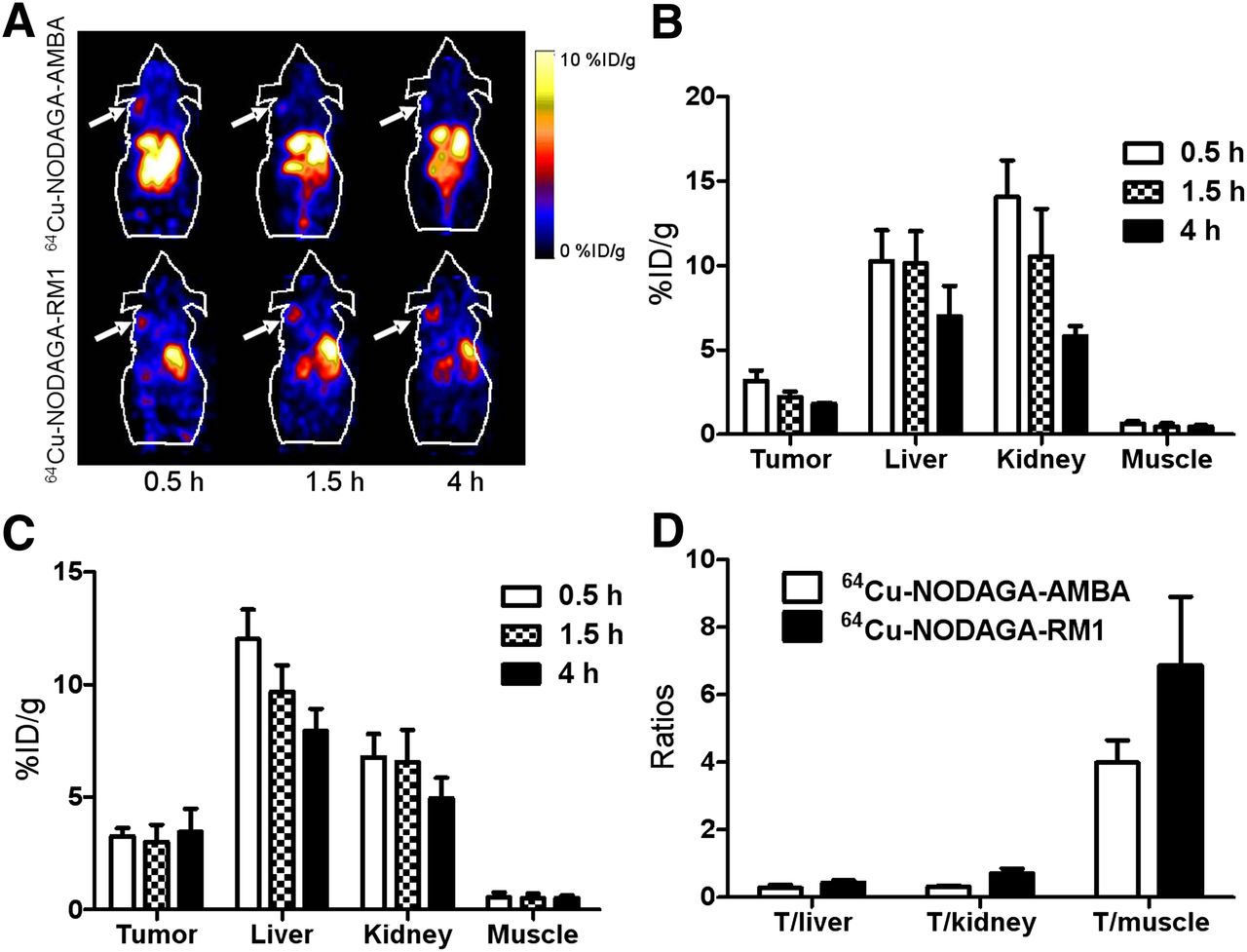
Representative decay-corrected coronal images of static scans at different time points after injection are shown in Figures 3–5. The PC3 tumors were clearly visualized using all of the PET probes. The PC3 tumors were visualized using 64Cu-NODAGA-RM1 and 64Cu-NODAGA-AMBA, with favorable tumor-to-background contrast, even at 0.5 h (Fig. 3A). Interestingly, 64Cu-NODAGA-RM1 exhibited persistent accumulation within the tumor until 4 h after injection, whereas 64Cu-NODAGA-AMBA exhibited more rapid tumor clearance and significantly reduced signal within the tumor at 4 h after injection. Further image quantification analysis confirmed the visualization results. The uptake of 64Cu-NODAGA-RM1 within the PC3 tumors was 3.3 ± 0.38, 3.0 ± 0.76, and 3.5 ± 1.0 %ID/g at 0.5, 1.5, and 4 h after injection, respectively. In contrast, 64Cu-NODAGA-AMBA exhibited only 3.2 ± 0.60, 2.2 ± 0.33, and 1.8 ± 0.10 %ID/g tumor uptake at 0.5, 1.5, and 4 h after injection, respectively (Figs. 3B and 3C). Moreover, both PET probes exhibited significant accumulation in both the liver and the kidneys (>10 %ID/g), indicating their clearance through both the hepatobiliary and the renal systems. Quantification analysis also revealed that 64Cu-NODAGA-RM1 exhibited significantly reduced kidney uptake at 0.5 and 1.5 h, compared with 64Cu-NODAGA-AMBA (P < 0.05), whereas no significant differences were observed between their levels of either kidney uptake at 4 h after injection or liver uptake at all of the time points (Figs. 3B and 3C). Increased tumor–to–normal-tissue ratios (tumor to liver, tumor to kidney, and tumor to muscle) were observed for 64Cu-NODAGA-RM1, compared with those for 64Cu-NODAGA-AMBA (Fig. 3D). Overall, 64Cu-NODAGA-RM1 exhibited superior in vivo performance, compared with the 64Cu-NODAGA-AMBA probe.
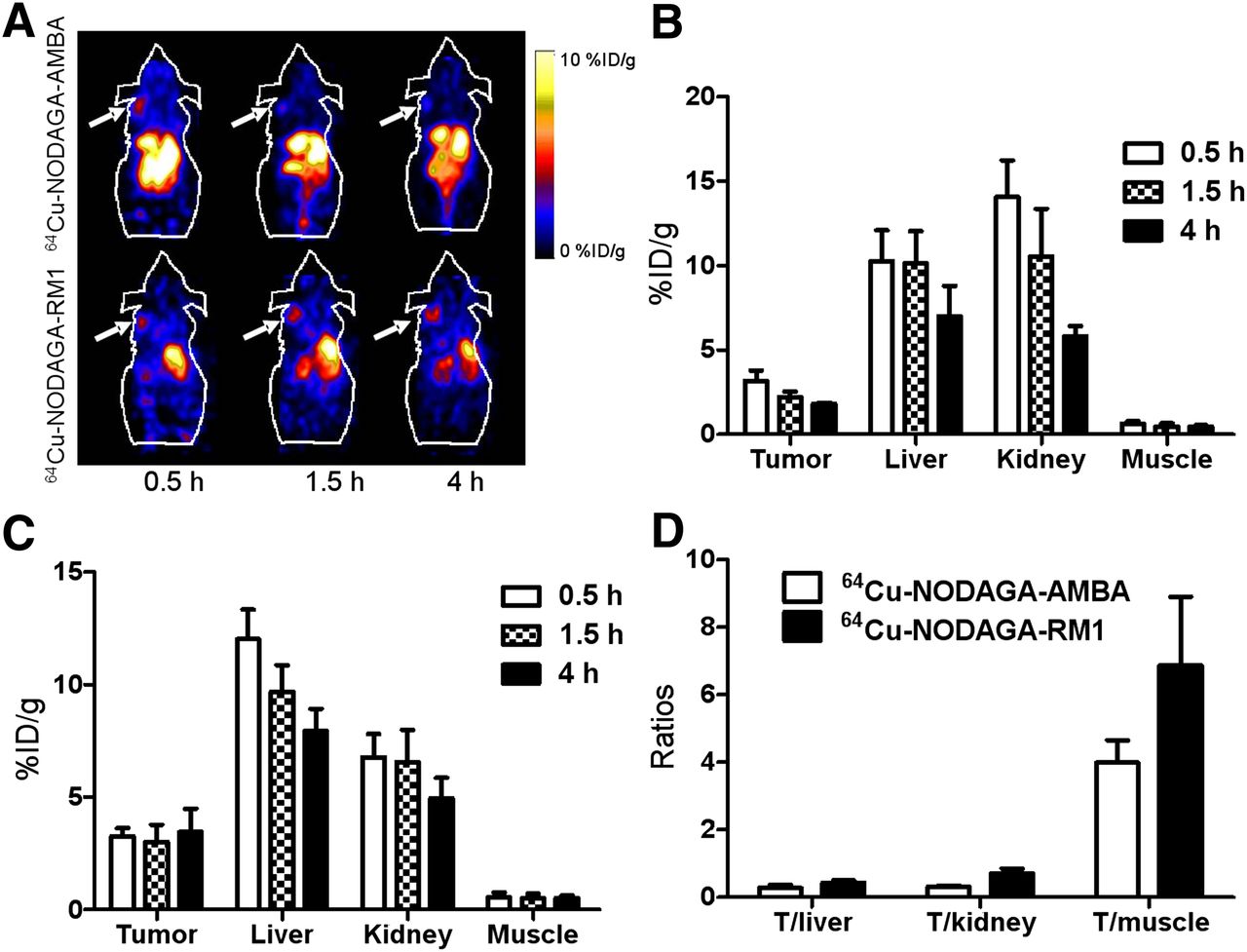
(A) Decay-corrected whole-body coronal small-animal PET images of PC3 tumor–bearing male nude mice from static scan at 0.5, 1.5, and 4 h after injection of 64Cu-NODAGA-AMBA and 64Cu-NODAGA-RM1. Tumors are indicated by arrows. (B and C) Small-animal PET quantification of tumors and major organs at 0.5, 1.5, and 4 h after injection of 64Cu-NODAGA-AMBA (B) or 64Cu-NOTAGA-RM1 (C). (D) Comparison of tumor–to–normal-tissue (muscle, kidney, and liver) ratios of 64Cu-NODAGA-RM1 and 64Cu-NODAGA-AMBA at 4 h after injection.
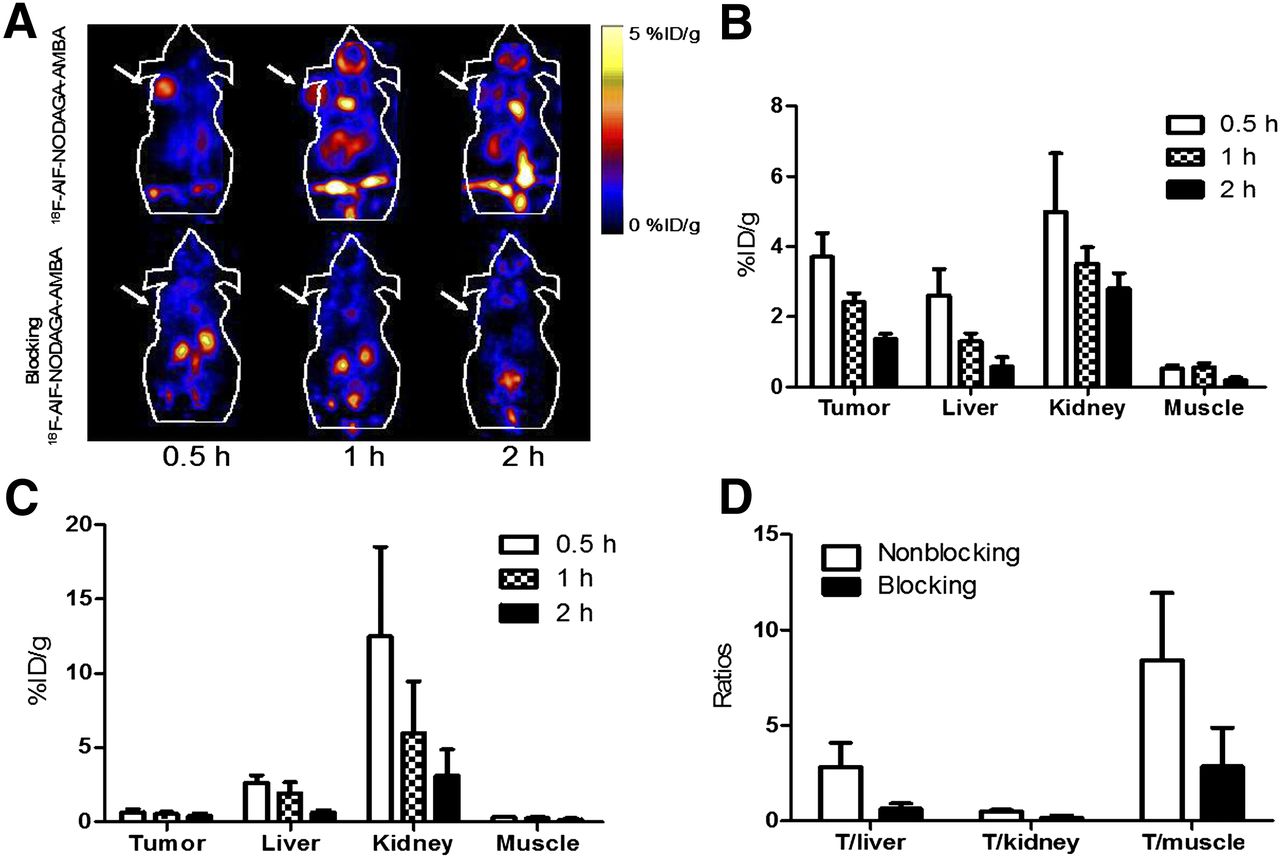
(A) Decay-corrected whole-body coronal small-animal PET images of PC3 tumor–bearing male nude mice from static scan at 0.5, 1, and 2 h after injection of 18F-AlF-NODAGA-AMBA without (top) or with (bottom) coinjection of AMBA as blocking agent (10 mg/kg of body weight). Tumors are indicated by arrows. (B and C) Small-animal PET image quantification of tumors and major organs at 0.5, 1, and 2 h after injection of 18F-AlF-NODAGA-AMBA without (B) or with (C) AMBA as blocking agent (10 mg/kg of body weight). (D) Comparison of tumor–to–normal-tissue (muscle, kidney, and liver) ratios of 18F-AlF-NODAGA-AMBA without or with AMBA at 2 h after injection.

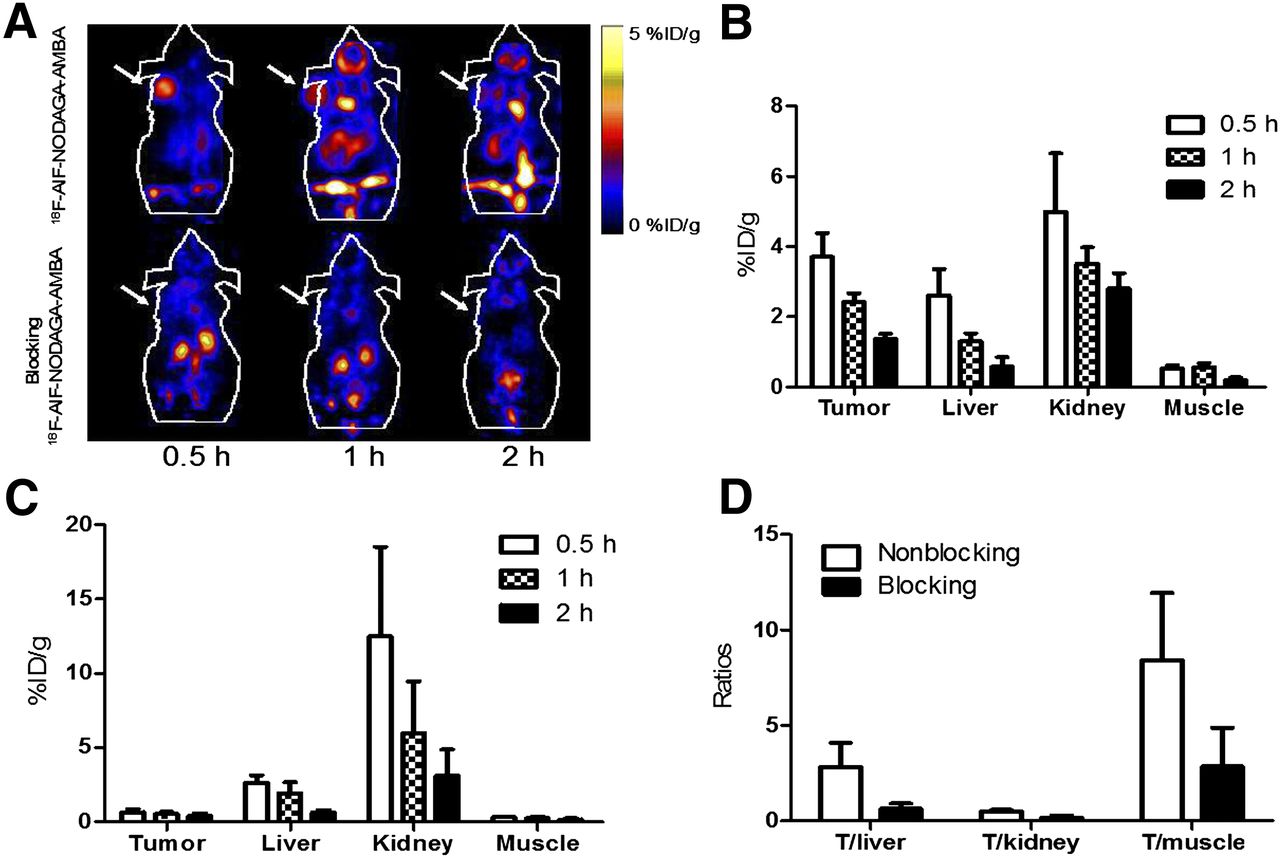
(A) Decay-corrected whole-body coronal small-animal PET images of PC3 tumor–bearing male nude mice from static scan at 0.5, 1, and 2 h after injection of 18F-AlF-NODAGA-RM1 without (top) or with (bottom) AMBA as blocking agent (10 mg/kg of body weight). Tumors are indicated by arrows. (B and C) Small-animal PET quantification of tumors and major organs at 0.5, 1, and 2 h after injection of 18F-AlF-NODAGA-RM1 without (B) or with (C) AMBA as blocking agent (10 mg/kg of body weight). (D) Comparison of tumor–to–normal-tissue (muscle, kidney, and liver) ratios of 18F-AlF-NODAGA-RM1 without or with AMBA at 2 h after injection.
The small-animal PET images and quantification analyses for 18F-AlF-NODAGA-AMBA and 18F-AlF-NODAGA-RM1 are shown in Figures 4 and 5, respectively. As clearly shown in the PET images (Figs. 4A and 5A, top), both probes accumulated rapidly within the tumor, and favorable tumor-to-background imaging contrasts were achieved at 0.5 h after injection. Furthermore, the coinjection of the unlabeled GRPR-targeting peptide AMBA significantly reduced the uptake of both probes (P < 0.05), resulting in significantly reduced tumor-to-background contrast in vivo (Figs. 4A and 5A, bottom). These results clearly demonstrated the excellent in vivo targeting ability and specificity of both probes. Moreover, similar to the 64Cu-labeled peptides, 18F-AlF-NODAGA-AMBA exhibited more rapid tumor clearance than 18F-AlF-NODAGA-RM1 (Figs. 4A and 5A, top). Quantification analysis indicated that the tumor uptake of 18F-AlF-NODAGA-AMBA was 3.7 ± 0.70, 2.4 ± 0.24, and 1.4 ± 0.13 %ID/g at 0.5, 1, and 2 h, respectively (Fig. 4B), whereas the tumor uptake of 18F-AlF-NODAGA-RM1 was 4.6 ± 1.5, 4.0 ± 0.87, and 3.9 ± 0.48 %ID/g at 0.5, 1, and 2 h, respectively (Fig. 5B). At 1 and 2 h after injection, the tumor uptake of 18F-AlF-NODAGA-RM1 was significantly increased, compared with that of 18F-AlF-NODAGA-AMBA (P < 0.05). The coinjection of 18F-AlF-NODAGA-AMBA or 18F-AlF-NODAGA-RM1 with blocking doses of AMBA also significantly reduced their tumor uptake (Figs. 4B and 5B). The tumor uptake was 0.66 ± 0.19, 0.55 ± 0.15, and 0.39 ± 0.18 %ID/g for 18F-AlF-NODAGA-AMBA and 2.5 ± 0.36, 1.6 ± 0.34, and 0.93 ± 0.14 %ID/g for 18F-AlF-NODAGA-RM1 at 0.5, 1, and 2 h after injection, respectively. Lastly, consistent with their corresponding 64Cu-labeled counterparts, both 18F-AlF-NODAGA-AMBA and 18F-AlF-NODAGA-RM1 were excreted through both the hepatobiliary and the renal systems. Taken together, these results clearly suggest 18F-AlF-NODAGA-RM1 to be a more promising probe than 18F-AlF-NODAGA-AMBA.
Biodistribution Studies
Next, we performed a biodistribution study of 64Cu-NODAGA-RM1 and 18F-AlF-NODAGA-RM1. The results are shown in Table 1. The tumor uptake for 64Cu-NODAGA-RM1 at 2 and 24 h after injection were 4.30 ± 0.98 and 2.62 ± 0.32 %ID/g, respectively. 64Cu-NODAGA-RM1 also showed relatively low kidney and liver uptake.
Biodistribution Data of 64Cu-NODAGA-RM1 and 18F-AlF-NODAGA-RM1 in PC3 Tumor–Bearing Mice
Without the presence of AMBA peptide as a blocking agent at 2 h after injection, the tumor, liver, and kidney uptake for 18F-AlF-NODAGA-RM1 (~0.37 MBq [10 μCi]/mouse, n = 5) was 5.2 ± 0.84, 2.5 ± 0.96, and 4.6 ± 1.9 %ID/g, respectively. In contrast, the same organ uptake for the blocking group was 1.8 ± 0.30, 1.9 ± 0.19, and 7.3 ± 2.1 %ID/g, which is consistent with the PET imaging results. Increased pancreatic uptake in the control tumor mice (5.1 ± 1.4 %ID/g), which is in contrast with reduced pancreatic uptake in the blocking mice (0.54 ± 0.13 %ID/g), was also observed. Again, the significantly reduced uptake of the probe in the GRPR-positive tissues, including the PC3 tumor and pancreas, confirmed the receptor-targeting specificity of the probes.
DISCUSSION
In this study, 2 important BBN peptides, AMBA and RM1, were labeled with 18F and used for PET imaging of PCa for the first time. These 2 peptides were first modified with the chelator NODAGA to render the bioconjugates more easily labeled with 64Cu and 18F-AlF to form stable radiocomplexes. After modification with NODAGA, the half maximal inhibitory concentration of each of the conjugated peptides was still within the nanomolar range (Fig. 2). Therefore, they were further radiolabeled with 64Cu and 18F-AlF and tested in vivo.
The radiolabeling of NODAGA-AMBA and NODAGA-RM1 with 64Cu is a relatively simple and straightforward process. Al-F chelation chemistry has been used for 18F labeling, as pioneered by McBride et al., and this strategy has many advantages, such as short labeling time and water compatibility (31). We previously synthesized 18F-AlF-NOTA-RGD2 as a PET probe for tumor angiogenesis imaging (33). The favorable in vivo performance of several 18F-AlF–labeled peptides reported thus far, together with their short routes of radiosynthesis, highlight the potential accelerated clinical translation of 18F-labeled peptides using this novel radiofluorination strategy. Thus, we explored this straightforward, single-step and powerful labeling strategy to radiofluorinate AMBA and RM1. Interestingly, compared with the yield produced by the 18F-AlF radiolabeling of NOTA-RGD2 (17.9%), the yields produced by NODAGA-RM1 and NODAGA-AMBA were approximately 5% decay-corrected, which was likely caused by the different chelating moieties used (36). But these labeling yields are still sufficient for producing enough PET probes for further in vitro and in vivo evaluation.
According to the in vitro serum stability assay and in vivo imaging results, both the 64Cu- and the 18F-labeled NODAGA-AMBA probes were less stable than the NODAGA-RM1 probes. Moreover, the 64Cu- and 18F-labeled NODAGA-RM1 probes exhibited more favorable in vivo tumor retention and imaging quality than the 64Cu- and 18F-labeled NODAGA-AMBA probes (Figs. 3–5). BBN peptide antagonists and agonists exhibited different stabilities and in vivo behavior. Schroeder et al. reported that the antagonist demobesin-1 exhibits superior in vivo stability, increased tumor uptake and retention, and faster pancreatic and renal clearance than the other 4 GRPR agonists (37). Mansi et al. also compared the performance of the radioantagonist 111In-RM1 with the radioagonist 111In-AMBA (24). 111In-RM1 demonstrated relatively lower affinity for GRPR but more favorable pharmacokinetics and targeting properties, as supported by increased tumor uptake and tumor–to–normal-tissue ratios. 111In-RM1 appeared to be superior to AMBA for in vivo SPECT imaging and for the targeted radiotherapy of GRPR-positive tumors. Consistent with these findings, our study using 64Cu and 18F-AlF demonstrated that antagonist RM1-based PET probes exhibit significantly increased stability and more optimal imaging performance than the agonist AMBA-based probes.
Recently, NOTA-8-Aoc-BBN(7-14)-NH2 was labeled with 18F using the Al-18F method described by Dijkgraaf et al., and the results showed that the AlF method does not affect the in vivo behavior of the peptide (38). For example, the uptake of 18F-labeled NOTA-8-Aoc-BBN(7-14)-NH2 in the PC3 tumors was 2.15 ± 0.55 %ID/g, which is similar to the 64Cu-labeled NOTA-8-Aoc-BBN(7-14) conjugate (3.59 ± 0.70 %ID/g) at 1 h after injection (18). In our study, 18F-AlF-NODAGA-RM1 also exhibited uptake similar to 64Cu-NODAGA-RM1 (4.6 ± 1.5 vs. 3.3 ± 0.38 at 0.5 h and 4.0 ± 0.87 vs. 3.0 ± 0.76 at 1 h, respectively). More importantly, 18F-AlF-NODAGA-RM1 exhibited increased tumor uptake, compared with 18F-AlF-NOTA-8-Aoc-BBN (7-14)NH2 (4.0 ± 0.87 vs. 2.15 ± 0.55), highlighting the advantages of using RM1 for PET probe development. Taken together, on the basis of their favorable in vitro serum stability and in vivo tumor imaging properties (Figs. 3 and 5), 64Cu-NODAGA-RM1 and 18F-AlF-NODAGA-RM1 warrant further investigation for clinical translation. In particular, the radioantagonist 18F-AlF-NODAGA-RM1 demonstrated wide availability, easy radiosynthesis, increased tumor uptake, and tumor-to-background contrast. Moreover, considering the different physical properties and availability of 64Cu and 18F, 18F-AlF-NODAGA-RM1 holds significant promise for imaging PCa in clinical applications.
CONCLUSION
NODAGA-RM1 and NODAGA-AMBA have been successfully prepared and radiolabeled with 64Cu and 18F. The radiolabeling methods are simple and straightforward. In vitro assay and in vivo studies showed that 64Cu- and 18F-labeled NODAGA-AMBA are limited by certain undesirable properties, such as relatively low stability and rapid tumor clearance. However, both 64Cu-NODAGA-RM1 and 18F-AlF-NODAGA-RM1 exhibit excellent in vitro serum stability and in vivo tumor imaging properties. In particular, 18F-AlF-NODAGA-RM1 exhibited increased tumor uptake and favorable pharmacokinetics, representing a highly promising probe for the PET imaging of PCa in clinical applications.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was partially supported by DOD-PCRP-NIA PC094646, NCI 5R01 CA119053, and the In vivo Cellular Molecular Imaging Center (ICMIC) grant P50 CA114747. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Nov. 6, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication February 11, 2013.
- Accepted for publication August 2, 2013.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A mass spectrometry-based method for the determination of in vivo biodistribution of tumor targeting small molecule-metal conjugates
- Development of Improved Tumor-Residualizing, GRPR-Targeted Agents: Preclinical Comparison of an Endolysosomal Trapping Approach in Agonistic and Antagonistic Constructs
- 18F-AlF-Labeled Biomolecule Conjugates as Imaging Pharmaceuticals
- Dual-Modality Imaging of Prostate Cancer with a Fluorescent and Radiogallium-Labeled Gastrin-Releasing Peptide Receptor Antagonist
- Bombesin-Targeted PET of Prostate Cancer
- Preclinical Comparison of Al18F- and 68Ga-Labeled Gastrin-Releasing Peptide Receptor Antagonists for PET Imaging of Prostate Cancer
- N-Terminal Modifications Improve the Receptor Affinity and Pharmacokinetics of Radiolabeled Peptidic Gastrin-Releasing Peptide Receptor Antagonists: Examples of 68Ga- and 64Cu-Labeled Peptides for PET Imaging
- Effect of Chelate Type and Radioisotope on the Imaging Efficacy of 4 Fibrin-Specific PET Probes