


Abstract
Tariquidar, a potent, nontoxic, third-generation P-glycoprotein (P-gp) inhibitor, is a possible reversal agent for central nervous system drug resistance. In animal studies, tariquidar has been shown to increase the delivery of P-gp substrates into the brain by severalfold. The aim of this study was to measure P-gp function at the human blood–brain barrier (BBB) after tariquidar administration using PET and the model P-gp substrate (R)-11C-verapamil. Methods: Five healthy volunteers underwent paired (R)-11C-verapamil PET scans and arterial blood sampling before and at 2 h 50 min after intravenous administration of tariquidar (2 mg/kg of body weight). The inhibition of P-gp on CD56-positive peripheral lymphocytes of each volunteer was determined by means of the 123Rh efflux assay. Tariquidar concentrations in venous plasma were quantified using liquid chromatography/mass spectrometry. Results: Tariquidar administration resulted in significant increases (Wilcoxon test for paired samples) in the distribution volume (DV, +24% ± 15%) and influx rate constant (K1, +49% ± 36%) of (R)-11C-verapamil across the BBB (DV, 0.65 ± 0.13 and 0.80 ± 0.07, P = 0.043; K1, 0.034 ± 0.009 and 0.049 ± 0.009, P = 0.043, before and after tariquidar administration, respectively). A strong correlation was observed between the change in brain DV after administration of tariquidar and tariquidar exposure in plasma (r = 0.90, P = 0.037). The mean plasma concentration of tariquidar achieved during the second PET scan (490 ± 166 ng/mL) corresponded to 100% inhibition of P-gp function in peripheral lymphocytes. Conclusion: Tariquidar significantly increased brain penetration of (R)-11C-verapamil–derived activity due to increased influx. As opposed to peripheral P-gp function, central P-gp inhibition appeared to be far from complete after the administered tariquidar dose.
The multidrug efflux transporter P-glycoprotein (P-gp) is highly expressed at the luminal endothelium of the human blood–brain barrier (BBB). Because of its high transport capacity and abundance, P-gp limits entry into the central nervous system (CNS) for many CNS-active drugs and is believed to contribute to patient-to-patient variability in response to CNS pharmacotherapy (1). Limited brain entry of therapeutic drugs mediated by active efflux transport is believed to contribute to the phenomenon of drug resistance in neurologic disorders, such as epilepsy and depression (transporter hypothesis of drug resistance) (1). A promising strategy to enhance drug penetration across the BBB and thereby overcome drug resistance is the administration of new-generation P-gp inhibitors, which were originally developed as an adjunctive therapy for drug-resistant cancers and are characterized by a high potency in the nanomolar range, lack of toxicity, and lack of cytochrome P450 interactions (2).
Animal studies with the third-generation P-gp inhibitors zosuquidar, elacridar, and tariquidar demonstrated substantially increased brain levels of cytostatics, antiepileptics, virostatics, and other CNS drugs (3–5). However, in most animal studies higher than clinically used doses of P-gp inhibitors were given, resulting in chemical knockout of P-gp and thereby an overestimation of the extent of clinically relevant P-gp inhibition in humans. In addition, considerable species differences in cerebral P-gp functionality have been described (5,6). Few studies have so far investigated the magnitude of P-gp inhibition at the human BBB. In a proof-of-concept study in 8 healthy volunteers, Sadeque et al. showed that the administration of loperamide caused respiratory depression, a CNS pharmacologic effect, when the P-gp inhibitor quinidine was coadministered (7). A follow-up clinical study using reduction in pupil diameter as a surrogate marker for central opioid effect, on the other hand, suggested that CNS activity of loperamide in humans was not affected by the administration of tariquidar at 2 mg/kg of body weight (BW) (8). However, the approach used in that study might suffer from lack of sensitivity in the measurement of moderate levels of P-gp inhibition, as expected after the administration of clinically used doses of P-gp modulators. PET with the radiolabeled P-gp substrate (R)-11C-verapamil is a validated method to measure P-gp function at the human BBB (9). It has been shown that the distribution volume (DV) of (R)-11C-verapamil is inversely related to cerebral P-gp activity (10). A study in nonhuman primates showed that brain uptake of racemic 11C-verapamil was increased by 4.61-fold after infusion of the second-generation P-gp inhibitor valspodar at a dose of 20 mg/kg/2 h (11). A similarly conducted PET study in healthy human volunteers revealed a significant but less pronounced (∼90%) increase of brain activity uptake after cerebral P-gp inhibition with cyclosporine A, although higher than clinically recommended doses of cyclosporine A were given (12).
Tariquidar, which has been developed up to clinical phase 3, is one of the most potent and selective third-generation P-gp inhibitors known to date (13). Tariquidar is a noncompetitive inhibitor of P-gp and not a substrate of it (14); tariquidar also inhibits the breast cancer resistance protein but at higher concentrations than it inhibits P-gp. However, unlike first- and second-generation P-gp inhibitors tariquidar does not inhibit multidrug resistance–associated proteins (MRPs), such as MRP1 (15). Preclinical studies in monkeys have shown that tariquidar is rapidly excreted via the hepatobiliary pathway, with an elimination half-life of 5 h. In monkey plasma, most of the tariquidar-related material (>95%) was found to exist as unchanged parent compound (16). Tariquidar has been tested up to a dose of 2 mg/kg of BW in 48 healthy subjects and more than 500 cancer patients in the course of 9 clinical trials (13). In some of these studies, in addition to the clinical response of cancer patients, the inhibition of 123Rh efflux from CD56-positive lymphocytes and reduction in clearance of 99mTc-sestamibi from the liver were assessed as surrogate efficacy markers of tariquidar-induced inhibition of P-gp in the periphery (17,18). Interestingly, greater resistance of P-gp at the BBB to inhibition, compared with other tissues, has been suggested (3,8).
For translating the concept of P-gp modulation for improving CNS delivery of drugs to the clinic, studies measuring surrogate markers of transport activity and inhibition at the human BBB are needed to validate P-gp modulation at the BBB, define the magnitude and time frame of inhibition, and—in an ultimate step—assess the clinical effect of P-gp modulation at the human BBB on CNS pharmacotherapy (19). In an ideal setting, a selective P-gp modulator would only transiently enhance BBB penetration of a targeted therapeutic drug.
We have previously performed a (R)-11C-verapamil small-animal PET study in rats showing that tariquidar given at a dose of 15 mg/kg resulted in a 12-fold increase of the (R)-11C-verapamil DV (10). In this study, we translated the set-up used in the previous small-animal PET study to human subjects to investigate, for the first time to our knowledge, the effect of tariquidar on the functional activity of P-gp at the human BBB.
MATERIALS AND METHODS
The study was performed at the Department of Clinical Pharmacology at the Medical University of Vienna. It was performed as a single-dose pilot clinical study in 5 healthy male subjects with a mean age ± SD of 32 ± 8 y and a mean BW ± SD of 74.2 ± 5.4 kg. Healthy volunteers were free of any medication known to interfere with cytochrome P450 enzymes or P-gp function.
The study protocol was approved by the local Ethics Committee and was performed in accordance with the Declaration of Helsinki (1964) in the revised version of 2000 (Edinburgh), the Guidelines of the International Conference of Harmonization, the Good Clinical Practice Guidelines, and the Austrian drug law (Arzneimittelgesetz). All subjects were given a detailed description of the study, and their written consent was obtained before they enrolled in the study.
PET and Experimental Procedures
On study day, each study participant underwent 2 consecutive PET scans of 120- and 40-min duration, with an interval of 2 h between the 2 scans (Fig. 1). During PET, each subject was positioned supine on the imaging bed of the PET camera, with the head in a fixation device to avoid movement artifacts. PET images were acquired with an Advance PET scanner (GE Medical Systems) run in 3-dimensional mode. To correct for tissue attenuation of photons, a 5-min transmission scan using two 68Ge (400-MBq) pin sources was recorded before radiotracer injection. One venous and one arterial catheter were placed on 1 arm for drug infusion, and a second venous catheter was placed on the other arm to obtain blood samples for quantification of tariquidar concentrations in plasma. At the start of each PET scan, 384 ± 13 MBq of (R)-11C-verapamil (5.2 ± 0.36 MBq/kg), synthesized from (R)-norverapamil (ABX Advanced Biochemical Compounds) and 11C-methyl triflate and dissolved in a volume of 10 mL of physiologic saline solution/ethanol (9/1, v/v), was intravenously injected over 20 s. Dynamic PET and arterial blood sampling were started at the time of radiotracer injection. The following frame sequence was used for PET: 1 × 15, 3 × 5, 3 × 10, 2 × 30, 3 × 60, 2 × 150, 2 × 300, and 10 × 600 s for scan 1 and 1 × 15, 3 × 5, 3 × 10, 2 × 30, 3 × 60, 2 × 150, 2 × 300, and 2 × 600 s for scan 2. During both scan 1 and scan 2, arterial blood samples were drawn at intervals of 7 s during the first 3 min after radiotracer injection and subsequently at 3.5, 5, 10, 20, 30, 55, 70, and 100 min after radiotracer injection (scan 1) and at 3.5, 5, 10, 20, 30, and 40 min after radiotracer injection (scan 2).
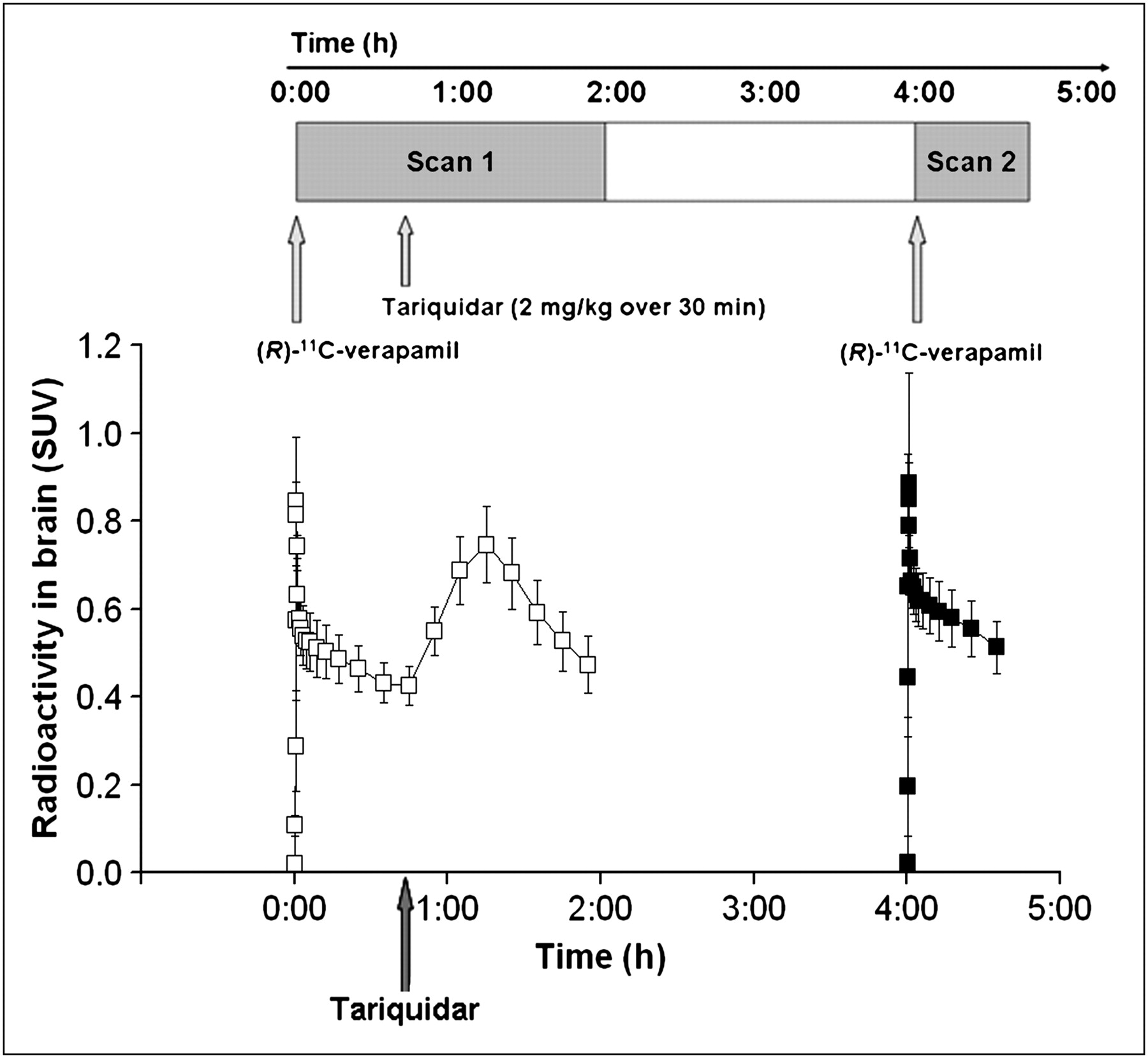
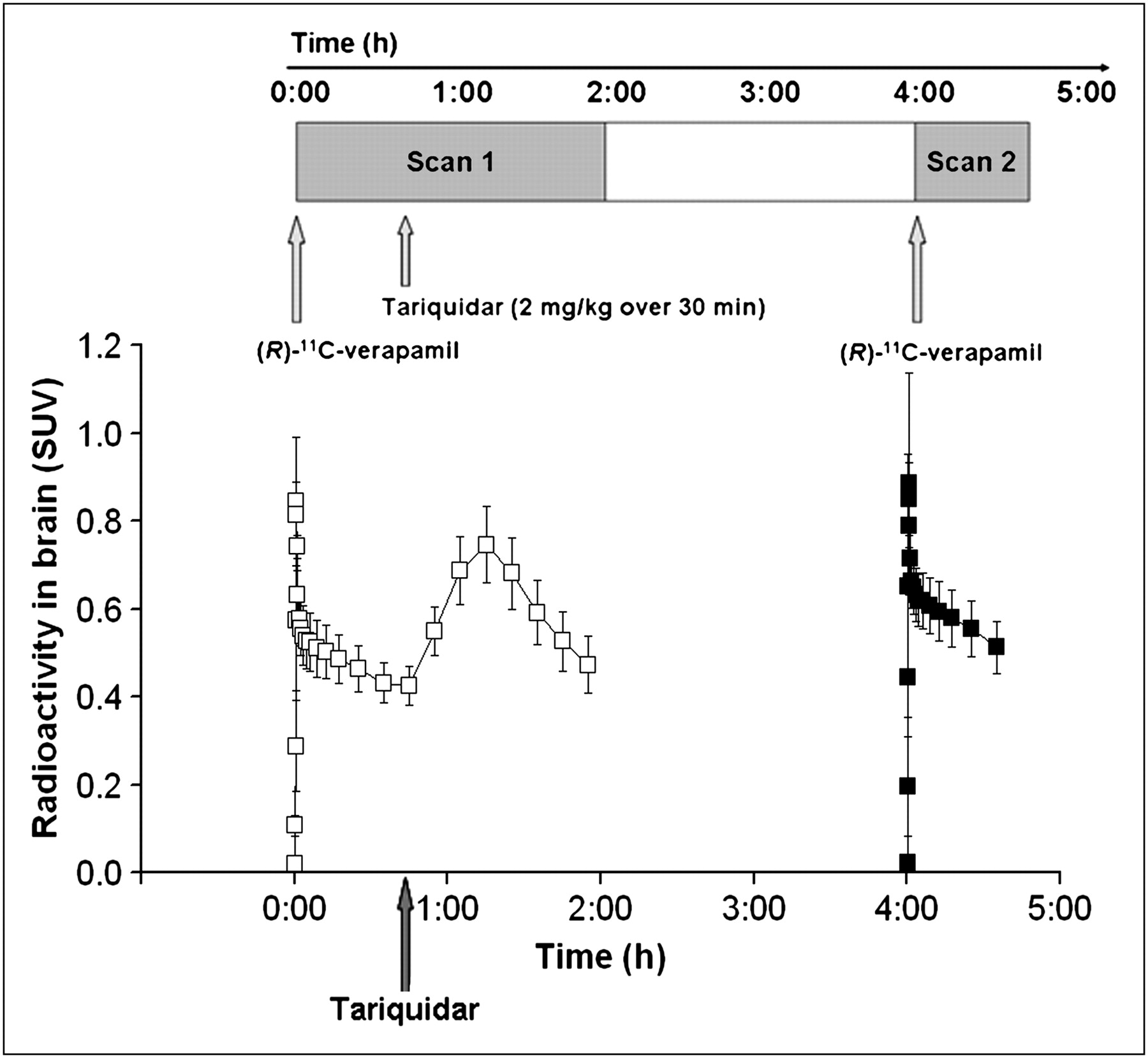
Time–activity curves (mean SUV ± SD) of (R)-11C-verapamil in whole-brain gray matter for paired PET scans. Tariquidar (2 mg/kg of BW) was administered as intravenous infusion over 30 min at 40 min after start of PET scan 1 (see timeline at top of figure). Scan 1 = □; Scan 2 = ▪.
During scan 1 (i.e., at 40 min after tracer injection), tariquidar was administered at a dose of 2 mg/kg of BW as an intravenous infusion over 30 min. Scan 2 was performed at 2 h 50 min after the end of tariquidar infusion (Fig. 1). Four-milliliter venous blood samples were drawn before (predose) and at 5, 15, 30, 60, 120, 200, 220, 240, 360, 480, 600, 720, and 1,440 min after the start of infusion of tariquidar.
Study Medication
Tariquidar was administered at a dose of 2 mg/kg of BW. Ten-milliliter vials of tariquidar for intravenous infusion containing 7.5 mg of tariquidar freebase per milliliter of 20% ethanol/80% propylene glycol were provided by AzaTrius Pharmaceuticals Pvt Ltd. The volume (mL) of concentrate for injection was calculated as BW (kg) × 2/7.5. Aqueous dextrose solution (5%, g/v) was added to yield a final volume of 250 mL, which was administered over 30 min.
PET Data Analysis
PET data were reconstructed by means of iterative reconstruction using ordered-subset expectation maximization with 28 subsets and 2 iterations. The loop filter (gaussian) was set to a full width at half maximum of 4.3 mm, and a postfiltering algorithm of 6.00 mm in full width at half maximum was applied. Attenuation correction was performed using the manufacturer's segmentation algorithm for transmission data. T1-weighted MRI scans had been recorded within 1 mo before the PET scan. MRI and PET data were processed with Analyze 8.0 (Biomedical Imaging Resource, Mayo Foundation) and SPM5 (Wellcome Department of Imaging Neuroscience, UCL) software as published previously (20). A whole-brain gray matter region of interest was defined using the Hammersmith n30r83 3-dimensional maximum probability atlas of the human brain (21). Radioactivity concentrations (kBq/g of tissue) were normalized to the injected radiotracer amount and expressed as standardized uptake values (SUVs). The radioactivity concentration data were combined to provide time–activity curves for the whole observation period.
Blood and Metabolite Analysis
Radioactivity counts in aliquots of arterial blood and plasma were measured with a γ-counter. Plasma samples were analyzed for radiolabeled metabolites of (R)-11C-verapamil using a previously described combined solid-phase extraction (SPE)/high-performance liquid chromatography (HPLC) assay (22,23). The polar radiolabeled metabolites of (R)-11C-verapamil (11C-formaldehyde and related species) were determined by SPE, and the lipophilic radiolabeled metabolites of (R)-11C-verapamil (i.e., the 11C-labeled N-dealkylation products D-617 and D-717), which are also P-gp substrates, were measured with HPLC. The 10-, 20-, 30-, and 40-min plasma samples of both scan 1 and scan 2 were analyzed for radiolabeled metabolites of (R)-11C-verapamil using the SPE/HPLC assay. Because of time constraints due to the short half-life of 11C, the 3.5-, 5-, 55-, 70-, and 100-min plasma samples were analyzed by SPE only. Plasma protein binding of (R)-11C-verapamil was determined by incubating plasma samples obtained before and after the administration of tariquidar with (R)-11C-verapamil during 30 min at 37°C, followed by ultrafiltration using Amicon Microcon YM-10 centrifugal filter devices (Millipore Corp.). The total concentration of radioactivity in whole blood was used for vascular correction of the PET data, assuming that blood constitutes 5% of brain volume.
Kinetic Modeling of (R)-11C-Verapamil
PET datasets (uncorrected for blood activity) from 0 to 40 min for scan 1 and scan 2 were used for data analysis. A standard 1-tissue 2-rate-constant (1T2K) or 2-tissue 4-rate-constant (2T4K) compartment model was fitted to the (R)-11C-verapamil time–activity curves in whole-brain gray matter (9,20) to estimate the influx and efflux rate constants of activity across the BBB (K1 and k2, respectively) and the distribution volume (DV). The arterial input function of (R)-11C-verapamil was constructed by correcting total decay-corrected activity concentrations in arterial plasma (kBq/mL) for the fraction of polar radiolabeled metabolites of (R)-11C-verapamil, as determined by SPE, and by subsequent interpolation of the activity data. Fits were performed using the weighted nonlinear-least-squares method as implemented in the Optimization Toolbox of MATLAB (The MathWorks). Goodness of fit was assessed by the visual inspection of observed and predicted concentrations versus time, correlation between observed and predicted concentrations, randomness of the residuals (runs test), and estimation of parameter uncertainties (variances) from the inverse of the appropriate Fisher information matrix. To obtain a model-independent estimate of DV, Logan graphical analysis was applied to the PET and arterial plasma data using MATLAB (24). To analyze the time course of the effect of tariquidar administration on brain activity during scan 1, an indirect-response pharmacokinetic–pharmacodynamic (PK–PD) model was used (25) (see supplemental data; supplemental materials are available online only at http://jnm.snmjournals.org).
Statistical Analysis
All statistical analysis was performed using SPSS software (version 16 for Windows; SPSS Inc.). Model parameters of (R)-11C-verapamil in the brain (K1, k2, DV) before and after the administration of tariquidar were compared using the Wilcoxon test for paired samples. Correlations between pharmacokinetic parameters of tariquidar in plasma and model parameters of verapamil in the brain were calculated using the Spearman rank coefficient.
Analysis of Tariquidar Concentration in Plasma
Tariquidar was assayed in human plasma using combined liquid chromatography tandem mass spectrometry (LC/MS). Plasma samples (100 μL) were spiked with elacridar as an internal standard. The drug was extracted from plasma samples using XAD4 polymeric adsorbent beads. The analytes were eluted from the beads with ethyl acetate/methanol (1/1, v/v). The eluate was concentrated to dryness, redissolved in methanol/water (1/9, v/v), and injected into the LC/MS system. LC/MS analysis was performed using a Thermo Scientific Surveyor HPLC system coupled to a Thermo Finnigan TSQ Quantum Discovery MAX mass spectrometer, with the flow rate maintained at 200 μL/min. Separation was performed using an Agilent Zorbax XDB C18 Eclipse column (2.1 × 50 mm; 3.5 μm). A gradient method using a mixture of methanol containing 0.1% aqueous formic acid (solvent A) and 0.2% aqueous formic acid (solvent B) was used. From the tariquidar concentration–time profiles in plasma, pharmacokinetic parameters were estimated as described in the supplemental data.
Measurement of Tariquidar-Induced Inhibition of P-gp Activity in Peripheral White Blood Cells by 123Rh Efflux Assay
Native fresh whole-blood samples (18 mL) were drawn from each volunteer before the administration of tariquidar. The assay for inhibition of P-gp in CD56-positive lymphocytes was performed with slight modifications according to previously published methods (17). Suspensions of isolated white blood cells (1 mL) were incubated at 37°C with 123Rh (0.2 μg/mL) and 10 different dilutions of tariquidar in dimethlysulfoxide (10 μL), resulting in tariquidar concentrations from 0.01 nM to 1 μM (0.0084–839 ng/mL). All data points were measured in duplicate. Accumulation levels of 123Rh were measured by fluorescence-activated cell sorting (FACS) analysis using a FACS-Calibur system (Becton Dickinson). Mean fluorescence units per cell directly correlated with the amount of cell-associated 123Rh. Hyperbolic dose response curves were fitted through the data points to estimate half-maximum effect concentration (EC50) values for tariquidar.
RESULTS
At the administered dose of 2 mg/kg of BW, tariquidar was well tolerated without occurrence of severe or serious adverse events. Mild adverse events, possibly related to the administration of the study medication, were mild hypotension, headache, and nausea in 1 patient and dizziness in 2 patients.
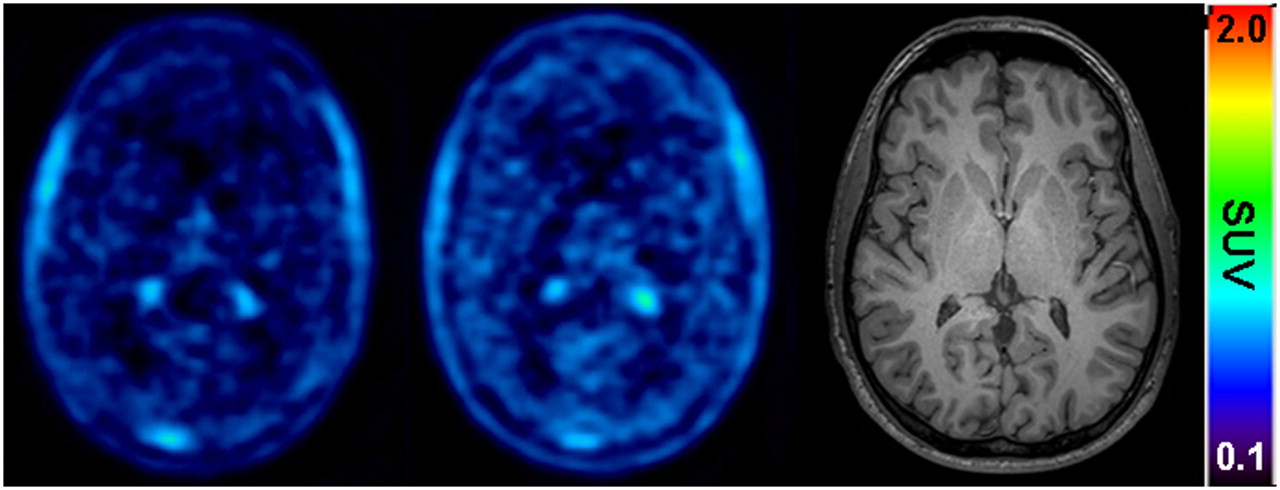
Figure 1 shows mean time–activity curves for the paired PET scans. The time points of (R)-11C-verapamil injection and tariquidar infusion are indicated in the figure. As a response to the start of tariquidar infusion during scan 1, brain radioactivity started to rise. A maximum increase of +74% was reached at 5 min after the end of tariquidar infusion (t = 75 min), as compared with baseline values (i.e., before the start of tariquidar infusion [t = 40 min]). At 35 min after the end of tariquidar infusion (t = 115 min), this effect had decreased to +10% on average. An indirect-response PK–PD model was fitted to the time course of brain activity after tariquidar infusion. Model fits and estimated outcome parameters are summarized in Supplemental Figures 2 and 3 and Supplemental Table 2. Figure 2 directly compares brain time–activity curves (corrected for blood activity) during the first 40 min of scan 1 and scan 2 (i.e., before and after tariquidar administration). Figure 3 shows PET summation images (scan 1 and 2) from the subject with the greatest increase in brain radioactivity after the administration of tariquidar (42% increase in DV during scan 2, when compared with the first 40 min of scan 1).

Time–activity curves (mean SUV ± SD), corrected for radioactivity in vasculature, of (R)-11C-verapamil in whole-brain gray matter from 0 to 40 min for PET scan 1 (before administration of tariquidar, □) and PET scan 2 (after administration of tariquidar, ▪).
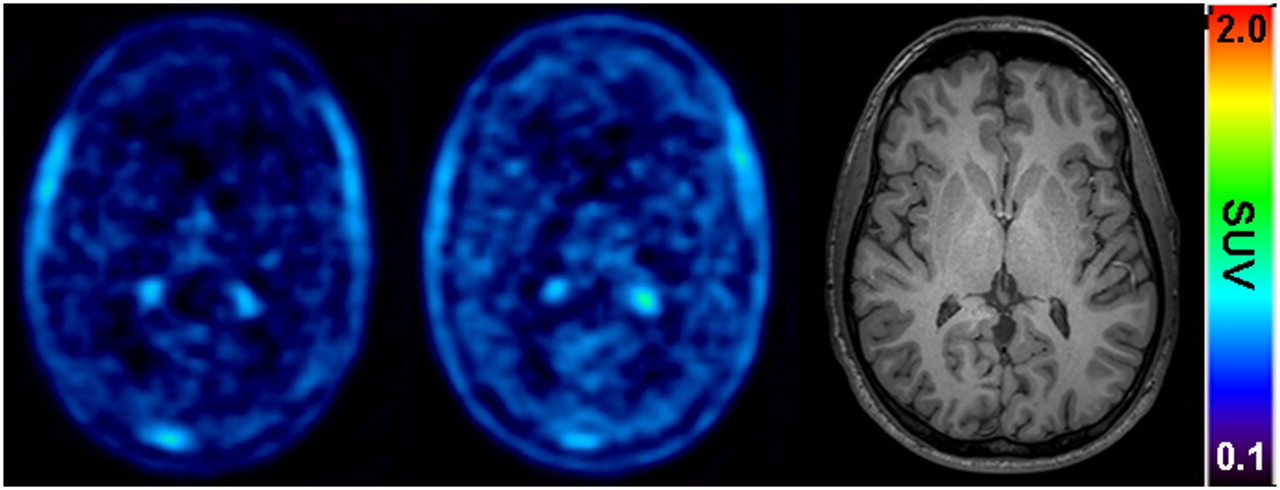
Transaxial PET summation pictures (10–40 min) of volunteer (subject 2) with greatest increase in (R)-11C-verapamil–derived brain activity, before (left picture) and after (middle picture) administration of tariquidar (2 mg/kg of BW). Activity concentration is expressed as SUV, and radiation scale is set from 0.1 to 2.0. Right picture shows transaxial T1-weighted MR image of same subject. Intense foci in PET images represent activity in choroid plexus and venous sinus.
Figure 4 displays mean time–activity curves of unchanged (R)-11C-verapamil in plasma during scan 1 and 2. (R)-11C-verapamil concentrations were slightly decreased after the administration of tariquidar (area under the curve from time zero to 40 min [AUC0–40], 38.3 ± 5.2 min and 34.3 ± 3.5 min, before and after administration of tariquidar, respectively, P = 0.043). The administration of tariquidar did not significantly influence the metabolism of (R)-11C-verapamil. Figure 5 displays the time course of the fractions of lipophilic and polar metabolites of (R)-11C-verapamil during scans 1 and 2. The AUC0–40 values for the fractions of lipophilic and polar metabolites of (R)-11C-verapamil in plasma were 7.3 ± 1.4 and 8.4 ± 2 min (P = 0.14) and 6.1 ± 1.3 and 6.1 ± 1.3 min (P = 0.89) before and after administration of tariquidar, respectively. At time points greater than 40 min in scan 1, the combined fraction of unchanged (R)-11C-verapamil and its lipophilic metabolites remained almost constant (0.74 ± 0.04 at t = 40 min, 0.72 ± 0.03 at t = 70 min, and 0.66 ± 0.01 at t = 100 min) (data not shown in Fig. 5). Total plasma activity levels also remained almost unchanged from 40 to 100 min of scan 1 (mean SUVs, 0.71 ± 0.07 at t = 40 min, 0.67 ± 0.06 at t = 70 min, and 0.66 ± 0.06 at t = 100 min).
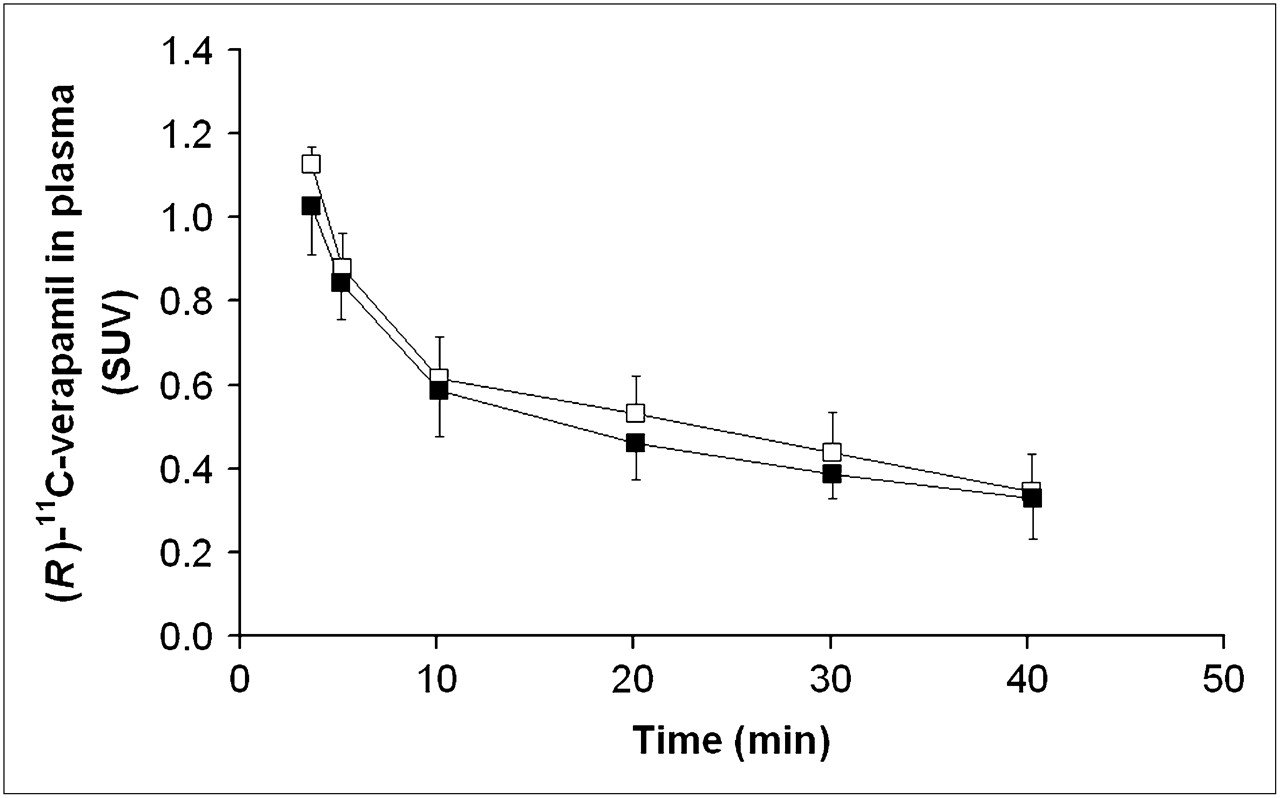
Time–activity curves (mean SUV ± SD) of unchanged (R)-11C-verapamil in arterial plasma (3–40 min) before (□) and after (▪) administration of tariquidar (2 mg/kg of BW).

Fractions of total plasma radioactivity (mean ± SD) of lipophilic (squares) and polar (circles) radiolabeled metabolites of (R)-11C-verapamil over time (10–40 min) before (open symbols) and after (filled symbols) administration of tariquidar (mean ± SD).
The plasma protein-bound percentage of (R)-11C-verapamil was not significantly altered by the administration of tariquidar (93% ± 2% vs. 94.% ± 2% before and after administration of tariquidar, respectively, P = 0.07). The plasma-to-blood ratio of activity was slightly decreased after the administration of tariquidar (1.41 ± 0.03 vs. 1.35 ± 0.05 before and after tariquidar, respectively, P = 0.04).
As previously described for (R)-11C-verapamil distribution in the rat brain after P-gp modulation (10), the 2T4K model provided better fits of the PET data in scan 2 than did the 1T2K model (mean Akaike information criterion, −28.6 vs. −36.5 for 1T2K vs. 2T4K model). For PET scan 1 (i.e., before P-gp modulation), both models provided fits of comparable quality (Akaike information criterion, −22.2 vs. −20.3 for 1T2K vs. 2T4K model). Parameter estimates obtained from the 2T4K model are displayed in Table 1. The DV of (R)-11C-verapamil and the influx rate constant of activity (K1) across the BBB increased significantly after the administration of tariquidar (DV, +24% ± 15%, P = 0.043; K1, +49% ± 36%, P = 0.043). There was no significant difference in efflux of activity across the BBB. Compartmental-model derived DV values agreed well with DVs estimated by Logan graphical analysis.
Outcome Parameters of 2T4K Model
Figure 6 shows the mean concentration–time profiles of tariquidar in venous plasma. Pharmacokinetic parameters of tariquidar are summarized in Supplemental Table 1. Table 2 shows the area under the curve from time zero to 4 h (AUC0–4) (i.e., end of scan 2) values of tariquidar in plasma of individual study subjects along with (R)-11C-verapamil DVs before and after tariquidar administration. A positive correlation was observed between the percentage change in (R)-11C-verapamil DV after the administration of tariquidar and the AUC0–4 (r = 0.74, P = 0.16) and AUC from time zero to 24 h ([AUC0–24]; r = 0.90, P = 0.037).
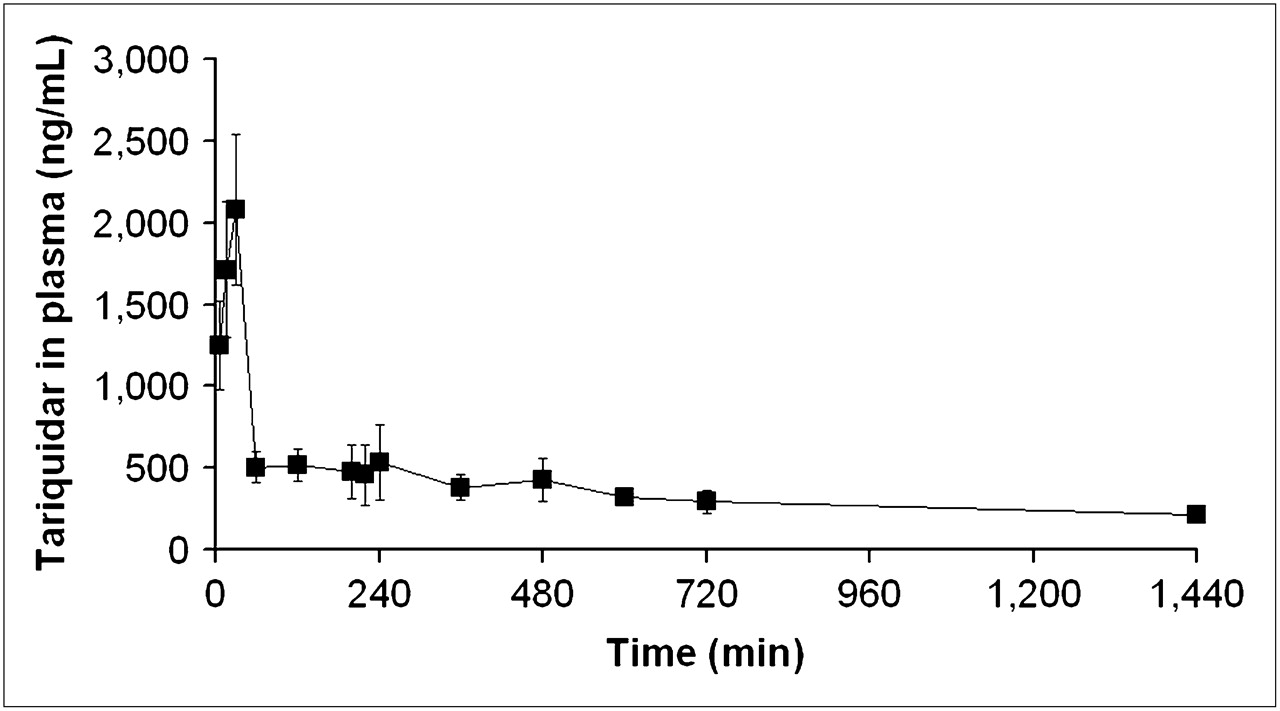
Concentration–time profile (ng/mL) of tariquidar (mean ± SD) in venous plasma.
Plasma AUCs of Tariquidar and (R)-11C-Verapamil DVs (2T4K Model) Before and After Tariquidar Administration in Individual Subjects
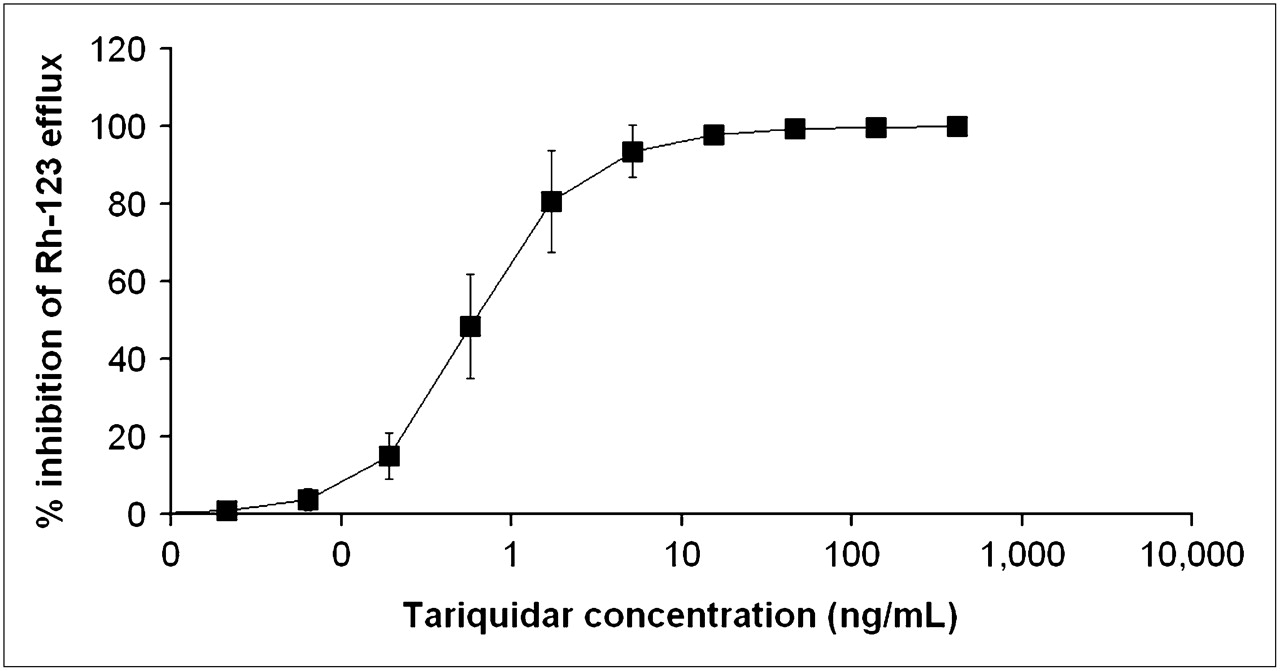
In peripheral white blood cells, the ex vivo addition of different tariquidar concentrations led to a dose-dependent increase in cellular 123Rh accumulation. Figure 7 shows mean concentration–effect curves of tariquidar. Half-maximum inhibition of 123Rh efflux from white blood cells was observed at an EC50 of 0.5 ± 0.2 ng/mL.

Inhibition of P-gp–mediated 123Rh efflux from peripheral white blood cells by different tariquidar concentrations added ex vivo. x-Axis is in logarithmic scale. Data present mean ± SD of results obtained with whole blood from same 5 volunteers who underwent paired PET scans. Mean EC50 was 0.5 ± 0.2 ng/mL.
DISCUSSION
(R)-11C-verapamil, in combination with PET, has previously proved suitable for studying P-gp activity at the BBB (9,10). In this pilot PET study, we investigated, for the first time to our knowledge, the ability of a potent, third-generation P-gp modulator—tariquidar—to inhibit P-gp function at the human BBB, at a dose that has been shown to decrease transporter activity in peripheral tissues (17,18). The percentage increase in the DV of (R)-11C-verapamil in the brain after the administration of tariquidar was used as a surrogate parameter of changes in P-gp function in the BBB (9). The paired-scan paradigm that we used in this study was in fact identical to a set-up that we had previously used in a small-animal PET study in rats (10). A previous study in healthy volunteers had shown excellent test–retest variability of paired (R)-11C-verapamil PET scans, as were used in this study (∼4% test–retest variability for DV values) (9).
The mean increase in DV of (R)-11C-verapamil in whole-brain gray matter after tariquidar administration was +24% ± 15% (Table 2), almost 10-fold less than the increase determined in the previous rat small-animal PET study (10). However, in the small-animal PET study, rats were administered 15 mg of tariquidar per kilogram (resulting in plasma tariquidar concentrations of about 1,400 ng/mL at the time of the PET scan)—that is, 7-fold higher doses than we administered to the volunteers in the present study. In rats, plasma tariquidar levels comparable to those achieved in the present PET study in humans (about 500 ng/mL) resulted in 3- to 4-fold increases of (R)-11C-verapamil DVs (26), suggesting species differences in tariquidar-induced cerebral P-gp inhibition or differences in P-gp expression and function between rats and humans (6).
The modest increase in brain DV that we observed was strongly correlated with individual plasma exposure of tariquidar and could, therefore, most likely be attributed to tariquidar-induced P-gp inhibition. Moreover, tariquidar affected neither the plasma protein binding nor the metabolism of (R)-11C-verapamil (Fig. 5), thus ruling out the possibility that these factors confounded the increase in brain DV of (R)-11C-verapamil. However, a previous study had shown that at 60 min after injection of (R)-11C-verapamil into rats, about 50% of brain activity was composed of polar radiolabeled metabolites, which presumably crossed the BBB independent of P-gp function (23). Although the polar fraction of radioactivity in plasma is about 3-fold lower in humans than in rats (23), it still cannot be excluded that part of the brain PET signal measured in this study was attributable to polar radiolabeled metabolites, which might have masked, to some extent, the P-gp inhibitory effect of tariquidar. In addition to the formation of polar metabolites, (R)-11C-verapamil is also metabolized to lipophilic radiolabeled metabolites, which are supposed to be substrates of P-gp and were therefore included into the arterial input function (9). Because it cannot be excluded that the lipophilic metabolites of (R)-11C-verapamil behaved differently from the parent tracer, their inclusion into the input function might lead to inaccurate measures of P-gp function with this radiotracer. Moreover, verapamil has been shown to be a substrate of MRP1 (27), which is also expressed at the BBB. However, because tariquidar does not inhibit MRP1 function (15), it is unlikely that the change in brain PET signal observed after tariquidar administration was related to changes in MRP1 transport of the radiotracer.
Distribution of unbound drug across the BBB is determined by influx versus efflux. As previously described for P-gp inhibition at the BBB by tariquidar (10) and other P-gp inhibitors (28,29), the increase in DV after the administration of tariquidar in our study was associated with a significant increase (+49% ± 36%) in the influx rate constant K1 (Table 1). This increase in K1 suggests that in the presence of an inhibitor, this balance is shifted in favor of drug influx and underlines the gate-keeper function of P-gp at the BBB.
By administering tariquidar during scan 1 rather than between scan 1 and scan 2 (timeline, Fig. 1), we attempted to better define the time window of P-gp modulation, to optimize a possible time point of therapeutic drug administration relative to the administration of the inhibitor drug. The time–activity curves shown in Figure 1 describe the time course of tariquidar-induced P-gp inhibition at the BBB. Surprisingly, brain activity started to rise significantly in immediate response to tariquidar infusion. Plasma analysis had shown that total activity counts and the fraction of (R)-11C-verapamil and its lipophilic metabolites, which are also P-gp substrates, were almost constant during the time course of tariquidar infusion, thereby suggesting that tariquidar-induced P-gp blockade facilitated brain entry of activity that had remained in the circulation. In contrast, because perfusion was not determined in the present study and the effect of tariquidar on cerebral blood flow is unknown at present, it cannot be excluded that at least part of the immediate rise in activity after tariquidar infusion can be attributed to increased perfusion rather than decreased P-gp activity. However, (R)-11C-verapamil is a low-extraction radiotracer (K1 = 0.049 after P-gp modulation in scan 2, corresponding to an extraction fraction [E] of 0.098 when assuming an absolute blood flow [F] of 0.5 mL·g−1·min−1 according to the relationship K1 = F·E) (28). Therefore, brain uptake of (R)-11C-verapamil can be considered to be insensitive to changes in cerebral blood flow under conditions of moderate P-gp inhibition, as used in this study. The mean maximum increase in activity concentrations in the brain was observed at 5 min after the end of tariquidar infusion. Forty minutes later, activity concentrations in the brain had significantly declined, suggesting that the effect of tariquidar was reversible. However, during scan 2, which was obtained at about 3 h after tariquidar administration, moderate P-gp inhibition was still evident. Brain activity influx after tariquidar infusion was analyzed by simple PK–PD models (supplemental data), suggesting that during the time course of tariquidar action the modulation of P-gp function entails both influx enhancement and efflux inhibition of activity at the BBB.
Our findings are quite in opposition to previous data showing that in vitro the P-gp inhibitory effect of tariquidar lasts for up to 22 h after the removal of tariquidar from the incubation medium (15) and that in vivo tariquidar fully inhibits P-gp mediated 123Rh efflux from peripheral lymphocytes for more than 24 h (3,8,17), at plasma concentrations similar to the ones used in this study. The concentrations of tariquidar in plasma during the second PET scan (200–240 min after the start of tariquidar infusion) and at 24 h after tariquidar infusion were 490 ± 166 ng/mL and 212 ± 24 ng/mL (Fig. 6)—that is, 980- and 424-fold higher than the EC50 for tariquidar-induced inhibition of 123Rh efflux from peripheral white blood cells (Fig. 7), respectively. In humans, tariquidar is bound to 99.5% to plasma proteins (Ulrich Elben, written communication, 2008). When correcting plasma concentrations for protein binding, the concentration values for unbound tariquidar at the time of the second scan (2.45 ± 0.83 ng/mL) were still about 5-fold higher than the in vitro EC50. The fact that P-gp at the BBB exhibits greater resistance to inhibition than P-gp in lymphocytes has previously been pointed out by others (3,8). For instance, Choo et al. found that the EC50 of tariquidar to enhance brain penetration of loperamide in mice was 25-fold higher than the EC50 for inhibition of 123Rh efflux from lymphocytes (3). Although the mechanisms underlying this differential sensitivity have not yet been elucidated, a higher transporter density of P-gp at the BBB and structural distinctions in transporter proteins expressed in brain and lymphocytes have been suggested (3,30,31).
CONCLUSION
Using the P-gp substrate (R)-11C-verapamil as a PET tracer, we showed that tariquidar, administered at a well-tolerated dose of 2 mg/kg of BW, induced a modest but significant increase in brain DV, most likely due to inhibition of P-gp activity in the BBB. This finding is in contrast to a previous study (8) that had failed to demonstrate a significant effect of the same dose of tariquidar on central opioid effects of the P-gp substrate loperamide in humans, thus underlining the greater sensitivity of direct measurement of CNS drug concentrations by PET as compared with surrogate pharmacologic markers of CNS drug penetration. Tariquidar-induced P-gp modulation at the human BBB appeared to be transient, and its magnitude was directly proportionate to serum drug exposure. P-gp appeared to be more resistant to inhibition in the BBB than in peripheral white blood cells. Whether higher drug plasma concentrations can safely be obtained in the clinic, to overcome this resistance and achieve higher levels of cerebral P-gp inhibition, needs to be investigated.
Acknowledgments
We thank Divya Maheshwari from AzaTrius Pharmaceuticals Pvt Ltd (London, U.K.) for providing us with vials of tariquidar for intravenous infusion. This study would not have been possible without the excellent technical support of Rainer Bartosch (Department of Nuclear Medicine), Research Nurse Edith Lackner, Johann Stanek, and Aiman Abrahim (Department of Clinical Pharmacology). Rebecca Brauner from the Department of Chemical Analytics at Austrian Research Centers GmbH-ARC is gratefully acknowledged for performing the LC/MS analysis of tariquidar plasma concentrations. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement 201380 (“Euripides”) and from the Austrian Science Fund (FWF) project Transmembrane Transporters in Health and Disease (SFB F35).
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication February 13, 2009.
- Accepted for publication August 28, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- ABCB1 and ABCG2 Regulation at the Blood-Brain Barrier: Potential New Targets to Improve Brain Drug Delivery
- Characterization of P-Glycoprotein Humanized Mice Generated by Chromosome Engineering Technology: Its Utility for Prediction of Drug Distribution to the Brain in Humans
- Clinical Applications of Simultaneous PET/MR Imaging Using (R)-[11C]-Verapamil with Cyclosporin A: Preliminary Results on a Surrogate Marker of Drug-Resistant Epilepsy
- Mapping the Binding Site of the Inhibitor Tariquidar That Stabilizes the First Transmembrane Domain of P-glycoprotein
- Increased Permeability-Glycoprotein Inhibition at the Human Blood-Brain Barrier Can Be Safely Achieved by Performing PET During Peak Plasma Concentrations of Tariquidar
- Role of (Drug) Transporters in Imaging in Health and Disease
- In Vivo Imaging as a Pharmacodynamic Marker
- Interaction of 11C-Tariquidar and 11C-Elacridar with P-Glycoprotein and Breast Cancer Resistance Protein at the Human Blood-Brain Barrier
- Retrospective Analysis of P-Glycoprotein-Mediated Drug-Drug Interactions at the Blood-Brain Barrier in Humans
- Modeling Cyclosporine A Inhibition of the Distribution of a P-Glycoprotein PET Ligand, 11C-Verapamil, into the Maternal Brain and Fetal Liver of the Pregnant Nonhuman Primate: Impact of Tissue Blood Flow and Site of Inhibition
- Behavioral Effects and Central Nervous System Levels of the Broadly Available {kappa}-Agonist Hallucinogen Salvinorin A Are Affected by P-Glycoprotein Modulation In Vivo
- A Novel Positron Emission Tomography Imaging Protocol Identifies Seizure-Induced Regional Overactivity of P-Glycoprotein at the Blood-Brain Barrier