


Abstract
The permeability-glycoprotein (P-gp) efflux transporter is densely expressed at the blood–brain barrier, and its resultant spare capacity requires substantial blockade to increase the uptake of avid substrates, blunting the ability of investigators to measure clinically meaningful alterations in P-gp function. This study, conducted in humans, examined 2 P-gp inhibitors (tariquidar, a known inhibitor, and disulfiram, a putative inhibitor) and 2 routes of administration (intravenous and oral) to maximally increase brain uptake of the avid and selective P-gp substrate 11C-N-desmethyl-loperamide (dLop) while avoiding side effects associated with high doses of tariquidar. Methods: Forty-two 11C-dLop PET scans were obtained from 37 healthy volunteers. PET was performed with 11C-dLop under the following 5 conditions: injected under baseline conditions without P-gp inhibition, injected 1 h after intravenous tariquidar infusion, injected during intravenous tariquidar infusion, injected after oral tariquidar, and injected after disulfiram. 11C-dLop uptake was quantified with kinetic modeling using metabolite-corrected arterial input function or by measuring the area under the time–activity curve in the brain from 10 to 30 min. Results: Neither oral tariquidar nor oral disulfiram increased brain uptake of 11C-dLop. Injecting 11C-dLop during tariquidar infusion, when plasma tariquidar concentrations reach their peak, resulted in a brain uptake of the radioligand approximately 5-fold greater than baseline. Brain uptake was similar with 2 and 4 mg of intravenous tariquidar per kilogram; however, the lower dose was better tolerated. Injecting 11C-dLop after tariquidar infusion also increased brain uptake, though higher doses (up to 6 mg/kg) were required. Brain uptake of 11C-dLop increased fairly linearly with increasing plasma tariquidar concentrations, but we are uncertain whether maximal uptake was achieved. Conclusion: We sought to increase the dynamic range of P-gp function measured after blockade. Performing 11C-dLop PET during peak plasma concentrations of tariquidar, achieved with concurrent administration of intravenous tariquidar, resulted in greater P-gp inhibition at the human blood–brain barrier than delayed administration and allowed the use of a lower, more tolerable dose of tariquidar. On the basis of prior monkey studies, we suspect that plasma concentrations of tariquidar did not fully block P-gp; however, higher doses of tariquidar would likely be associated with unacceptable side effects.
The efflux transporter P-glycoprotein (P-gp) is a component of the blood–brain barrier (BBB) and a proposed mechanism of drug resistance (1). Under physiologic conditions, P-gp acts at high capacity, completely blocking entry into the brain of avid substrates, including radiolabeled substrates. More than 50% of P-gp transporters may need to be inhibited pharmacologically to increase uptake of radiolabeled substrates into a range measurable by PET (2). Notably, the P-gp inhibitor tariquidar has been shown to increase brain uptake of the substrate PET radiotracers 11C-N-desmethyl-loperamide (11C-dLop) and 11C-(R)-verapamil 2- to 3-fold, compared with baseline (3–6).
To detect pathologic overexpression of P-gp using PET, normal P-gp function (which is already of high capacity) must be distinguished from P-gp overexpression (which has even higher capacity). Treating subjects with a fixed dose of tariquidar before PET imaging would hypothetically increase radiotracer uptake into a dynamic range at which differential response to inhibition—caused by differential P-gp function—could be measured.
To achieve increases in radiotracer brain uptake into this range, however, past studies have required high intravenous doses (>2 mg/kg) of tariquidar that were associated with adverse drug reactions (4,7,8). In addition, because these studies administered the PET radiotracer after completion of tariquidar infusion, they may have missed the window of peak P-gp inhibition and, thus, the most reliable measurement of increased P-gp inhibition. Although tariquidar is the only third-generation P-gp inhibitor that has been found to be safe for human administration, other potential methods of pharmacologic inhibition of P-gp include oral administration of tariquidar (9) or oral administration of disulfiram, a drug whose metabolites have been shown to inhibit P-gp in vitro (10–12).
This study sought to determine whether increased inhibition of P-gp at the human BBB could be safely achieved and reliably measured by changing the timing or route of administration of tariquidar or using disulfiram. 11C-dLop PET was performed on healthy volunteers under baseline conditions or after P-gp inhibition. For the P-gp blocked condition, 11C-dLop was injected 1 h after intravenous tariquidar infusion, during intravenous tariquidar infusion, after oral tariquidar, or after disulfiram.
MATERIALS AND METHODS
Thirty-seven healthy volunteers (mean age ± SD, 29.1 ± 6.9 y; 18 women, 19 men) were included. Volunteers with serious medical or psychiatric illness, those using illicit drugs, and those taking medications other than oral contraceptives were excluded. The study was approved by the National Institutes of Health Combined Neurosciences Institutional Review Board. All participants gave written informed consent before entering the study. Subjects were monitored for changes in blood pressure, temperature, heart rate, and respiration rate before and after tariquidar infusion. These parameters, as well as electrocardiogram tracing, were similarly monitored after 11C-dLop injection. Blood and urine laboratory tests were repeated within 24 h of study completion. Plasma was drawn to measure total tariquidar concentration for 16 subjects and free fraction of tariquidar for 5 subjects. Methods for measuring total and free concentration of tariquidar are given in the supplemental materials (supplemental materials are available at http://jnm.snmjournals.org).
11C-dLop Imaging
11C-dLop was prepared by the methylation of its primary amide precursor with 11C-iodomethane, as described previously (13). The radiotracer was obtained in high radiochemical purity (100%) and with a specific activity of 101 ± 50 GBq/μmol at the time of injection.
Fourteen 11C-dLop scans were obtained without P-gp inhibition (baseline).The remaining 28 scans were obtained with P-gp inhibition. Methods for PET imaging and tariquidar preparation are given in supplemental materials.
Intravenous Tariquidar with Delayed Radiotracer Injection
Scan data from 7 subjects who received either 4 mg/kg or 6 mg/kg of intravenous tariquidar in a previously published study (4) were included in this study for comparison. For these subjects, 11C-dLop was injected approximately 60 min after completion of the tariquidar infusion.
Intravenous Tariquidar with Concurrent Radiotracer Injection
Twelve subjects received intravenous tariquidar at doses of either 2 mg/kg (n = 10; 4 women; body weight, 71.2 ± 18.7 kg) or 4 mg/kg (n = 2; 1 woman; body weight, 72.8 ± 19.4 kg). Subjects had 2 intravenous catheters placed (1 in each antecubital vein) before imaging. Under both dose conditions, tariquidar was infused at a rate of 4 mg/kg/h through 1 catheter. Halfway through the tariquidar infusion (at 30 min for the 4 mg/kg group and at 15 min for the 2 mg/kg group), 11C-dLop was injected through the other catheter. Arterial sampling was not performed.
Oral Tariquidar Administration
To determine the dose strategy for oral tariquidar, we performed pharmacokinetic studies in healthy volunteers (supplemental materials). On the basis of the results, 3 additional subjects (all men; body weight, 76.1 ± 16.5 kg) received oral tariquidar at a dose of 1,500 mg 10–12 h before 11C-dLop injection. Arterial sampling was not performed.
Oral Disulfiram Administration
Disulfiram was administered at 2 doses: 500 mg of oral disulfiram as a 1-time dose 10 h before 11C-dLop injection (n = 3; all men; body weight, 83.4 ± 12.0 kg) or 2.5 g over 4 d before 11C-dLop injection (n = 3; all women; body weight, 78.1 ± 24.6 kg). Subjects were given written and oral instructions to abstain from alcohol-containing products for 2 d prior to and 2 wk after disulfiram administration. Arterial sampling was performed.
11C-dLop Image Analysis
Reconstructed PET images were realigned and normalized to stereotactic space using Pixelwise Modeling Software 3.0 (PMOD Technologies Ltd.). The Montreal Neurologic Institute template was used to define a region of composite neocortex by creating a weighted average of activity from the frontal, parietal, occipital, and temporal cortices. Regions were manually adjusted when necessary to exclude cerebrospinal fluid space and choroid plexus.
The concentration of radioactivity was expressed as standardized uptake value (SUV) = (measured activity per cm3 brain/injected activity)⋅(g of body weight). The area under the time–activity curve from 10 to 30 min (AUC10–30) for each composite brain region was calculated using the trapezoidal method.
For those subjects with metabolite-corrected plasma data, rate constants (K1 and k2) and total distribution volume (VT) were calculated using a 1-tissue-compartmental model. The total concentration of radioactivity in whole blood was used for vascular correction, assuming that blood constitutes 5% brain volume.
Because the posterior pituitary lacks a BBB and prior baseline PET scans have shown high uptake of 11C-dLop in this organ (6), we measured activity in the pituitary gland to estimate brain uptake of 11C-dLop in complete absence of P-gp (supplemental materials).
Statistical Analysis
Statistical analysis was performed using SPSS 17.0 (IBM). Group differences in 11C-dLop uptake were evaluated using the Mann–Whitney U test. To determine whether the tariquidar–11C-dLop dose–response curve best fit a linear or nonlinear model, we used the Curve Estimation function in SPSS to perform linear and nonlinear (quadratic, cubic, and sigmoidal) regressions. The estimated half maximum effective concentration (EC50) value and its 95% confidence interval were determined with the R software package (www.r-project.org) using a lognormal dose–response model.
RESULTS
Safety Analysis
Tariquidar was generally well tolerated in subjects who received the lowest doses (2 mg/kg intravenously and 1,500 mg orally). Two subjects who received 2 mg/kg intravenously reported a mild metallic taste during the tariquidar infusion. Two subjects who received 4 mg of intravenous tariquidar per kilogram experienced light-headedness and conjunctival injection, and 1 of these subjects had a brief syncopal episode immediately after the PET scan that resolved without sequelae. One subject who received 6 mg of intravenous tariquidar per kilogram complained of a metallic taste accompanied by nausea that resolved once tariquidar infusion was stopped.
Disulfiram was well tolerated by all subjects. One subject experienced a mild, transient disulfiram–ethanol reaction 3 d after the PET scan due to unwittingly consuming food prepared with wine.
Imaging Results
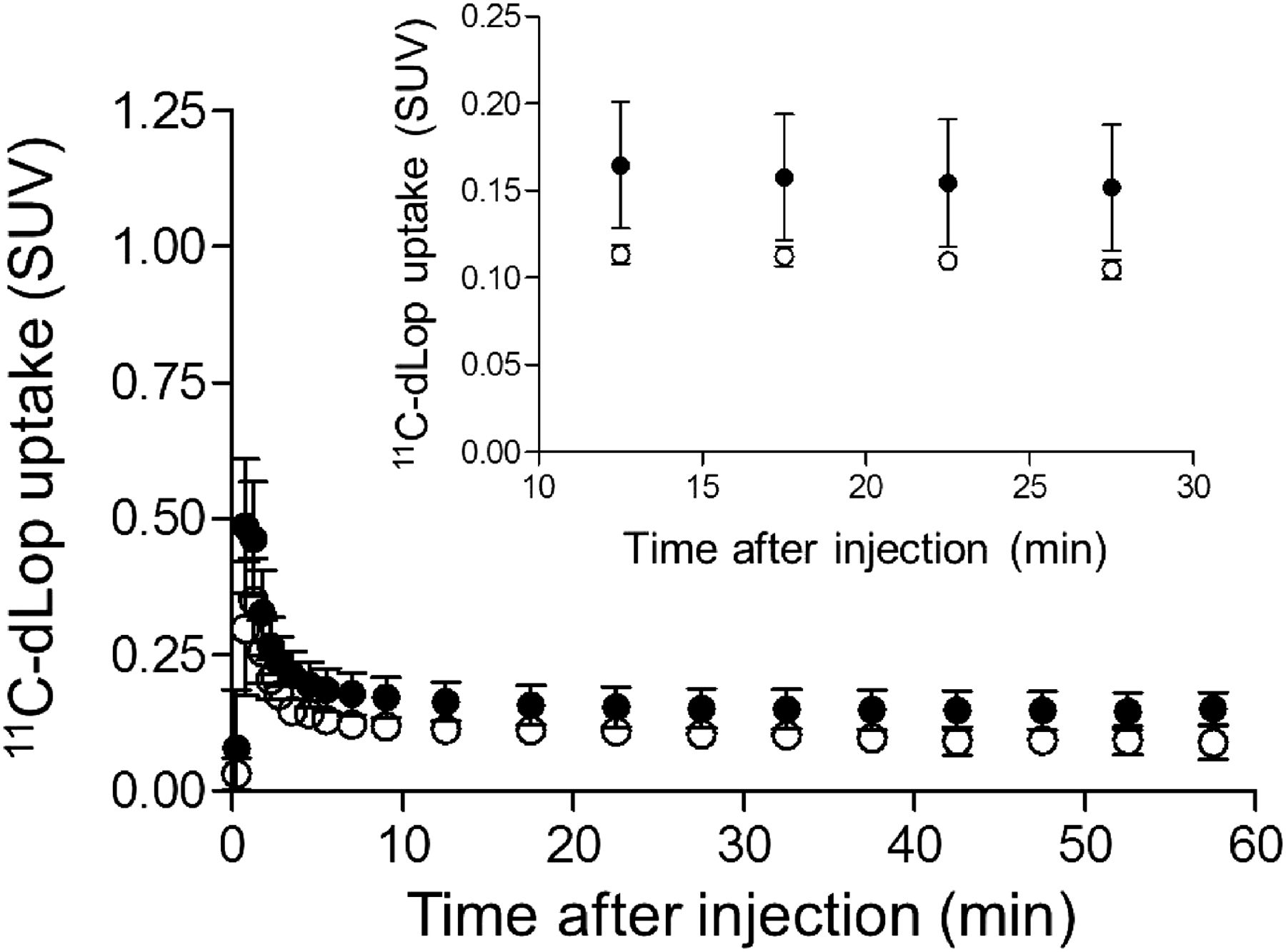
When 11C-dLop was injected in the baseline condition, uptake of activity in the brain was negligible, in agreement with previous studies. Brain time–activity curves reached a plateau at less than 0.2 SUV (Fig. 1). Both K1 and k2 values were low, resulting in VT values of 1.20 ± 0.16. AUC10–30 values for composite neocortex were 2.94 ± 0.66 SUV⋅min (Table 1). Individual region-of-interest values are shown in Supplemental Table 1.

Composite neocortex time–activity curves showing brain uptake of radioactivity after 11C-dLop injection. 11C-dLop was injected at baseline (●) and during intravenous infusion of tariquidar at 2 (○) and 4 mg/kg (▲). Error bars denote SD.
Uptake of 11C-dLop in Composite Neocortex at Baseline and After Pharmacologic Inhibition of P-Glycoprotein
When 11C-dLop was injected approximately 60 min after tariquidar infusion, brain uptake of 11C-dLop was approximately 2- to 4-fold greater than at baseline (Table 1). Brain time–activity curves reached plateaus of approximately 0.3 and approximately 0.6 SUV during delayed administration of tariquidar at 4 and 6 mg/kg intravenously, respectively. The resulting AUC10–30 values for composite neocortex were 1.7–3.4 times greater than baseline.
When 11C-dLop was injected during infusion of tariquidar, brain uptake of 11C-dLop was approximately 5-fold greater than at baseline (Figs. 1 and 2; Table 1). Brain time–activity curves reached plateaus of 0.4–1.4 and 0.7–1.1 SUV during concurrent administration of tariquidar at 2 and 4 mg/kg intravenously, respectively. The resulting AUC10–30 values for composite neocortex were approximately 5 times greater than baseline, approximately 3 times greater than delayed administration of 4 mg of intravenous tariquidar per kilogram, and approximately 1.5 times greater than delayed administration of 6 mg of intravenous tariquidar per kilogram. At baseline, 11C-dLop uptake in the pituitary was 17 times greater than in the composite neocortex (Supplemental Fig. 1). After P-gp inhibition, uptake in the pituitary was 3.5 times greater than in composite neocortex. 11C-dLop uptake in the pituitary was similar at baseline and after P-gp inhibition (∼2.7 vs. ∼2.5 SUV).

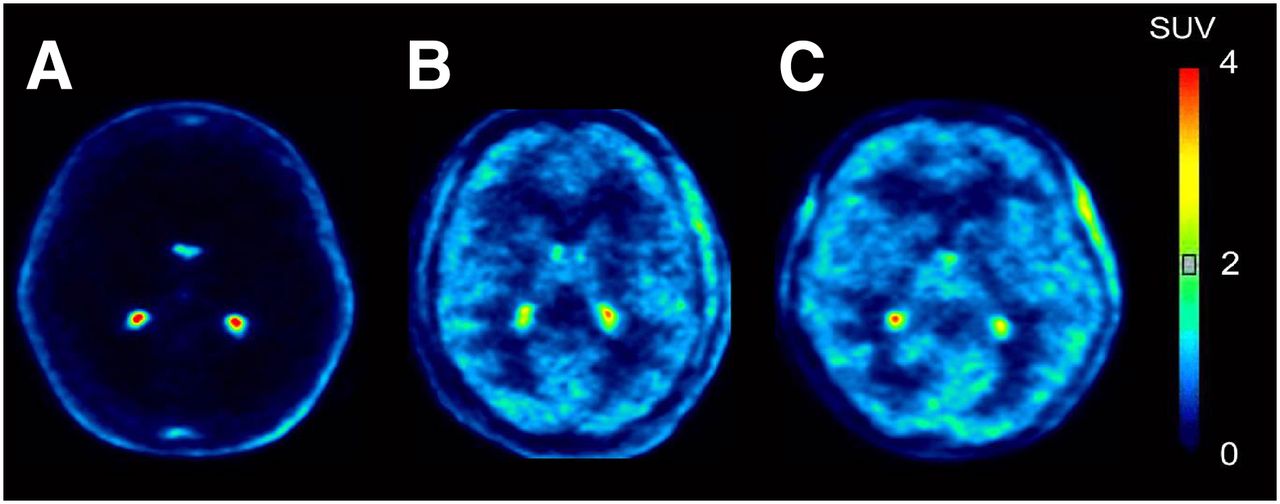
Representative images from 11C-dLop PET scans. Images are summed from 10 to 30 min after injection. Radioligand was injected either at baseline (A) or during intravenous infusion of tariquidar at doses of 2 (B) and 4 mg/kg (C).
Oral tariquidar did not increase brain uptake of 11C-dLop. Measured values for AUC10–30 did not differ from those in baseline scans (Fig. 3; Table 1). Results from the pharmacokinetic study for oral tariquidar are given in the supplemental materials.
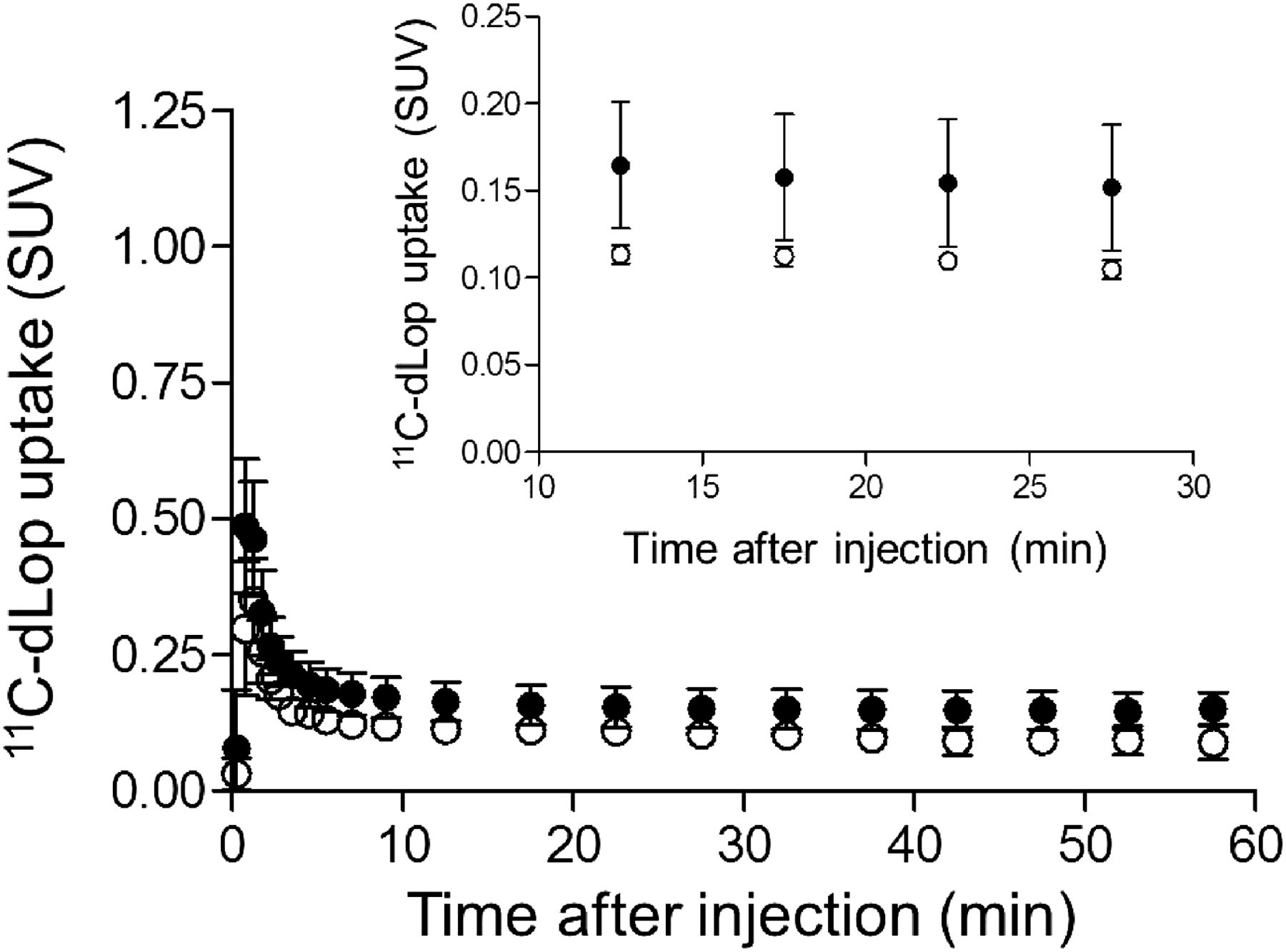
Composite neocortex time–activity curves showing brain uptake of radioactivity after 11C-dLop injection at baseline (●) and after 1,500 mg of oral tariquidar (○). Error bars denote SD.
Neither low- nor high-dose disulfiram administration was associated with 11C-dLop uptake into brain greater than baseline (Fig. 4; Table 1). Disulfiram did not result in changes in VT, K1, k2, or AUC10–30. The plasma-free fraction (fp) of 11C-dLop was slightly lower in disulfiram-treated subjects than in subjects who had 11C-dLop under baseline conditions (12% ± 1% vs. 15% ± 2%; P = 0.0426, Mann–Whitney U test). However, VT/fp values did not differ between disulfiram-treated and baseline subjects (P = 0.2284).
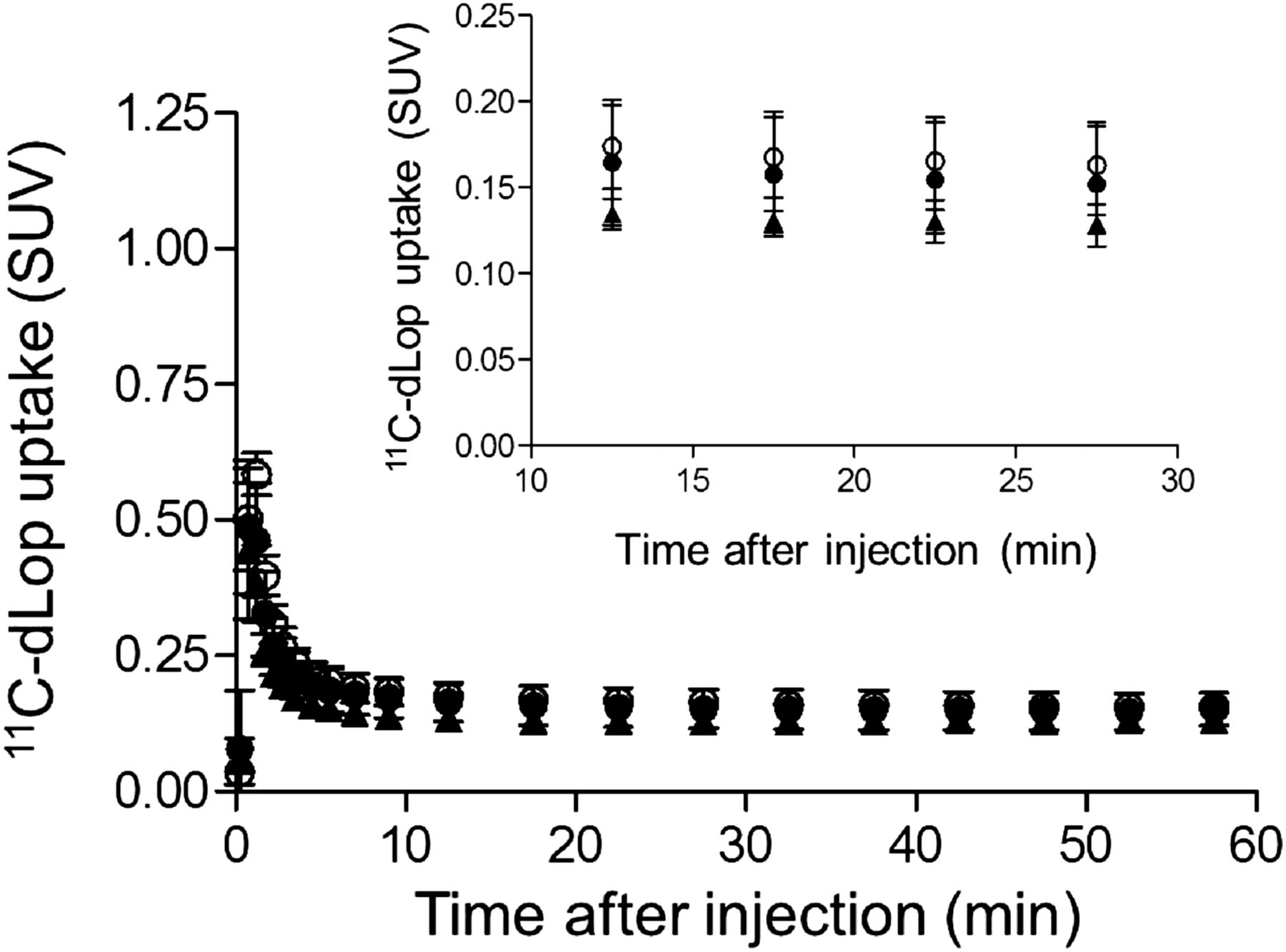
Composite neocortex time–activity curves showing brain uptake of radioactivity after injection of 11C-dLop at baseline (●) and after either 1-time dose of 500 mg of disulfiram (○) or 2.5 g of disulfiram over 4 d (▲). Error bars denote SD.
Dose–Response of Tariquidar on 11C-dLop Uptake
We measured the plasma concentrations of tariquidar for subjects who had concurrent radiotracer injection, oral tariquidar, and 1 subject who had delayed radiotracer injection. For the concurrent group, plasma concentrations of tariquidar were 1,040 ± 360 ng/mL (1,609 ± 556 nM) for the 2 mg/kg dose and 1,438 ± 528 ng/mL (2,224 ± 817 nM) for the 4 mg/kg dose. Plasma concentrations of tariquidar after oral administration were 133 ± 46 ng/mL (205 ± 71 nM), or only 13% of the concentration achieved with concurrent administration of 2 mg/kg intravenously. The subject who had intravenous tariquidar (4 mg/kg) with delayed radiotracer injection had plasma concentrations of 232 ng/mL (358 nM), or 22% of the mean value for subjects who had concurrent injection of radiotracer at the same dose of tariquidar. The free fraction of tariquidar was 0.0054 ± 0.0002 for the 5 subjects who had 11C-dLop imaging and 0.0053 ± 0.0003 for all 21 subjects who had free fraction measured.
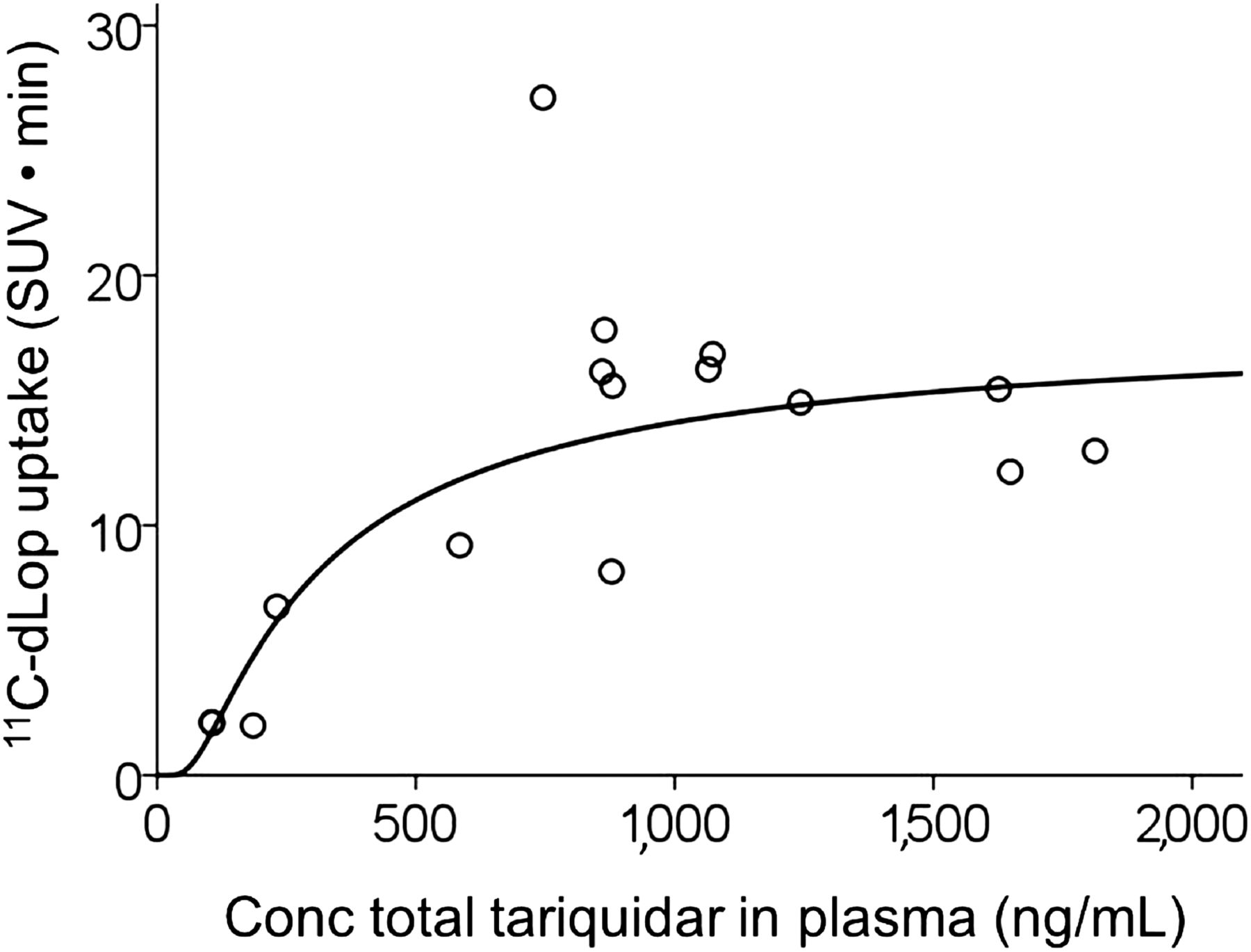
11C-dLop uptake into the brain increased linearly, with the total concentration of tariquidar at low and moderate concentrations of the inhibitor; however, at higher concentrations, the PET response appeared to plateau (Fig. 5). Goodness of fit was greater for the sigmoidal model (r2 = 0.81) than linear (r2 = 0.30), quadratic (r2 = 0.65), or cubic (r2 = 0.66) models. The estimated EC50 value and its 95% confidence interval were 306 ng/mL and 20–592, respectively. The estimated maximum for 11C-dLop uptake and its 95% confidence interval were 15 SUV·min and 12–19, respectively. This maximum corresponds to peak uptake of 0.8 SUV in the brain. Brain uptake of the radiotracer did not correlate with the free concentration of tariquidar in plasma (r = −0.61, P = 0.28); however, free fraction was measured in only 5 subjects.

Dose–response curve for tariquidar and uptake of 11C-dLop in composite neocortex. Conc = concentration.
DISCUSSION
Performing 11C-dLop PET imaging during peak plasma concentrations of tariquidar resulted in greater P-gp inhibition at the BBB than injecting radiotracer after a delay. This concurrent administration strategy allowed use of a lower dose of tariquidar, expected to result in fewer adverse events. In contrast, neither 1,500 mg of oral tariquidar nor oral disulfiram inhibited P-gp. These results suggest that the ability of tariquidar to inhibit P-gp at the BBB depends on the concentration of tariquidar in plasma. These results also suggest that oral tariquidar and disulfiram are not viable alternatives to intravenous tariquidar for inhibiting P-gp.
In a previous study from our laboratory, we injected 11C-dLop approximately 1 h after tariquidar infusion was completed, and other studies using 11C-(R)-verapamil injected the radiotracer approximately 1–3 h after tariquidar (7,8). One of the studies using 11C-(R)-verapamil showed that plasma concentrations of tariquidar declined rapidly after infusion and were followed by a long terminal phase, remaining stable at approximately 500 ng/mL (773 nM) for several hours (8). Therefore, in our prior study using 11C-dLop, we presumed that this terminal phase represented the concentration of tariquidar required to effectively inhibit P-gp at the BBB, given that plasma concentrations between 150 and 200 ng/mL (230–310 nM) inhibit P-gp activity on leukocytes (9). However, subsequent studies demonstrated that greater concentrations of tariquidar are required to block P-gp activity at the BBB than on leukocytes (14,15). Therefore, by administering 11C-dLop after completion of the tariquidar infusion, we may have missed the time window of maximal P-gp inhibition at the BBB. No previously published report or study has tested the efficacy of injecting substrate radiotracers during the tariquidar infusion, when plasma concentrations of this inhibitor peak (∼1,000 ng/mL or 1,500 nM during a 2 mg/kg infusion). In this study, 11C-dLop PET imaging was performed during infusion of 2 or 4 mg of tariquidar per kilogram. We observed brain uptake comparable to that seen when performing 11C-dLop PET at 1 h after infusion of 6 mg of tariquidar per kilogram (4). These results suggest that concurrent administration of radiotracer allows the use of lower doses of tariquidar while yielding a similar degree of P-gp inhibition. This method also reduces the amount of time required to administer tariquidar and obtain the 11C-dLop PET scan.
Studies have been conducted in drug-resistant cancer patients investigating the use of tariquidar to improve the efficacy of chemotherapy (16,17). These studies have failed to increase penetration of chemotherapy drugs, perhaps because tariquidar was given at too low a dose (<4 mg/kg intravenously) with too long an interval between administration of tariquidar and chemotherapy. The results from this study suggest that low-dose tariquidar has the potential to effectively inhibit P-gp and increase penetration of substrate medications when both the inhibitor and the active drug are simultaneously administered intravenously. Although it would be ideal to infuse tariquidar over the entire time course of the 11C-dLop PET scan, we infused at a fast rate over a short interval to achieve high plasma concentrations while avoiding toxicity. Peak uptake of 11C-dLop occurs within the first few minutes after injection with little washout thereafter (6). Therefore, we don’t expect the short tariquidar infusion time to have caused large variance in the AUC10–30 data. Rapid clearance of tariquidar from plasma may alter the kinetics of P-gp substrates that reversibly bind in the brain, however. Therefore, longer infusion time may be preferred for P-gp radiotracers such as 11C-(R)-verapamil.
The adverse events observed using high doses of tariquidar (4–6 mg/kg intravenously) may have been caused by high amounts of the excipient, propylene glycol, delivered alongside tariquidar. In support of this theory, infrequent, mild side effects were observed when lower amounts of propylene glycol were delivered (intravenous tariquidar, 2 mg/kg), and no side effects were observed when no propylene glycol was delivered at all (1,500 mg of oral tariquidar). However, oral tariquidar may be more tolerable simply because of its low bioavailability.
Oral tariquidar did not increase brain uptake of 11C-dLop, most likely because oral administration resulted in low plasma concentrations. Repeated administration of high oral doses of tariquidar could theoretically result in stable, high plasma concentrations over time. Whether this strategy would effectively inhibit P-gp and improve penetration of substrate medications in a clinical setting remains to be seen.
Similarly, disulfiram did not increase 11C-dLop brain uptake, even at a dose of 2.5 g over 4 d. Disulfiram metabolites irreversibly inhibit P-gp in vitro (10), making this drug, which is inexpensive and commercially available, an attractive alternative to tariquidar for P-gp inhibition in a clinical setting. However, high doses of disulfiram have been associated with hepatic, neurologic, and dermatologic side effects (18). Our results suggest that either disulfiram metabolites do not inhibit P-gp at the BBB in vivo or doing so would require doses so large they would likely be toxic.
We found that the tariquidar–11C-dLop dose–response curve best fit a sigmoidal curve, suggesting that the PET response plateaued at high concentrations of tariquidar. Bauer et al. similarly showed that brain uptake of 11C-(R)-verapamil did not increase with doses of tariquidar greater than 6 mg/kg intravenously (7). Although these results suggest that plasma concentrations of tariquidar greater than 1,000 ng/mL completely inhibit P-gp at the BBB, we do not know whether P-gp was maximally blocked in these experiments. We suspect that that the apparent plateau in 11C-dLop uptake was caused by noise and variability of the biologic response. In our earlier study, we estimated the first-pass extraction of 11C-dLop to be approximately 40% in monkey brain when the PET scan was performed after P-gp inhibition with (2R)-anti-5-{3-[4-(10,11-dichloromethanodibenzo-suber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (5). Therefore, we would expect complete blockade of P-gp to result in high 11C-dLop uptake in the brain. Instead, the mean uptake was less than 1 SUV in the concurrent tariquidar group (Fig. 1), suggesting that P-gp was still preventing 11C-dLop from entering the brain to some extent at the highest concentrations of tariquidar achieved in this study.
Because both 11C-dLop and tariquidar are lysosomotropic (19), the apparent plateau in 11C-dLop uptake could have been due to tariquidar outcompeting 11C-dLop in lysosomes at higher concentrations of tariquidar. This possibility is speculative as we have no evidence that tariquidar enters the brain. Tariquidar is not only an inhibitor of P-gp but also a substrate for the breast cancer resistance protein (20), which thereby blocks its entry into the brain. Further studies are required to determine whether tariquidar enters the brain at high plasma concentrations.
11C-dLop uptake in the pituitary did not increase after P-gp inhibition, suggesting that the lack of BBB around the pituitary allows entry of 11C-dLop independent of P-gp. That pituitary uptake was severalfold greater than brain uptake after tariquidar supports our conclusion that P-gp was not completely blocked in this study.
Although we didn’t measure plasma radioactivity in most subjects in this study, we believe that increased brain radioactivity was due to P-gp inhibition, not increased 11C-dLop in plasma. We previously showed that intravenous tariquidar with delayed injection of radioligand increased brain radioactivity without increasing 11C-dLop in plasma (4). Also, we recently performed concurrent tariquidar–11C-dLop PET in a separate study, and peak plasma concentration of 11C-dLop was similar (7.6 SUV) to reported values for subjects at baseline (4).
One limitation of this study is that we did not perform genetic testing to correct for the presence of known polymorphisms on the ABCB1 gene. Some of these polymorphisms decrease P-gp expression or function (21) and may contribute to interindividual differences in PET response to tariquidar. Another limitation is that most PET scans were analyzed by calculating the AUC10–30 to avoid the need for arterial sampling. Full quantification of the PET data may have resulted in less variance in 11C-dLop uptake values. However, we do not think the simplified AUC10–30 measure introduces a large amount of error because our previous report showed a strong correlation between AUC10–30 and K1 values for 11C-dLop (4). Moreover, because 11C-dLop rapidly clears from plasma (6), avoiding use of the arterial input function may actually reduce error in quantifying uptake of 11C-dLop in the brain. Another limitation is that free fraction of tariquidar was measured in only a subset of subjects. Because only tariquidar that is not bound to proteins in plasma is available to bind to and inhibit P-gp, only the free concentration of tariquidar would be expected to correlate with 11C-dLop uptake into the brain. However, we found little variance in the free fraction of tariquidar (coefficient of variation, 6%), suggesting we would have seen a similar dose–response relationship using the free concentration of tariquidar.
CONCLUSION
Performing 11C-dLop PET imaging during peak plasma concentrations of tariquidar resulted in increased P-gp inhibition at the human BBB and allowed use of lower doses of the inhibitor. On the basis of prior studies in monkeys, we suspect that plasma concentrations of tariquidar did not fully block P-gp; however, higher doses of tariquidar would likely be associated with unacceptable side effects. This paradigm of concurrent administration of tariquidar and P-gp substrate drug may have potential for treating diseases associated with P-gp overexpression (e.g., multidrug resistant cancer).
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH) under clinicaltrials.gov identifier NCT00605254 (protocol 08-M-0062). No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank AzaTrius Pharmaceuticals for supplying tariquidar; Judith Starling, George Grimes, and Gerald Overman for formulating tariquidar; Jinsoo Hong and Yi Zhang for assistance in the production of radiotracer; David Luckenbaugh and Seonjoo Lee for assistance with statistical analysis; Ioline Henter for excellent editorial assistance; and Maria D. Ferraris-Araneta, Denise Rallis-Frutos, Yulin Chu, Barbara Scepura, Emily Page, Gerald Hodges, Robert Gladding, and the NIH PET Department for successfully completing the PET studies. The internal standard used in the liquid chromatography–tandem mass spectrometry method was synthesized by Sanjay Telu.
Footnotes
Published online Dec. 11, 2014.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 6, 2014.
- Accepted for publication November 13, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Imaging the Activity of Efflux Transporters at the Blood-Brain Barrier in Neurologic Diseases: Radiotracer Selection Criteria
- Imaging P-Glycoprotein Induction at the Blood-Brain Barrier of a {beta}-Amyloidosis Mouse Model with 11C-Metoclopramide PET
- Impact of P-Glycoprotein Function on the Brain Kinetics of the Weak Substrate 11C-Metoclopramide Assessed with PET Imaging in Humans
- A Proof-of-Concept Study to Inhibit ABCG2- and ABCB1-Mediated Efflux Transport at the Human Blood-Brain Barrier
- P-Glycoprotein (ABCB1) Inhibits the Influx and Increases the Efflux of 11C-Metoclopramide Across the Blood-Brain Barrier: A PET Study on Nonhuman Primates
- Strategies to Inhibit ABCB1- and ABCG2-Mediated Efflux Transport of Erlotinib at the Blood-Brain Barrier: A PET Study on Nonhuman Primates