


Abstract
Routine application of 18F-labeled peptides for quantitative in vivo receptor imaging of receptor-expressing tissues and quantification of receptor status using PET is limited by the lack of appropriate radiofluorination methods for routine large-scale synthesis of 18F-labeled peptides. To satisfy this demand, a new 18F-labeling methodology based on the chemoselective oxime formation between an unprotected aminooxy-functionalized peptide and an 18F-labeled aldehyde or ketone was investigated and optimized with respect to peptide conjugation. Methods: 4-[18F]Fluorobenzaldehyde ([18F]FB-CHO) was prepared from the 4-formyl-N,N,N-trimethylanilinium precursor via direct no-carrier-added 18F-fluorination (dimethyl sulfoxide, 60°C, 15 min) and purified using a cation-exchange/reversed-phase cartridge system. Radiochemical yields (RCYs) of N-(4-[18F]fluorobenzylidene)oxime ([18F]FBOA) formation with various aminooxy-modified peptides such as minigastrin, RGD, and octreotate analogs were investigated as a function of reaction time and temperature, peptide concentration, and pH. Biodistribution studies were performed with an [18F]FBOA-RGD dimer ((c(RGDfE)HEG)2-K-Dpr-[18F]FBOA, 60 and 120 min after injection) and a gylcosylated [18F]FB-Tyr3-octreotate (Gluc-S-Dpr([18F]FBOA)TOCA), 10 and 60 min after injection) using M21 and M21L human melanoma and AR42J rat pancreatic tumor-bearing nude mice, respectively. Results: [18F]FB-CHO was obtained in a nonoptimized RCY of 50% within 30 min. At low peptide concentrations (0.5 mmol/L), optimal [18F]FBOA-labeling efficiencies (60%–80%) were obtained within 15 min at 60°C and pH 2–3, independently of the peptide used, affording the [18F]FBOA-peptides in overall RCYs of up to 40% (from end of bombardment) after purification. Both (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA and Gluc-S-Dpr([18F]FBOA)TOCA showed pharmacokinetics suitable for early (≤60 min) high-contrast PET imaging, high tumor uptake (2.48 ± 0.15 %ID/g [RGD] and 21.8 ± 1.4 %ID/g [TOCA] at 60 min after injection, where %ID/g = percentage injected dose per gram), and tumor-to-organ ratios that compared well with the corresponding [18F]fluoropropionyl analogs [18F] Galacto-RGD and Gluc-Lys([18F]FP)TOCA, which are prepared via multistep procedures. Conclusion: Oxime formation between aminooxy-functionalized peptides and an 18F-labeled aldehyde or ketone—in this case, [18F]FB-CHO—combines fast 1-step, high-yield synthesis of an 18F-labeled prosthetic group stable against in vivo defluorination with rapid, 1-step chemoselective conjugation to unprotected peptides under mild conditions. Thus, it allows fast and straightforward large-scale production of 18F-labeled peptides for clinical routine PET application. Furthermore, it opens new perspectives to peptide radiohalogenation in general, permitting labeling of the same precursor both with diagnostic (18F, 124I, 120gI, 123I) and therapeutic (211At, 131I) radiohalogens.
The fact that many human tumors overexpress a variety of receptors for regulatory peptides and peptide hormones (1) constitutes the basis of peptide receptor–targeted diagnostic imaging in nuclear medicine. Intense research has therefore been directed toward the development of small radiolabeled neuropeptide analogs with nuclear and pharmacokinetic properties suitable for high-contrast localization of human neoplasms. Of the imaging techniques available, PET represents the standard of excellence with respect to sensitivity, resolution, and the possibility for quantification. However, although several peptide analogs labeled with SPECT radionuclides such as 99mTc and 111In have entered clinical studies (2,3) or even clinical routine (4,5), only a limited number of receptor-targeted peptides has been labeled with PET radionuclides such as 18F, 68Ga, 64Cu, and also 86Y (for dosimetry); of these, only a low fraction has been applied for in vivo receptor imaging in patients (6–9).
18F with its half-life of 109.7 min and low β+-energy (0.64 MeV) represents the ideal radionuclide for PET (10). However, in contrast to radiometallation, rapid and direct no-carrier-added (n.c.a.) 18F-labeling of complex biomolecules such as peptides is not possible. Thus, various 18F-labeled prosthetic groups have been developed (11,12) and applied for conjugation labeling of biomolecules. The most frequently used method is 18F-acylation (13–15), which has been applied for 18F-labeling of octreotide (16,17), α-melanocyte-stimulating hormone (18), (Arg15,Arg21)vasoactive intestinal polypeptide (19), an RGD-containing glycopeptide (20), human C-peptide (21), and neurotensin(8–13) (22). In most cases, this methodology entails time-consuming, moderate-yield, multistep radiosyntheses of the 18F-labeled prosthetic group. So far, only one 1-step preparation of an n.c.a. 18F-fluoroacylation agent has been described (23). However, comparably low yields of N-succinimidyl 4-([18F]fluoromethyl)benzoate (18%–25% within 30 min) as well as defluorination (24) prevented routine application of this 18F-labeled prosthetic group.
A further disadvantage of the 18F-fluoroacylation methodology consists of the necessity to use protected peptide precursors for conjugation with the prosthetic group, which in turn requires subsequent deprotection.
Besides these practical drawbacks, the 18F-labeled peptide analogs developed so far often showed unsuitable biodistribution—that is, high hepatic and intestinal uptake (16,17), low tumor accumulation and retention (16), or low in vivo stability (22). This is not the case for Gluc-Lys([18F] FP)TOCA, the only 18F-labeled somatostatin analog used for in vivo sst quantification in patients so far (9), whose applicability for clinical routine—despite excellent imaging properties—is limited only by the disadvantages of the 18F-fluoroacylation methodology.
To allow fast and straightforward large-scale production of 18F-labeled peptides for clinical routine application, a new strategy ideally allowing both 1-step, high-yield synthesis of an 18F-labeled prosthetic group with stability against in vivo defluorination and fast, 1-step, chemoselective conjugation with unprotected peptides, preferably in aqueous media, was needed.
Oxime formation between an aminooxy- and a carbonyl- functionality has already been explored for the preparation of 14C-labeled analogs of putrescine, cadaverine, and diaminohexane for application in radiometric assays by reaction of the aminooxy-functionalized amines with 2-[14C] acetone (25) and for radioiodination of antibodies (26). Recently, it has been proposed as a suitable methodology for the synthesis of multimeric cyclic RGD peptides using solid-phase peptide synthesis and chemoselective oxime ligation (27). Chemoselective oxime formation has also been applied for the synthesis of glycopeptides (28,29), oligonucleotide-peptide conjugates (30,31), and complex proteins (from peptide fragments) (32). Furthermore, it was used for conjugation of oligoribonucleotides and proteins with metal chelates (33). Overall, the major advantages of this synthetic approach are its high chemoselectivity, the use of unprotected aminooxy precursors, and the fact that coupling with the carbonyl component can be performed in aqueous media (pH 4–7).
Based on these results, chemoselective oxime formation (Fig. 1) was applied for prosthetic group radiofluorination of various small peptides with relevance for clinical PET receptor imaging using 4-[18F]fluorobenzaldehyde ([18F]FB-CHO) (34,35). The new 18F-labeling strategy was evaluated in detail and optimized with respect to peptide labeling efficiency using various analogs of minigastrin, RGD, and octreotate functionalized with aminooxyacetic acid (Fig. 2). Furthermore, the suitability of 2 representative RGD and octreotate analogs for in vivo αvβ3 integrin and sst mapping, respectively, was also evaluated in vivo in M21 and M21L melanoma (RGD analog) and AR42J (octreotate derivative) tumor-bearing nude mice.

Radiosynthesis of [18F]FB-CHO and conjugation to aminooxy-functionalized peptides via oxime formation. Reaction conditions are (1) [K⊂2.2.2.]+ 18F−, 15 min at 60°C in dimethyl sulfoxide, followed by cartridge purification (1, SCX; 2, RP-18) and elution of [18F]FB-CHO with MeOH, and (2) aminooxy peptide (0.5 mmol/L) in H2O, MeOH, and trifluoroacetic acid (pH 2.5), 15 min at 60°C. [18F]FBOA = N-(4-[18F]fluorobenzylidene)oxime.
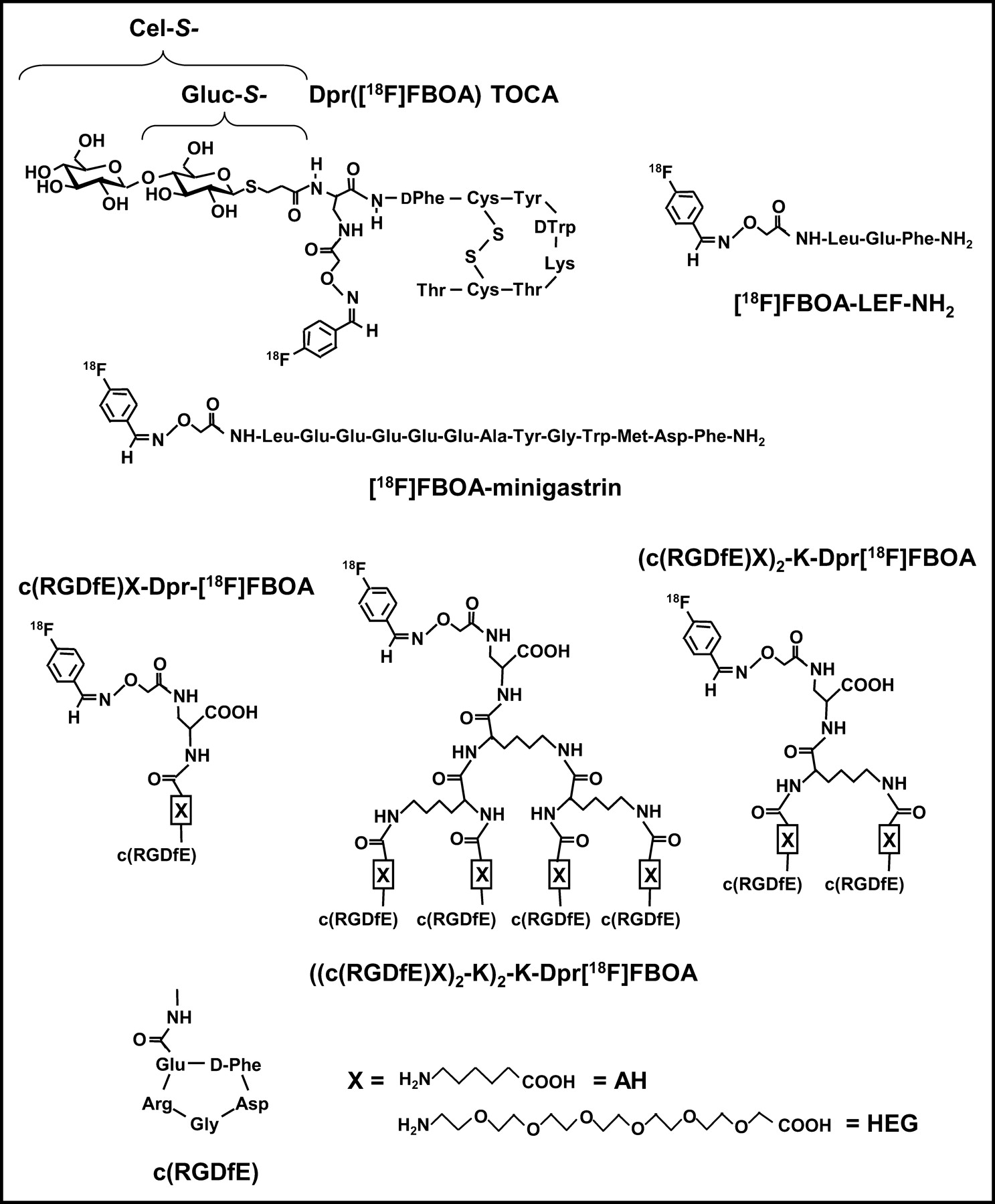
Structures of all [18F]FBOA-labeled peptides investigated in this study.
MATERIALS AND METHODS
General Conditions
Fmoc-amino acids (Fmoc = 9-fluoroenylmethoxycarbonyl-) as well as N-Boc-aminooxyacetic acid ((Boc)Aoa) and Rink Amide resin AM (4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl)phenoxyacetamido-norleucylaminomethyl resin) were purchased from Novabiochem. Solvents and all other organic reagents were purchased from Merck Eurolab, Alexis, Aldrich, or Fluka.
Solid-phase peptide synthesis was performed manually using a flask shaker (St. John Associates Inc.).
Analytic reversed-phase high performance liquid chromatography (RP-HPLC) was performed on a Nucleosil 100 C18 (5 μm, 125 × 4.0 mm) column using a Sykam gradient HPLC System. The peptides were eluted applying different gradients of 0.1% trifluoroacetic acid (TFA) (v/v) in H2O (solvent A) and 0.1% TFA (v/v) in acetonitrile (solvent B) at a constant flow of 1 mL/min (specific gradients are cited in the text). Ultraviolet detection was performed at 220 nm using a UVIS 200 photometer (Linear Instrument Corp.). Retention times (tR) and the capacity factor (K′) are cited in the text.
Preparative RP-HPLC was performed on the same HPLC system using a YMC RP 18 s, 4-μm, 80-Å column (150 × 20 mm; YMC Europe GmbH) at a constant flow of 10 mL/min.
Mass spectra were recorded on the LC-MS system LCQ from Finnigan using the Hewlett Packard series 1100 HPLC system.
Peptide Synthesis
Aminooxyacetyl-Leu-Glu-Phe-NH2 (Aoa-LEF-NH2).
Dry Rink Amide resin (0.74 mmol of Fmoc- groups) was suspended is 15 mL 1-methyl-2-pyrrolidone. After removal of the N-terminal Fmoc-protecting group with 20% piperdine in N,N-dimethylformamide (DMF) (v/v), the peptide sequence Fmoc-Leu-Glu(OtBu)-Phe- was assembled on the resin according to a standard Fmoc protocol using 1.5 equivalent (eq) of Fmoc-amino acid with 1.5 eq of N-hydroxybenzotriazole and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate, respectively, as coupling reagents and N-ethyldiisopropylamine (DIPEA) as a base. N-terminal derivatization with N-Boc-aminooxyacetic acid ((Boc)Aoa) was performed using 10 eq of (Boc)Aoa, 1-hydroxy-7-azabenzotriazole, N,N′-diisopropylcarbodiimide, respectively, and DIPEA as a base. Cleavage from the solid support and concomitant removal of the acid-labile protecting group was performed using a mixture of 95% TFA, 2.5% triisobutylsilane, and 2.5% H2O (v/v/v). After purification via preparative HPLC, Aoa-LEF-NH2 was obtained in 13% yield.
HPLC (15%–50% solvent B in 20 min): tR = 9.55, K′ = 6.02.
Calculated monoisotopic mass for Aoa-LEF-NH2 (C22H33N5O7): 479.2.
Found: m/z = 463.3 [M-NH2]+, 480.2 [M+H]+, 502.4 [M+Na]+, 959.2 [2M+H]+.
Aminooxyacetyl-Minigastrin (Aoa-Minigastrin).
The peptide sequence Fmoc-Leu-Glu(OtBu)-Glu(OtBu)-Glu(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Tyr(tBu)-Gly-Trp(Boc)-Met-Asp(OtBu)-Phe- was synthesized on Rink Amide resin as described for Aoa-LEF-NH2. Coupling of (Boc)Aoa and subsequent cleavage from the resin were also performed as outlined. Yield after preparative HPLC was 12%.
HPLC (15%–50% solvent B in 20 min): tR = 17.03, K′ = 11.52.
Calculated monoisotopic mass for Aoa-minigastrin (C76H102N16O28S): 1,718.7.
Found: m/z = 860.7 [(M+2H)/2]+, 879.7 [(M+H+K)/2]2+, 1,719.4 [M+H]+, 1,741.7 [M+Na]+.
The syntheses of the monomeric (c(RGDfE)-Dpr-Aoa (Dpr = 2,3-diaminopropionic acid), c(RGDfE)-AH-Dpr-Aoa, and c(RGDfE)-HEG-Dpr-Aoa); dimeric ((c(RGDfE)AH)2-K-Dpr-Aoa and (c(RGDfE)HEG)2-K-Dpr-Aoa); and tetrameric (((c(RGDfE)AH)2-K)2-K-Dpr-Aoa and ((c(RGDfE)HEG)2-K)2-K-Dpr-Aoa) cyclo-RGD derivatives and of the glycosylated octreotide analogs Gluc-S- and Cel-S-Dpr(Aoa)TOCA investigated in this study (Fig. 2) are described in detail elsewhere (28,36).
Synthesis of Precursors and Chromatographic Standards
Synthesis of 4-Formyl-N,N,N-Trimethylanilinium Triflate (37).
To a solution of 4-N,N-dimethylaminobenzaldehyde (66 mg, 0.44 mmol) in dichloromethane (7 mL), methyltrifluoromethansulfonate (54 μL, 0.48 mmol) was added via syringe. After stirring overnight, the crude product was precipitated by the addition of Et2O and recrystallized from dichloromethane/diethyl ether. 4-Formyl-N,N,N-trimethylanilinium triflate was obtained as white needles (100 mg; yield, 71%).
1H-NMR (250 MHz, (CD3)2SO): δ(ppm) 3.63 (9 H, s), 8.12–8.22 (4 H, m), 10.11 (1H, s) (NMR = nuclear magnetic resonance).
Conjugation of Aminooxyacetic Acid with 4-Fluorobenzaldehyde.
Aminooxyacetic acid hemihydrochloride (5.2 mg, 23.8 μmol) was dissolved in 48 μL H2O. Then, 4-fluorobenzaldehyde (5.6 μL, 52.4 μmol) and 1 μL TFA (2.5%) were added, and the mixture was heated to 60°C for 30 min. After cooling, the product was purified using RP-HPLC.
Calculated monoisotopic mass for FBOA-OH (C9H8NO3F) (FBOA = N-(4-[18F]fluorobenzylidene)oxime): 197.05.
Found: m/z = 198.1 [M+H]+, 220.0 [M+Na]+.
1H-NMR (250 MHz, CDCl3): δ(ppm) 4.79 (2H, s, CH2), 7.11 (2H, t, J = 8.7 Hz, arom.), 7.58–7.64 (2H, m, arom.), 8.22 (1H, s, CH=N).
Synthesis of 19F-Reference Compounds
Generally, 26.2 nmol of peptide (1 eq) were dissolved in 47 μL of H2O and MeOH (v/v) (21:79), and 28.7 nmol 4-fluorobenzaldehyde (1.1 eq) were added. After 30 min at 60°C, the product was purified using analytic RP-HPLC.
FBOA-LEF-NH2.
Calculated monoisotopic mass for FBOA-LEF-NH2 (C29H36N5O7F): 585.2.
Found: m/z = 569.2 [M-NH2]+, 586.2 [M+H]+, 608.4 [M+Na]+, 1171.2 [2M+H]+.
HPLC (15%–50% solvent B in 20 min, 50% constant solvent B 20–30 min): tR = 18.10, K′ = 12.31.
FBOA-Minigastrin.
Calculated monoisotopic mass for FBOA-minigastrin (C83H105N16O28SF): 1,824.7.
Found: m/z = 1,808.2 [M-NH2]+, 1,825.3 [M+H]+, 1,847.5 [M+Na]+.
HPLC (15%–50% solvent B in 20 min): tR = 22.12, K′ = 15.26.
c(RGDfE)-Dpr-FBOA.
Calculated monoisotopic mass for c(RGDfE)-Dpr-FBOA (C38H48N11O12F): 869.3.
Found: m/z = 870.4 [M+H]+, 892.3 [M+Na]+, 908.3 [M+K]+.
HPLC (15%–50% solvent B in 20 min): tR = 15.68, K′ = 10.53.
c(RGDfE)-AH-Dpr-FBOA.
Calculated monoisotopic mass for c(RGDfE)-AH-Dpr-FBOA (C44H59N12O13F): 982.4.
Found: m/z = 492.5 [(M+2H)/2]2+, 983.5 [M+H]+, 1,005.5 [M+Na]+, 1,021.4 [M+K]+.
HPLC (15%–50% solvent B in 20 min): tR = 16.22, K′ = 10.93.
(c(RGDfE)AH)2-K-Dpr-FBOA.
Calculated monoisotopic mass for (c(RGDfE)AH)2-K-Dpr-FBOA (C92H116N23O23F): 1,809.9.
Found: m/z = 906.4 [(M+2H)/2]2+, 917.2 [(M+H+Na)/2]2+, 1,810.9 [M+H]+.
HPLC (15%–50% solvent B in 20 min): tR = 16.33, K′ = 11.01.
((c(RGDfE)AH)2-K)2-K-Dpr-FBOA.
Calculated monoisotopic mass for ((c(RGDfE)AH)2-K)2-K-Dpr-FBOA (C158H230N45O43F): 3,464.7.
Found: m/z = 867.9 [(M+4H)/4]4+, 1,156.5 [(M+3H)/3]3+, 1,733.8 [(M+2H)/2]2+.
HPLC (15%–50% solvent B in 20 min): tR = 16.44, K′ = 11.09.
c(RGDfE)-HEG-Dpr-FBOA.
Calculated monoisotopic mass for c(RGDfE)-HEG-Dpr-FBOA (C52H75N12O19F): 1,190.6.
Found: m/z = 1,191.6 [M+H]+, 1,213.5 [M+Na]+, 1,229.4 [M+K]+.
HPLC (15%–50% solvent B in 20 min): tR = 16.32, K′ = 11.00.
(c(RGDfE)HEG)2-K-Dpr-FBOA.
Calculated monoisotopic mass for (c(RGDfE)HEG)2-K-Dpr-FBOA (C98H148N23O35F): 2,226.05.
Found: m/z = 743.4 [(M+3H)/3]3+, 1,114.4 [(M+2H)/2]2+, 1,125.4 [(M+H+Na)/2]2+.
HPLC (15%–50% solvent B in 20 min): tR = 17.13, K′ = 11.60.
((c(RGDfE)HEG)2-K)2-K-Dpr-FBOA.
Calculated monoisotopic mass for ((c(RGDfE)HEG)2-K)2-K-Dpr-FBOA (C190H294N45O67F): 4,297.1.
Found: m/z = 1,076.1 [(M+4H)/4]4+, 1,434.1 [(M+3H)/3]3+.
HPLC (15%–50% solvent B in 20 min): tR = 17.00, K′ = 11.50.
Gluc-S-Dpr(FBOA)TOCA (36).
Calculated monoisotopic mass for Gluc-S-Dpr(FBOA)TOCA (C70H90N13O21S3F):1563.5.
Found: m/z = 1564.5 [M+H]+, 1602.3 [M+K]+.
HPLC (15%–50% solvent B in 20 min): tR = 18.33, K′ = 12.48.
Cel-S-Dpr(FBOA)TOCA (36).
Calculated monoisotopic mass for Cel-S-Dpr(FBOA)TOCA (C76H100N13O26S3F): 1,725.6.
Found: m/z = 883.1[(M+K+H)/2]2+, 1,726.5 [M+H]+, 1,748.6 [M+Na]+.
HPLC (15%–50% solvent B in 20 min): tR = 18.20, K′ = 12.29.
Radiolabeling
Thin-layer chromatography (TLC) was performed using Kieselgel 60 F254 TLC plates from Merck. The solvent systems used are specified in the text. For detection of radiolabeled compounds, an automatic TLC analyzer, Tracemaster 20 (Bertold), was used.
HPLC was performed using a Sykam gradient HPLC System and an UVIS 200 photometer. For radioacitvity measurement, the outlet of the ultraviolet photometer was connected to a Na(Tl) well-type scintillation counter (Ace Mate 925-Scint; EG&G Ortec). The RP-HPLC columns and solvents used in this section are as follows.
System A.
Nucleosil 100 5-C18, 125 × 4.6 mm (Macherey and Nagel), 1 mL/min flow.
System B.
YMC RP 18 s, 4 μm, 80 Å, 150 × 20 mm, 10 mL/min flow, gradient = 0% solvent B for 5 min, then 15%–50% solvent B in 20 min.
System C.
Multospher 100 RP 18–5, 250 × 10 mm (Macherey and Nagel), 3 mL/min flow, gradient = 20%–70% solvent B in 20 min.
Production of 18F-Fluoride.
18F-Fluoride was produced via the 18O(p,n)18F nuclear reaction by bombardment of an isotopically enriched 18O-H2O target with an 11-MeV proton beam at the in-house RDS-112 cyclotron (Siemens/CTI).
General Procedure for Azeotropic Drying of 18F-Fluoride Cryptate Solution.
The aqueous potassium carbonate solution (34.6 mmol/L, 10–100 μL) containing 18F-fluoride (0.185–0.555 GBq) was added to a conical vial (Reactivial) containing 0.25 mL dry acetonitrile as well as Kryptofix 2.2.2. (Merck; 5 mg, 13.3 μmol). The solvents were evaporated at 110°C using a gentle stream of argon. The azeotropic drying step was repeated at least 3 times with 300-μL portions of acetonitrile.
[18F]FB-CHO.
The dry cryptate ([K⊂2.2.2.]+ 18F−) was resolubilized with a solution of 4-formyl-N,N,N-trimethylanilinium triflate (usually 2–3 mg, 6.4–9.6 μmol) in 150 μL of dry dimethyl sulfoxide (DMSO). The vial was closed and the mixture was stirred at 60°C for 15 min. Completion of the labeling reaction was verified via RP-HPLC (system A, 20%–70% solvent B in 20 min). For cartridge purification, the solution was then diluted with 2 mL of H2O, and [18F]FB-CHO was separated from unreacted 4-formyl-N,N,N-trimethylanilinium precursor using a LiChrolut SCX-cartridge (Waters). The eluted [18F]FB-CHO was then immobilized on a Sep-Pak C18 cartridge. The cartridge was washed with 4.5 mL MeCN and H2O (10:90, v/v) and 3 × 10 mL of H2O, and the [18F]FB-CHO was eluted with 0.5–1 mL of MeOH.
Alternatively, [18F]FB-CHO was purified using preparative RP-HPLC (system C). The product fraction was diluted with H2O to a volume of approximately 40 mL, and the [18F]FB-CHO was immobilized on a Strata X cartridge (33 μm, 60 mg; Phenomenex). After washing with 5 mL of H2O, the [18F]FB-CHO was eluted with 0.5–1 mL of MeOH.
Overall RCY: 52%–77%.
Product HPLC (system A, 20%–70% solvent B in 20 min): tR = 10.2 min, K′ = 6.50.
TLC (ethyl acetate and hexane, 1:1): Rf = 0.69.
General Procedure for Peptide Labeling with [18F]FB-CHO for Kinetic Studies.
A solution of peptide (0.026 μmol) in 10 μL H2O was added to 40 μL of the methanolic [18F]FB-CHO solution (1.48–4.44 MBq). TFA (2%, 1 μL) was added to adjust the pH to 2.5, and the reaction mixture was heated to 60°C. Samples were taken after various reaction times (from 1 to 40 min) and the peptide labeling yield via [18F]FBOA formation was analyzed using RP-HPLC (system A, 15%–50% solvent B in 20 min).
General Procedure for Peptide Labeling with [18F]FB-CHO for Biodistribution Studies.
A solution of peptide (0.11 μmol) in 100 μL H2O was added to 500 μL of the methanolic [18F]FB-CHO solution (92.5–170.2 MBq). TFA (2.5%, 8 μL) was added to adjust the pH to 2.5. After 10 min at 60°C, the 18F-labeled peptides were separated from unreacted precursor using preparative HPLC (system B). The solvents were removed in vacuo, and the radiopeptides were dissolved in phosphate-buffered saline (PBS) (pH 7.4) to yield solutions with activity concentrations of 1.11–1.85 MBq/100 μL.
Using Preparative HPLC.
Gluc-S-Dpr(Aoa)TOCA: tR = 14.4 min, K′ = 3.12.
Gluc-S-Dpr([18F]FBOA)TOCA: tR = 20.3 min, K′ = 4.79.
(c(RGDfE)HEG)2-K-Dpr-Aoa: tR = 14.0 min, K′ = 3.00.
(c(RGDfE)HEG)2-K-Dpr-[18F]FBOA: tR = 18.2 min, K′ = 4.20.
pH Dependence of [18F]FBOA Formation.
Samples (10 mL) of a 50 mmol/L potassium dihydrogen phosphate solution were buffered to various pH values in the range of 1–9 using either phosphoric acid (80%) (pH 1–4) or 1 mol/L KOH (pH 5–9). With these buffers, 2.6 mmol/L buffer solutions of the respective aminooxy peptide precursors for 18F-labeling were prepared. To 10 μL of each peptide solution, 40 μL of methanolic [18F]FB-CHO solution were added. The final pH of the reaction mixture was then determined using a glass electrode. The mixtures were incubated for 15 min at 60°C. [18F]FBOA formation as a function of pH was monitored via RP-HPLC.
Amino Acid Competition with [18F]FBOA Formation.
Equimolar amounts (approximately 12 μmol) of aminooxyacetic acid hemihydrochloride as well as lysine, cysteine, arginine, serine, and histidine were dissolved in 1,170 μL of a mixture of H2O and DMF (116:1, v/v). For the control, a solution containing only lysine, cysteine, arginine, serine, and histidine was prepared. Of these solutions, 10 μL were added to 40 μL of methanolic [18F] FB-CHO solution (1.48–4.44 MBq), respectively. After adjusting the pH to 4 using TFA (5%, 1 μL), the reaction mixtures were heated to 60°C for 25 min. The formation of 18F-labeled products was monitored using RP-HPLC.
Radiochemical yield (RCY) of N-(4-[18F]fluorobenzylidene)-O-carboxymethyl oxime: 90.5%.
HPLC (system A, 15%–50% solvent B in 20 min): tR = 16.75 min, K′ = 11.32.
Biodistribution Studies
Tumor Models.
For in vivo evaluation of the somatostatin receptor ligand Gluc-S-Dpr([18F]FBOA)TOCA, AR42J cells were used as a transplantable rat pancreatic tumor model with high sst2-somatostatin receptor expression. AR42J cells were obtained from the European Collection of Cell Cultures). Cells were maintained in RPMI 1640 medium (Seromed) supplemented with 10% fetal calf serum (Seromed) and 2 mmol/L l-glutamine (Gibco BRL Life Technologies).
Biodistribution of c(RGDfE)-derivatives was investigated in mice bearing 2 xenotransplanted human melanomas, M21 and M21-L. While the M21 melanoma cell line expresses the αvβ3 integrin, the melanoma M21-L cell line shows weak expression of the αvβ3 integrin and, therefore, was used as a negative control (38). M21 and M21-L cells were grown in RPMI 1640 medium supplemented with 10% fetal calf serum and gentamycin (Seromed).
All cultures were maintained at 37°C in a 5% CO2/humidified air atmosphere.
To establish tumor growth, cells were detached from the surface of the culture flasks using 1 mmol/L ethylenediaminetetraacetic acid in PBS, centrifuged, and resuspended in serum-free culture medium. In the case of the AR42J cells, the concentration of the cell suspension was 2.5–5 × 106 cells per 100 μL serum. Nude mice (female, 6–8 wk) were injected subcutaneously with 100 μL of the cell suspension into the flank. In the case of the melanoma cell lines, 5 × 106 M21 cells were injected subcutaneously in the right flank, and 1.5 × 107 M21-L cells were injected in the left flank of nude mice (female, 6–8 wk). Ten to 14 d after tumor transplantation, all mice showed solid palpable tumor masses (tumor weight, 200–900 mg) and were used for experiments.
Biodistribution.
For biodistribution studies, 1.48–2.22 MBq of the respective 18F-labeled radioligand in 100 μL PBS (pH 7.4) were injected intravenously into the tail vein of tumor-bearing nude mice. To investigate the extent of receptor-specific binding of the octreotide and RGD derivatives in sst- and αvβ3 integrin-expressing tissues, 0.4 mg Tyr3-octreotide/kg (10 μg per mouse) or 18 mg c(RGDfV)/kg (cyclo-(Arg-Gly-Asp-d-Phe-Val); 360 μg per mouse), respectively, were coinjected. The animals were killed at different times after injection (10 and 60 min after injection [octreotide analog] or 60 and 120 min after injection [RGD analog]) and the organs of interest were dissected. The radioactivity was measured in weighted tissue samples using a γ-counter. Results are expressed as the percentage injected dose per gram (%ID/g) (mean ± SD, 3–5 animals per group).
PET Imaging.
For PET imaging, the 18F-labeled peptide (2.37 MBq in 200 μL PBS, pH 7.4) was injected intravenously into the tail vein of AR42J tumor- bearing nude mice. For competition studies, 0.6 mg Tyr3-octreotide/kg (15 μg per mouse) were coinjected. After 60 min, mice were anesthesized using a combination of xylazine and ketamine. Static PET images (20 min, 5-min transmission, zoom 3.0) were acquired using a Siemens EXACT HR+ scanner. The axial resolution of the scanner at full width at half maximum) is approximately 5 mm; the transaxial resolution is approximately 8 mm. Images were reconstructed by iterative reconstruction (8 iterations, 4 subsets).
RESULTS
Radiolabeling
Synthesis of [18F]FB-CHO.
Direct 18F-fluorination of 4-formyl-N,N,N-trimethylanilinium triflate using dry cryptate ([K⊂2.2.2.]+ 18F−) was performed within 15 min at 60°C in DMSO. Subsequent purification of [18F]FB-CHO was performed using 2 alternative methods—either preparative RP-HPLC followed by cartridge immobilization of the product and subsequent elution with MeOH or using a cartridge system consisting of a strong cation-exchange material and an RP cartridge. For both purification methods, RCYs of [18F]FB-CHO were comparable (50%–55%) after overall synthesis times of 55 min (HPLC purification) and 30 min (cartridge purification), respectively. [18F]FB-CHO was obtained with high radiochemical purity (>98%).
Peptide Labeling via Oxime Formation with [18F]FB-CHO.
The reaction scheme of 18F-labeling of aminooxy-functionalized peptides via oxime formation with [18F] FB-CHO, leading to the corresponding N-(4-fluorobenzylidene)oxime ([18F]FBOA), is depicted in Figure 1. To optimize peptide 18F-labeling conditions with respect to peptide concentration, reaction time, and reaction temperature, one exemplary RGD analog, c(RGDfE)-Dpr-Aoa (Fig. 2), was incubated with methanolic [18F]FB-CHO at pH 2.5 under varying conditions (Fig. 3). At 20°C, peptide concentrations of ≥1.6 mmol/L are necessary to obtain yields of ≥93% of c(RGDfE)-Dpr-[18F]FBOA within 15 min. For a peptide concentration of 0.5 mmol/L, only 33% RCY was observed at 20°C within the same time. However, when the reaction temperature was raised to 60°C, yields of c(RGDfE)-Dpr-[18F]FBOA were increased to 90% within 15 min and to 98% within 30 min. In the case of the 2.2 mmol/L peptide solution, nearly quantitative yields were observed within only 5 min at 60°C. In contrast, reaction time had to be extended to 15 min under the same conditions to obtain a similar RCY of [18F]FBOA-minigastrin (data not shown).

RCY of c(RGDfE)-Dpr-[18F] FBOA as function of peptide concentration, reaction time, and reaction temperature (A, 20°C; B, 60°C). 18F-labeling was generally performed in H2O and MeOH (10:40, v/v) at pH 2.5.
Because of the high RCYs obtained with the 0.5 mmol/L peptide within 15 min at 60°C, these conditions were chosen as the standard conditions for further [18F]FBOA-labeling studies.
Figure 4 shows the RCYs of [18F]FBOA formation with a series of different Aoa-functionalized peptides as a function of reaction time (0.5 mmol/L peptide, 60°C, pH 2.5). Aoa-LEF-NH2 shows the fastest reaction kinetics with [18F] FB-CHO and an RCY of approximately 85% within 10 min. Reaction kinetics were slightly slower and nearly identical in the case of all RGD analogs investigated: the 2 monomeric derivatives c(RGDfE)AH-Dpr-[18F]FBOA and c(RGDfE)HEG-Dpr-[18F]FBOA; one dimeric analog, (c(RGDfE)AH)2-K-Dpr-[18F]FBOA (data not shown); and the tetrameric derivative ((c(RGDfE)HEG)2-K2)-K-Dpr-[18F]FBOA. Yields of 80%–90% were reached after reaction times of 15–20 min. For the somatostatin analog Gluc-S-Dpr([18F]FBOA)TOCA, reaction kinetics during the first 7 min were identical to those found for the RGD peptides. At later time points, however, RCYs of Gluc-S-Dpr([18F]FBOA)TOCA were lower (60% vs. 80%–85% after 15 min, 73% vs. 90%–93% after 40 min, respectively). As for all RGD analogs, RCYs of approximately 80% were reached within 15 min for the other octreotate derivative in this study, Cel-S-Dpr([18F] FBOA)TOCA (data not shown).
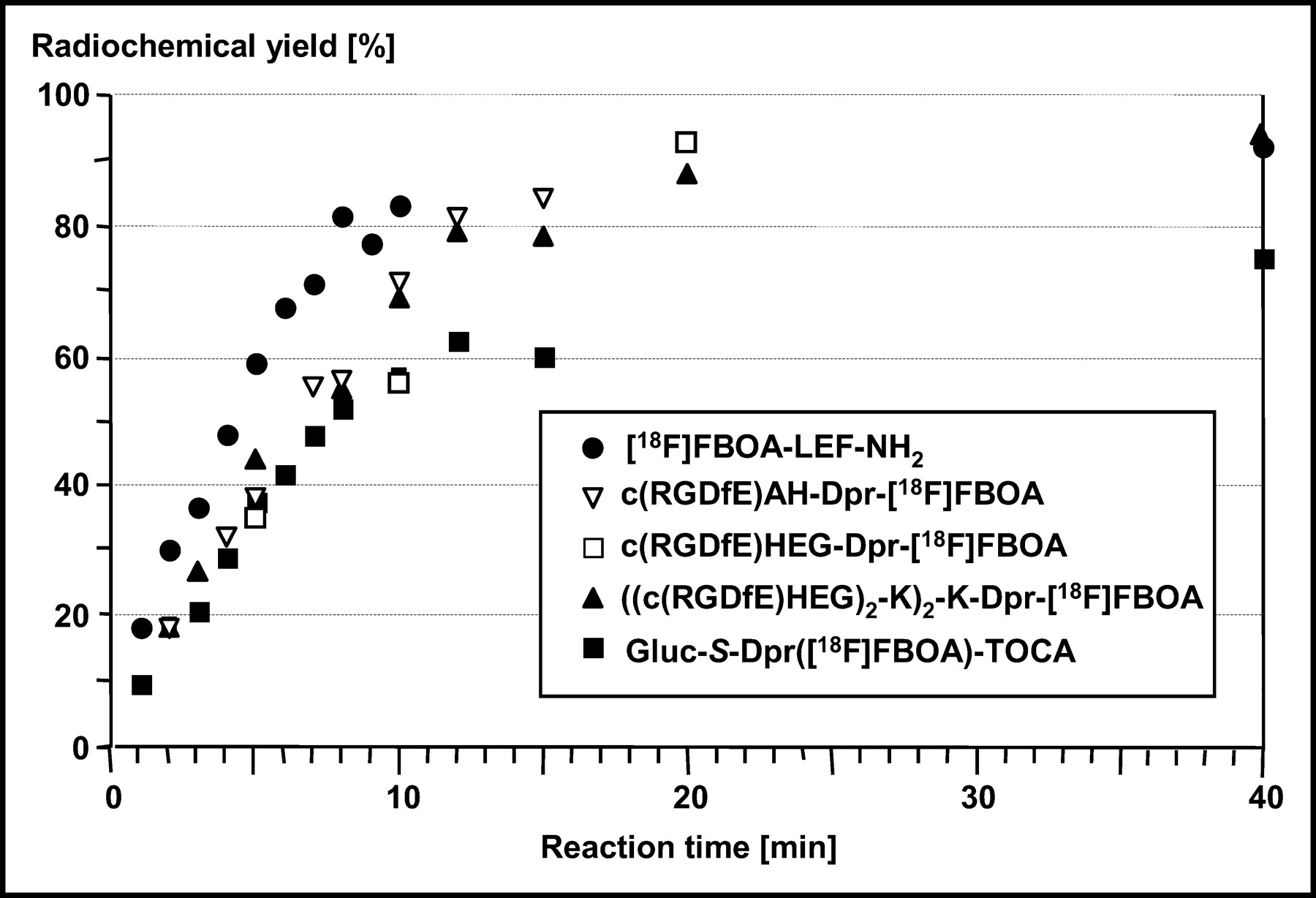
Reaction time dependence of RCY of oxime formation between [18F]FB-CHO and various aminooxy-functionalized peptides. 18F-labeling was generally performed using 0.5 mmol/L peptide solution in H2O and MeOH (10:40, v/v) at pH 2.5 (15 min, 60°C).
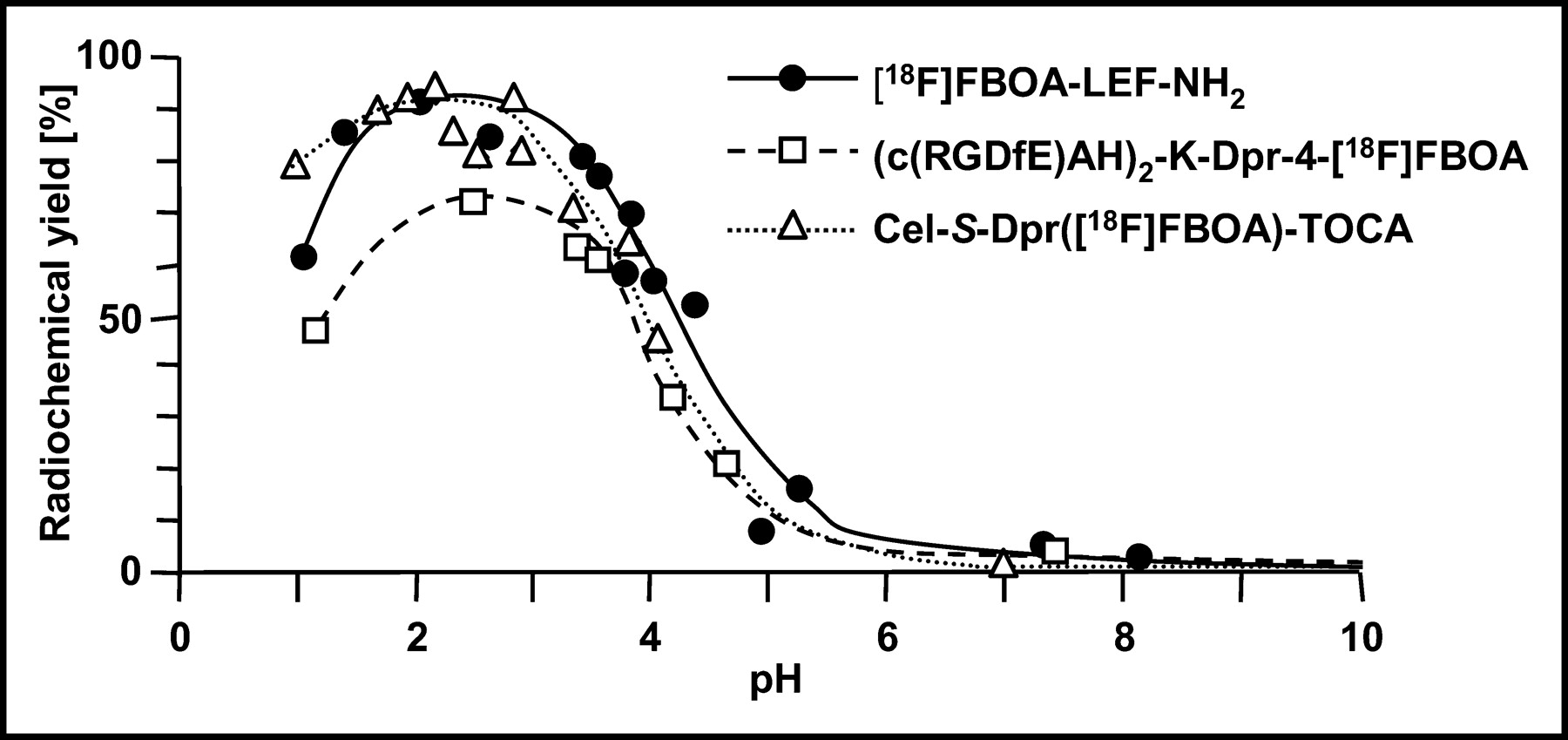
The pH dependence of [18F]FBOA formation was determined for Aoa-LEF-NH2, (c(RGDfE)AH)2-K-Dpr-Aoa, and Cel-S-Dpr(Aoa)TOCA (Fig. 5). For all peptides, maximum RCYs of [18F]FBOA labeling were observed in the pH range between 2 and 4. At pH values above 4, RCYs were already significantly decreased and were reduced to zero at physiologic pH. Although yields of [18F]FBOA-LEF-NH2 and the [18F]FBOA-RGD analog were also dramatically reduced at pH values below 1.5, the [18F]FBOA-labeling efficiency of Cel-S-Dpr(Aoa)TOCA was only slightly affected by decreasing pH.
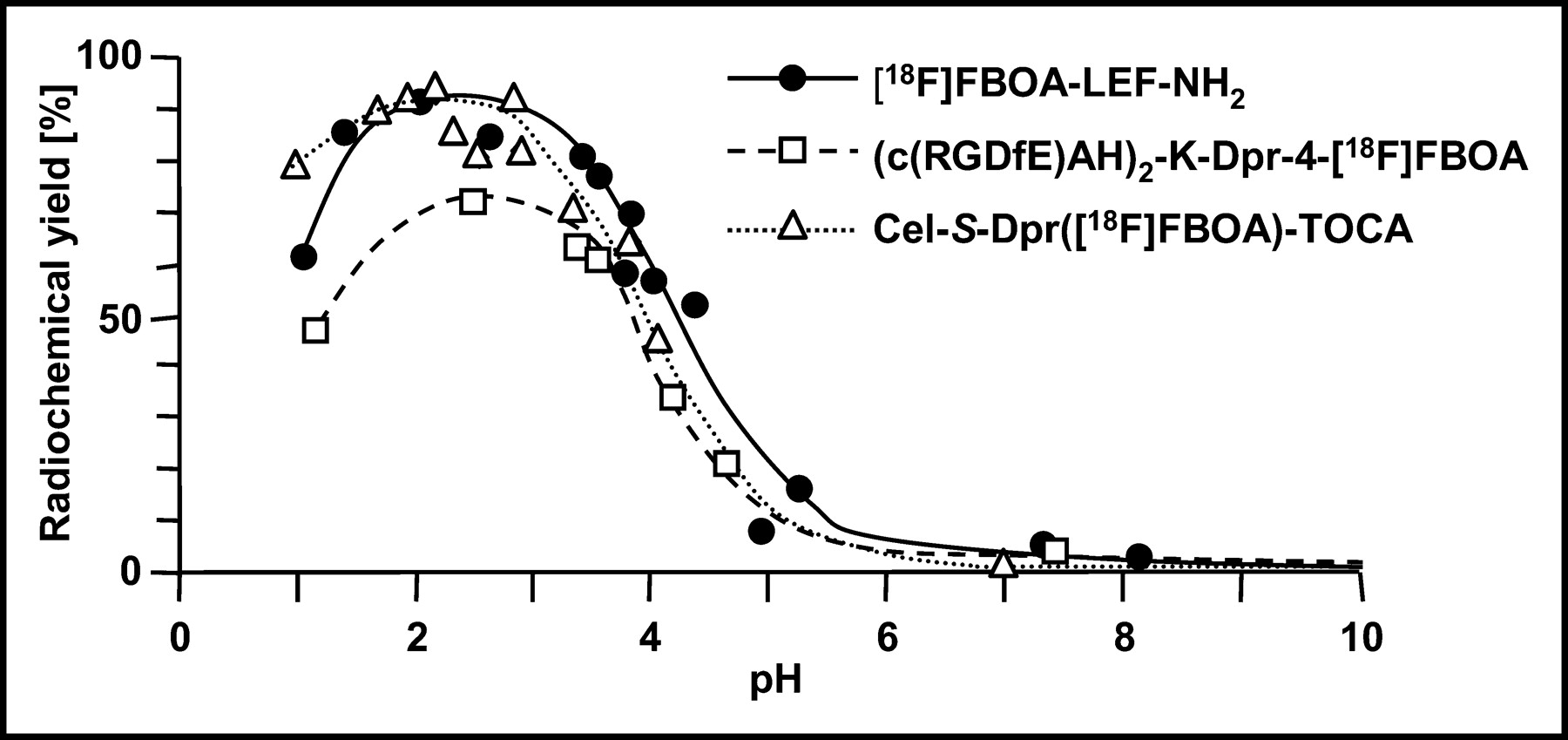
RCY of [18F]FBOA-LEF-NH2, (c(RGDfE)AH)2-K-Dpr-[18F]FBOA, and Cel-S-Dpr([18F]FBOA)TOCA as function of pH of reaction mixture. Peptides were dissolved in 50 mmol/L potassium dihydrogen phosphate solution buffered to various pH values with either phosphoric acid (pH 2–4) or KOH (pH 5–8). Labeling was generally performed under standard conditions (0.5 mmol/L peptide, 60°C, 15 min).
To determine the specificity of [18F]FBOA formation with the aminooxy group, an amino acid competition experiment was performed. Cysteine, arginine, histidine, serine, and lysine were incubated with [18F]FB-CHO both in the presence and in the absence of an equimolar amount of 2-aminooxyacetic acid under standard conditions (60°C, 25 min). In the latter case, 15% of the [18F]FB-CHO reacted to a radiolabeled conjugate, which has been identified as the reaction product with cysteine. However, in the presence of 2-aminooxyacetic acid, the RCY of [18F]FBOA-OH was 93% within 25 min in all cases, indicating a high specificity of oxime formation between [18F]FB-CHO and the aminooxy group.
Biodistribution Studies
To investigate the potential of [18F]FBOA labeling for the synthesis of 18F-labeled receptor-binding peptides with suitable pharmacokinetics for clinical application, one representative RGD and one TOCA analog, (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA and Gluc-S-Dpr([18F]FBOA)TOCA (Fig. 2), were evaluated in in vivo biodistribution studies. In the case of (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA, M21 melanoma-bearing nude mice expressing the αvβ3 integrin were used. For the negative control, M21L melanomas, known to show low αvβ3 integrin expression, were used. Gluc-S-Dpr([18F]FBOA)TOCA was evaluated in nude mice bearing an AR42J rat pancreatic acinar tumor with high sst2 expression.
(c(RGDfE)HEG)2-K-Dpr-[18F]FBOA shows rapid renal excretion and low activity levels in blood, liver, and intestine already at 60 min after injection. (Table 1). At this time point, kidney accumulation of (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA is still comparably high (1.72 ± 0.18 %ID/g) but is reduced to 0.70 ± 0.32 %ID/g after 120 min. At both 60 and 120 min after injection, the tumor uptake of (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA was higher by a factor of 5–6 than that in the negative control (2.48 ± 0.15 vs. 0.55 ± 0.02 %ID/g and 1.63 ± 0.13 vs. 0.29 ± 0.07 %ID/g at 60 and 120 min after injection, respectively), demonstrating specificity of uptake. This was further confirmed by coinjection of 360 μg unlabeled c(RGDfV) per mouse, which resulted in a reduction of M21 tumor uptake to 0.35 ± 0.11 %ID/g at 60 min after injection (data not shown)—that is, control levels—whereas activity uptake in the M21L tumor remained almost unaffected (0.40 ± 0.21 %ID/g at 60 min after injection).
Comparative Biodistribution of [18F]Galacto-RGD and (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA in M21 and M21L Melanoma-Bearing Nude Mice 60 and 120 Minutes After Injection
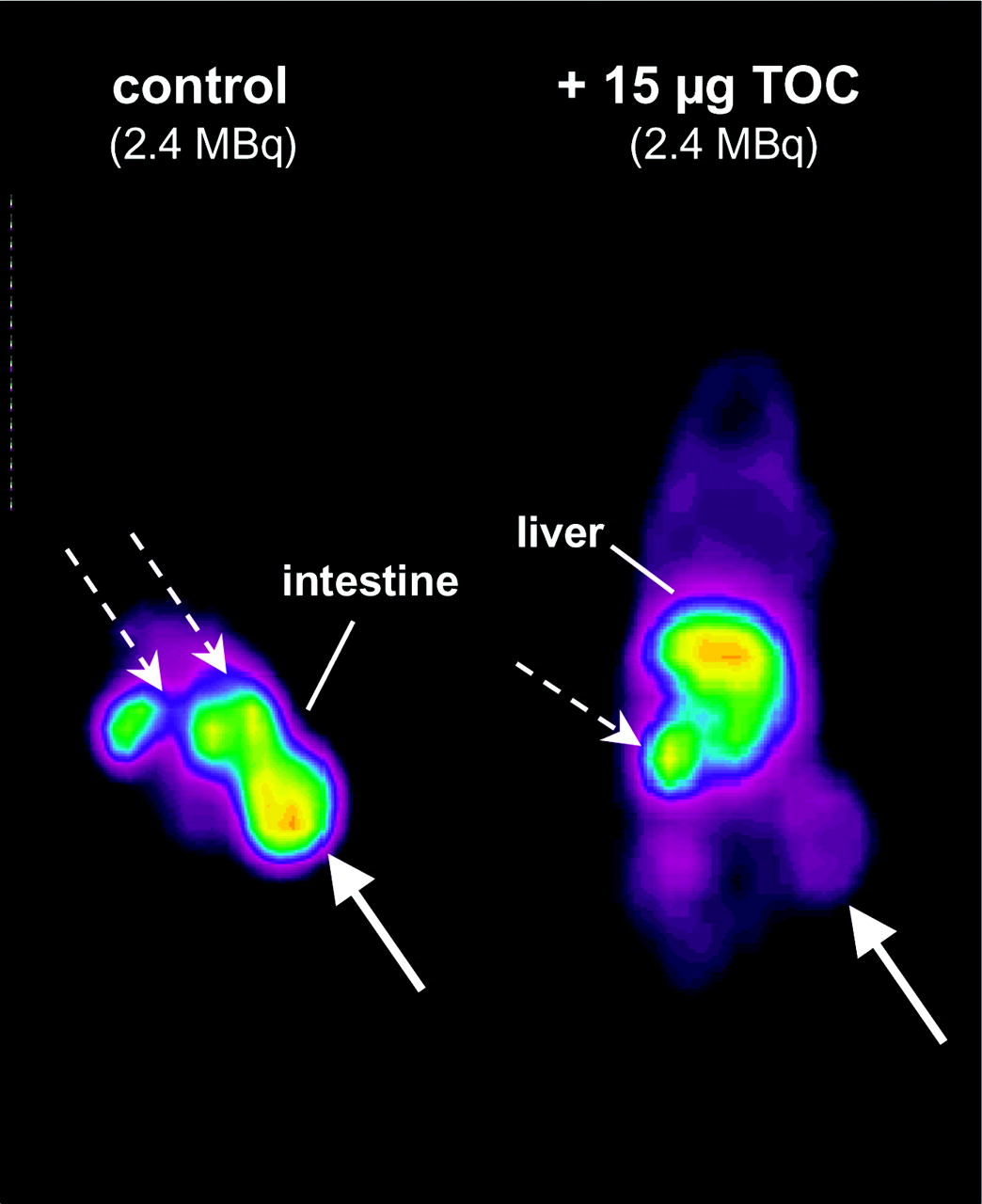
Gluc-S-Dpr([18F]FBOA)TOCA (Table 2) showed rapid blood clearance and was mainly excreted via the kidneys. However, the observed decrease in liver accumulation and the parallel increase in intestinal activity between 10 and 60 min after injection (liver, 16.96 ± 2.03 to 3.54 ± 0.88 %ID/g, and intestine, 2.69 ± 0.49 to 6.96 ± 1.22 %ID/g, at 10 and 60 min after injection, respectively) also indicate a certain contribution of hepatobiliary clearance to ligand excretion. Generally, accumulation of Gluc-S-Dpr([18F] FBOA)TOCA in sst-positive tissues was high. Interestingly, activity levels in stomach, pancreas, and adrenals decreased with time, whereas tumor uptake increased from 15.9 ± 2.2 to 21.8 ± 1.4 %ID/g between 10 and 60 min after injection. That uptake of Gluc-S-Dpr([18F]FBOA)TOCA in tumor and other sst-positive tissues was receptor mediated was demonstrated in a competition biodistribution experiment as well as in competition imaging studies using PET (Fig. 6). When 10 μg Tyr3-octreotide were coinjected with Gluc-S-Dpr([18F]FBOA)TOCA, tumor accumulation 1 h after injection was reduced to 26.4% ± 2.9% of control. Uptake in stomach and pancreas was reduced to 42.0% ± 18% and 48.9% ± 10% of control, respectively (data not shown).

PET competition study in AR42J tumor-bearing nude mice 50–70 min after injection of 2.4 MBq Gluc-S-Dpr([18F]FBOA)TOCA. Mice were either injected with radioligand only (control, left) or coinjected with 15 μg unlabeled TOC (right). Solid arrows indicate tumor; dashed arrows indicate kidneys.
Comparative Biodistribution of Gluc-Lys([18F]FP)TOCA and Gluc-S-Dpr([18F]FBOA)TOCA in AR42J Rat Pancreatic Tumor-Bearing Nude Mice 10 and 60 Minutes After Injection
For both peptides investigated, the low-activity accumulation in the femur—especially at later time points—indicates stability of the [18F]FBOA-moiety toward in vivo defluorination.
DISCUSSION
During the last decade, the use of small radiolabeled receptor-binding peptides has gained considerable impact on diagnostic nuclear medicine and peptide receptor radiotherapy. Although 18F-fluorine exhibits ideal nuclear characteristics for PET, and although PET offers unique features with respect to resolution and receptor quantification, only a few peptides have been labeled with 18F. This is mainly due to the current lack of straightforward, rapid, and high-yield 18F-labeling strategies. So far, most 18F-labeling approaches require time-consuming multistep syntheses of a prosthetic group and the use of protected peptide precursors, all of which entails low overall RCYs and, thus, limited practicability.
The chemoselective oxime ligation between an 18F-labeled aldehyde or ketone and an unprotected aminooxy-functionalized peptide introduced in this study represents a rapid, high-yield, 2-step 18F-labeling method for peptides and, thus, offers the unique opportunity to overcome the aforementioned limitations. The applicability of this new methodology crucially relies on the efficiency and ease of both the radiosynthesis of the 18F-labeled aldehyde or ketone—in this case, [18F]FB-CHO—and the chemoselective ligation of the [18F]aldehyde or ketone to the aminooxy-functionalized peptide. In this study, particular focus was directed toward the optimization of reaction conditions during peptide labeling. The conditions for the synthesis of [18F]FB-CHO applied in this study were based on previously reported protocols (35,37,39), and only minor adjustments of the reaction conditions and the subsequent cartridge purification step have been made so far to improve the labeling yield of the final 18F-labeled product. Under the nonoptimized conditions applied, the present methodology allows rapid (maximum, 30 min from end of bombardment) 1-step synthesis of [18F]FB-CHO in reasonable RCY and high radiochemical purities (>98%) and is suitable even for large-scale (2–3 GBq) production of [18F]FB-CHO.
In the only other 1-step radiosynthesis of an 18F-labeled prosthetic group reported so far, N-succinimidyl 4-([18F] fluoromethyl)benzoate (23), yields after an overall reaction time of 30 min were comparably low (18%). Furthermore, since this compound is an 18F-fluoroacylation agent, it can only be used for nonchemoselective 18F-labeling of at least partially protected precursors, which in turn necessitates an additional final deprotection step.
In contrast, and as demonstrated in an amino acid competition study, the oxime formation between [18F]FB-CHO and the aminooxy group is highly chemoselective. This circumstance allows 1-step [18F]FBOA labeling of any unprotected peptide without detectable formation of side products. The only exception in this study was Gluc-S-Dpr([18F] FBOA)TOCA, which showed the formation of a second 18F-labeled product eluting closely to the product peak during HPLC. Further investigation is required to determine whether this side product is an [18F]FBOA-isomer (cis- or trans-R-O-N=CH-Ar), resolved under the HPLC conditions applied, or whether it stems from precursor contamination with aminooxy-functionalized impurities.
Furthermore, 18F-labeling via oxime formation bears the advantage of being equally effective in a variety of solvents, including aqueous buffers and organic solvents such as DMSO, acetone, DMF, and MeOH as well as mixtures thereof (28–31,33). This allows 18F-labeling of peptides with limited solubility in either aqueous or organic solvents.
Independently of the type of peptide investigated, maximum RCYs were obtained between pH 2 and 4 (15 min, 60°C, 0.5 mmol/L peptide; Fig. 5). Almost no [18F]FBOA formation was observed at pH values of ≥5. These findings are in contradiction to data from other studies, which showed up to quantitative oxime formation at pH values between 4 and 7.5 (25,29,33,34). One possible explanation for the divergence is the substantial differences in aldehyde-to-aminooxy compound concentration ratios between the present n.c.a. case and those applied during organic synthesis.
Another factor with particular relevance for clinical routine 18F-labeling of peptides is the amount of unlabeled precursor needed for peptide labeling. Under the reaction conditions applied (60°C, 15 min, pH 2.5), only 25–100 nmol of peptide were needed (Fig. 3). This is a 15- to 25-fold higher peptide amount than that usually applied for kit radiolabeling and patient application of, for example, radiolabeled octreotide analogs (111In-Octreoscan, 10 nmol). Therefore, time-consuming HPLC separation of the [18F] FBOA-labeled peptides from unreacted aminooxy precursor is still indispensable. Further optimization studies are therefore focused on a further reduction of reaction volume and, thus, the peptide amount, which may then allow omission of the HPLC purification and a reduction of overall [18F] FBOA-labeling time from approximately 60 min at present to 40 min.
Even taking into account these minor restrictions, the oxime formation represents a, so far, unique methodology for fast, 2-step radiofluorination of small peptides. The applicability of this 18F-labeling strategy for the synthesis of peptide analogs with suitable pharmacokinetics has been demonstrated by the exemplary in vivo evaluation of the RGD analog (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA and the somatostatin analog Gluc-S-Dpr([18F]FBOA)TOCA (Figs. 6 and 7; Tables 1 and 2).

(A) Tumor-to-organ ratios 60 min after injection of [18F]Galacto-RGD and (c(RGDfE)HEG)2K-Dpr-4-[18F]FBOA in M21 melanoma-bearing nude mice (n = 3–5). (B) Tumor-to-organ ratios 60 min after injection of Gluc-Lys([18F]FP)TOCA and Gluc-S-Dpr([18F]FBOA)TOCA in AR42J rat pancreatic tumor-bearing nude mice (n = 4 or 5).
In the case of the RGD analog, tumor-to-nontumor ratios (Fig. 7) are comparable or even higher than those of [18F] Galacto-RGD, a glycosylated RGD analog that has already been successfully applied for animal PET imaging of αvβ3 integrin expression (20). In the case of Gluc-S-Dpr([18F] FBOA)TOCA, tumor accumulation 60 min after injection is increased by approximately 50% compared with the reference Gluc-Lys([18F]FP)TOCA (9). Due to the introduction of the comparably bulky and lipophilic [18F]FBOA group, however, lipophilicity of Gluc-S-Dpr([18F]FBOA)TOCA is significantly higher than that of the [18F]FP analog (log Pow [octanol water partition coefficient] = −1.24 ± 0.03 and −1.70 ± 0.03, respectively (36)). As illustrated in Figures 6 and 7, this leads to enhanced hepatic and intestinal accumulation and, thus, reduced tumor-to-liver and tumor-to-intestine ratios. It has been demonstrated previously that increasing the size of the sugar moiety can be used as a means to further reduce ligand lipophilicity and, consequently, the extent of hepatobiliary excretion (40). Based on this experience, Cel-S-Dpr([18F]FBOA)TOCA can be assumed to show optimized pharmacokinetics compared to the glucose analog and presumably even to Gluc-Lys([18F] FP)TOCA. A detailed comparative evaluation of this compound will be published elsewhere (36).
It can be anticipated that adaptation of the presented methods will also allow the production of other radiolabeled peptides, such as radioiodinated, radiobrominated, or radioastatinated peptides using appropriate aromatic aldehydes radiohalogenated with these nuclides.
CONCLUSION
The chemoselective formation of oximes, which has been evaluated for radiofluorination of peptides, represents a novel, innovative, and straightforward method that is superior to other strategies currently available. It allows rapid, high-yield conjugation of labeled aldehydes (e.g., [18F]FB-CHO) to aminooxy peptides in a chemoselective manner, thus permitting the use of fully deprotected peptides. Thus, this new method is suitable for large-scale production of 18F-labeled peptides for clinical PET imaging. Though initially devised as model compounds, the αvβ3 integrin antagonist (c(RGDfE)HEG)2-K-Dpr-[18F]FBOA and the somatostatin receptor ligand Gluc-S-Dpr([18F]FBOA)TOCA showed favorable pharmacokinetics and high tumor-to-nontumor ratios in vivo, demonstrating the suitability of [18F] FBOA-labeled peptides for PET applications.
Acknowledgments
The authors are indebted to Claudia Bodenstein, Brigitte Dzewas, and Coletta Kruschke for their excellent technical assistance. We acknowledge David A. Cheresh, The Scripps Research Institute, La Jolla, CA, for supplying the M21 and M21L melanoma cells, and Wolfgang Linke for supplying c(RGDfV) used in the in vivo competition studies. This work was supported by grants from the Deutsche Forschungsgemeinschaft, FOR 411/We 2386/2-1 and FOR 411/Ke 147/36-1.
Footnotes
Received Sep. 17, 2003; revision accepted Dec. 29, 2003.
For correspondence contact: Hans-Jürgen Wester, PhD, Nuklearmedizinische Klinik und Poliklinik, Klinikum rechts der Isar, Technische Universität München, Ismaninger Strasse 22, D-81675 München, Germany.
E-mail: h.j.wester{at}lrz.tum.de
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Production of diverse PET probes with limited resources: 24 18F-labeled compounds prepared with a single radiosynthesizer
- In Vivo PET Imaging of the Cancer Integrin {alpha}v{beta}6 Using 68Ga-Labeled Cyclic RGD Nonapeptides
- Preclinical Evaluation of a High-Affinity 18F-Trifluoroborate Octreotate Derivative for Somatostatin Receptor Imaging
- Three Methods for 18F Labeling of the HER2-Binding Affibody Molecule ZHER2:2891 Including Preclinical Assessment
- A Novel Facile Method of Labeling Octreotide with 18F-Fluorine
- Targeting of CCK-2 Receptor-Expressing Tumors Using a Radiolabeled Divalent Gastrin Peptide
- 18F-4V for PET-CT Imaging of VCAM-1 Expression in Atherosclerosis
- A Novel Method of 18F Radiolabeling for PET
- In vivo imaging of pyrrole-imidazole polyamides with positron emission tomography
- Preparation of a Promising Angiogenesis PET Imaging Agent: 68Ga-Labeled c(RGDyK)-Isothiocyanatobenzyl-1,4,7-Triazacyclononane-1,4,7-Triacetic Acid and Feasibility Studies in Mice
- Small-Animal PET Imaging of Human Epidermal Growth Factor Receptor Type 2 Expression with Site-Specific 18F-Labeled Protein Scaffold Molecules
- 18F-Labeled BBN-RGD Heterodimer for Prostate Cancer Imaging
- microPET of Tumor Integrin {alpha}v{beta}3 Expression Using 18F-Labeled PEGylated Tetrameric RGD Peptide (18F-FPRGD4)
- Use of a Peptide Derived from Foot-and-Mouth Disease Virus for the Noninvasive Imaging of Human Cancer: Generation and Evaluation of 4-[18F]Fluorobenzoyl A20FMDV2 for In vivo Imaging of Integrin {alpha}v{beta}6 Expression with Positron Emission Tomography
- Nuclear Imaging Probes: from Bench to Bedside
- A Thiol-Reactive 18F-Labeling Agent, N-[2-(4-18F-Fluorobenzamido)Ethyl]Maleimide, and Synthesis of RGD Peptide-Based Tracer for PET Imaging of {alpha}v{beta}3 Integrin Expression
- Molecular Targeting with Peptides or Peptide-Polymer Conjugates: Just a Question of Size?
- The Progress and Promise of Molecular Imaging Probes in Oncologic Drug Development
- Targeting Tumor Angiogenesis: Comparison of Peptide and Polymer-Peptide Conjugates