


Abstract
Gastrin/cholecystokinin subtype 2 receptors (CCK-2Rs) are overexpressed in several tumor types and are, thus, a potential target for peptide receptor radionuclide therapy (PRRT) of cancer. To improve the in vivo performance of CCK-2R binding peptides, we have previously synthesized and screened a series of divalent gastrin peptides for improved biochemical and biologic characteristics. In this study, we explore in more detail the most promising of these compounds and compare its performance with a previously described monomeric peptide. Methods: From six 111In-labeled 1,4,7,10-tetraazacyclododecane-N,N′,N″,N′″-tetraacetic acid (DOTA)–conjugated divalent gastrin peptides based on the C-terminal sequence of minigastrin, the maleimide-linked compound DOTA-GSC(succinimidopropionyl-EAYGWNleDF-NH2)-EAYGWNleDF-NH2 (MGD5) was selected. The in vitro stability, receptor binding, and internalization of 111In-MGD5 were studied and compared with those of monomer compound 111In-APH070. In vivo biodistribution and imaging using a SPECT/CT camera were also performed. Results: More than 90% of the labeled divalent peptide remained intact after 20 h of incubation in plasma. The inhibitory concentration of 50% of the divalent peptide was 1.0 versus 5.6 nM for the monomer, and the dissociation constant was 0.7 versus 2.9 nM. The rate of internalization of the divalent peptide was twice that of the monomer. Tumor uptake of the divalent peptide in vivo was about 6 times that of the monomer. The rate of washout of the divalent peptide from the tumor was lower than that of the monomer. Conclusion: Dimerization of the CCK-2R binding site results in an increase in binding affinity and an increase in tumor uptake both in vitro and in vivo. It is likely that these increases would result in improved tumor-targeting efficiency in patients with CCK-2R–positive tumors.
Because of its high expression in several tumor types, the gastrin/cholecystokinin subtype 2 receptor (CCK-2R) is of interest as a target for diagnostic imaging and peptide receptor radionuclide therapy (PRRT) of cancer. These tumor types include medullary thyroid carcinoma (MTC) (1), neuroendocrine tumors (2), small cell lung cancer, and colorectal cancers (in which increased expression of CCK-2 messenger RNA has been demonstrated) (3). In PRRT, a peptide is labeled with a particle-emitting radioisotope (such as a β- or an α-particle emitter). Ideally, specific uptake of the radiolabeled compound into tumor with rapid clearance from the rest of the tissues and organs leads to selective damage to tumor tissue (4). Although several studies have been performed using radiolabeled peptides that bind to the gastrin/CCK-2R (5), most of these have either demonstrated high kidney uptake (6) or modest tumor uptake (7,8), both of which make them unsuitable for PRRT. A recent study by Fröberg et al. (9) compared the uptake of 3 radiolabeled CCK/gastrin analogs in patients with MTC and demonstrated that peptides based on either the CCK-8 sequence or a truncated (des-pentaglu) gastrin sequence showed significantly lower tumor uptake than one containing the full minigastrin sequence. However, when peptides with this sequence are labeled with trivalent metallic radionuclides such as 90Y, they exhibit high renal uptake, which results in unacceptable renal toxicity in patients treated with this radiopharmaceutical.
In a previous study, we focused on the selection and optimization of monomeric 111In-labeled gastrin analogs based on the in vivo tumor-to-kidney ratio (10). This strategy produced a lead compound that showed low kidney retention but also relatively low tumor uptake in mice. A pilot clinical trial of this compound in patients with MTC confirmed the results of Fröberg—that is, that tumor uptake is low with peptides based on the truncated gastrin sequence (Peter van Rijk, oral communication, 2006). An alternative approach must, therefore, be found that increases tumor uptake but not at the expense of high renal retention.
It has been known for some time that multimerization of small molecules results in an increase in affinity to their complementary binding site (11), and multivalent arginine-glycine-aspartate (RGD) peptides have been used extensively to target αvβ3 integrin receptors with some success (12). The dimerization of peptides also has been used to increase enzymatic stability and is a means of improving selectivity without a concurrent increase in pharmacologic effect (13). In a previous study, we explored the effect of the dimerization of gastrin analogs as a means of increasing tumor localization while maintaining low levels of kidney retention. We synthesized several 1,4,7,10-tetraazacyclododecane-N,N′,N″,N′″-tetraacetic acid (DOTA)–peptide divalent peptides using various linkers and Nle/Met substitutions and evaluated them in vitro and in vivo (14). From this comparison, we have selected a maleimide-linked compound, DOTA-GSC(succinimidopropionyl-EAYGWNleDF-NH2)-EAYGWNleDF-NH2 (called MGD5), and compared its performance with that of a monomeric peptide, 111In-APH070 (10), by assessing their in vitro stability, receptor binding, internalization, tumor targeting, and in vivo biodistribution.
MATERIALS AND METHODS
Chemicals
All peptides were synthesized by piCHEM. The structures of MGD5 and APH070 are shown in Figure 1. High-performance liquid chromatography (HPLC)–grade acetonitrile was purchased from Sigma-Aldrich. HPLC-grade triethylammonium acetate (1 M solution) was purchased from Fluka. Disodium edetate (0.1 M) British Pharmacopoeia was purchased from Norwich NHS Trust. 111InCl3 in 0.05 M HCl was purchased from Tyco. All other chemicals were of analytic grade and were purchased from Sigma-Aldrich.

Chemical structures of APH070 (upper) and MGD5 (lower).
Cell Lines and Animal Models
AR42J rat pancreatic tumor cells were supplied by Cancer Research U.K. Cell Services. These were either used immediately or maintained in RPM1 (Cancer Research U.K. Cell Services) supplemented with 10% fetal bovine serum (FBS) (Gibco). For biodistribution studies, 5–7.5 million cells were injected subcutaneously into the left flank or shoulder of female CD1 nude mice (Charles River Laboratories) and grown for 7–10 d.
Radiolabeling and Radiochemical Analysis
The general labeling procedure was as follows: to a 2-mL polypropylene vial was added 15–80 μL of 111InCl3 (10–50 MBq) in 0.05 M HCl, 3–16 μL of gentisic acid (8.3 mg/mL) in 1 M ammonium acetate solution (pH 5.5) (one fifth the volume of 111InCl3), and 2.5–15 μL of peptide (1 mg/mL in water, 0.01 M phosphate buffer, pH 7.2 [APH070] or 50% phosphate-buffered saline [PBS]/water [MGD5]). The solution was heated at 98°C for 12 min (APH070) or 80°C for 15 min (MGD5) before 0.1 M ethylenediaminetetraacetic acid (EDTA) (one twentieth the volume of the reaction mixture) was added. The solutions were diluted with PBS, and radiochemical purity was determined using reversed-phase HPLC.
The radiolabeled peptides were analyzed using a Beckman system gold 128 solvent module and a 166 ultraviolet detector module (monitoring at 220 nm) combined with a GABi Star radiochemical detector (Raytest, GmbH). For method 1, compounds were separated on a 5-μm Jupiter 300 column (250 × 4.6 mm internal diameter; Phenomenex), with the following parameters: solvent A, 0.1% trifluoroacetic acid (TFA) in water; solvent B, 0.1% TFA in acetonitrile; and gradient, 0% B for 2 min, changing to 60% B over 20 min, then to 100% B over 5 min and back to 0% B over 5 min (flow rate, 1 mL/min).
Instant thin-layer chromatography (ITLC) was performed using ITLC silica gel (Pall Corp.) and the following 2 solvent systems: 50 mM EDTA in 0.1 M ammonium acetate, pH 5.5, and 3.5% (v/v) ammonia/methanol, 1:1. In the EDTA and ammonium acetate system, 111In-peptide remained at the origin (Rf = 0) and 111In-EDTA eluted to the solvent front (Rf = 1). In the ammonia and methanol system, 111In-peptide and 111In-EDTA both had an Rf of 1 and any colloidal material present in the reaction mixture remained at the origin.
Purification of Peptides
When required, the radiolabeled peptides were purified by HPLC. In the case of MGD5, the reaction mixture was diluted to 100–200 μL with 0.01 M phosphate buffer, pH 8.5, and the pH was adjusted to 8.5–9 with NaOH if necessary. The whole reaction mixture was purified by HPLC using method 2. For method 2, using an Agilent Eclipse XDB-C18 column (5-μm pore size, 4.6 × 150 mm), we separated the 111In-MGD5 from unlabeled MGD5 using a gradient of 0.1 M triethylammonium acetate (TEAA) (pH 7; solvent A) and acetonitrile (solvent B) as follows: 0% B for 2 min, increasing to 60% B over 17 min, then increasing to 95% B over 3 min and back to 0% B over 4 min (flow rate, 1 mL/min). The radioactive peak corresponding to 111In-MGD5 was collected, and the acetonitrile in the solvent was evaporated using a centrifugal evaporator (Speed Vac Plus SC110A; Savant). The pH was again increased to 8.5–9 to increase the solubility of 111In-MGD5.
To separate 111In-APH070 from unlabeled APH070, method 3, with the following parameters, was used: 0.01 M TEAA, 0.004% glacial acetic acid (w/v) (pH 5.5; solvent A) and acetonitrile (solvent B) on an Agilent Eclipse XDB-C18 column (5-μm pore size, 4.6 × 150 mm) with the gradient at 15% B for 2 min, increasing to 25% B over 20 min, then increasing to 60% B over 3 min and back to 15% B over 3 min (flow rate, 1.5 mL/min). The peak corresponding to 111In-APH070 was collected.
Plasma Stability Studies
The stability of 111In-MGD5 in mouse plasma and also in 0.1 M phosphate buffer, pH 8.5–9, and in PBS, pH 7.4, was compared. Whole mouse blood was centrifuged in a microfuge at 13,000 rpm, and the plasma supernatant was collected. Volumes of 200 μL of HPLC-purified radioligand were added to either 500 μL of plasma or 500 μL of phosphate buffer, pH 8.5–9, or 500 μL of PBS and incubated at 37°C. At various time points, 80 μL of the mixture were removed and mixed with 120 μL of acetonitrile to precipitate the plasma proteins. The sample was centrifuged for 10 min. Acetonitrile was again evaporated from the supernatant and the remainder analyzed by HPLC at each required time point using method A.
Cell Membrane Preparation
AR24J cell membranes were prepared as follows: adherently growing cells were harvested using versene (0.2% EDTA), diluted with ice-cold 20 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES), pH 7.3 (containing a 0.2 mg/mL concentration of bacitracin and 5 mM MgCl2) (buffer A), and centrifuged for 20 min at 1,800 rpm (500g), 4°C. The pellet was resuspended in 8 mL of buffer A and homogenized for 15 s using an Ultraturrax homogenizer. The cell homogenate was centrifuged for 10 min (1,800 rpm, 500g). The supernatant was transferred into a 30-mL ultracentrifuge tube and placed on ice. The pellet was resuspended in buffer A and the procedure repeated until 3 aliquots of cell supernatant had been collected. The combined supernatant was centrifuged for 45 min at 48,000g (20,000 rpm using a Beckman Coulter Avanti J-E centrifuge with a JA25.50 rotor). The resulting membrane pellet was resuspended in buffer A, and a Bio-Rad DC Protein Assay was performed in microplates (Bio-Rad Laboratories). The membranes were stored in aliquots at −80°C.
Receptor Binding Assays
Competition binding assays were performed on AR42J cell membranes using 111In-APH070 as the radioligand. APH070 was radiolabeled with 111InCl3 as described and purified from the unlabeled peptide by HPLC (method 3) to obtain a high-specific-activity radioligand, which was then diluted with PBS/1% bovine serum albumin (BSA). MGD5 or APH070 competitor was dissolved in 50% PBS/water and then serially diluted with PBS/1% BSA to give a concentration range of 1 × 10−5 to 1 × 10−10 M. Each well of a Multiscreen 96-well binding assay plate with glass fiber (type B) filters (Millipore) was prewet with 250 μL of 20 mM HEPES (pH 7.3) containing 1% (w/v) BSA, 5 mM MgCl2, and a 0.2 mg/mL concentration of bacitracin (buffer B) and filtered. To each well was added 50 μL of competitor solution (in buffer B at concentrations corresponding to between 0.0001 and 1,000 nM in the well mixture), 50 μL of radioligand solution (50,000–100,000 counts per minute diluted in buffer B), and 100-μL cell membranes (100 μg of protein diluted in 20 mM HEPES, pH 7.3, containing 5 mM MgCl2 and a 0.2 mg/mL concentration of bacitracin). The plate was incubated at room temperature for 2 h and then filtered under a vacuum; each well was washed twice with 250 μL of ice-cold 15 mM Tris and 139 mM NaCl (pH 7.4). The filters were counted in a γ-counter, and nonlinear regression was performed using Origin (version 6.1; Microcal) to obtain inhibitory concentration of 50% (IC50) values.
Saturation binding assays with 111In-APH070 and 111In-MGD5 were performed on whole adherent AR24J cells, which had been seeded in 6-well plates (1 × 106/well) and grown in a 5% CO2 atmosphere humidifying incubator at 37°C for 48 h. Radiolabeling was performed by heating 5.56 μg of MGD5 or 3.36 μg of APH070 (2 × 10−9 mol) with 9.5−13 MBq of 111In and 1 M ammonium acetate and 8.3 mg of gentisic acid per milliliter (pH 5.5) for 10 min. A 2-fold molar excess of stable isotope InCl3 was added and the mixture heated for a further 5 min. The reaction was quenched with EDTA (0.1 M, 25-fold molar excess more than InCl3) and diluted to 1 mL with 0.01 M phosphate buffer (pH 8.5–9; MGD5) or PBS (APH070) to give a 2,000 nM solution. Serial dilutions from 10 to 0.1 nM were made in PBS/1% BSA. Either unlabeled APH070 or MGD5 (1,000 nM in PBS/1% BSA) was used as the competitor. The assay was performed as follows: the medium was removed from each well and the cells washed with 1 mL of RPMI (1% FBS) before the addition of 1.2 mL of RPMI containing 1% v/v FBS and 0.1% (w/v) sodium azide to minimize internalization, 150 μL of either PBS/1%BSA (to measure total binding) or 150 μL of competitor (to measure nonspecific binding) in triplicate. This was followed by the addition to each well (in triplicate) of 150 μL of increasing concentrations of radioligand. Each concentration of radioligand (150 μL) was also pipetted in triplicate into counting tubes for measuring total counts. The plates were incubated at room temperature for 1.5 h, after which the incubation mixture was removed and the plates washed, and 1 mL of 1 M NaOH was added per well. This mixture was removed to counting tubes after 15 min along with washings of 2 × 1 mL of PBS. These and the total count tubes were counted in the γ-counter. Dissociation constant (Kd) and maximum numbers of binding sites were calculated with nonlinear regression using Origin (version 6.1).
Internalization Experiments
Adherently growing AR42J cells that had been seeded in 6-well plates (1 × 106/well) and maintained in a 5% CO2 atmosphere humidifying incubator at 37°C for 48 h were used in this assay. The medium was removed, and the cells were washed twice with ice-cold internalization medium (RPMI supplemented with 1% v/v FBS), and then 1.2 mL of internalization medium was added to each well. This was followed by the addition of 150 μL of radioligand in 1% (w/v) BSA/PBS buffer and either 150 μL of 1% (w/v) BSA/PBS alone (to measure total counts) or 150 μL of 10 μM competitor (either APH070 or MGD5) in 1% (w/v) BSA/PBS to measure nonspecific binding. The wells corresponding to total binding and nonspecific binding were pipetted in triplicate for each time point of 10 and 30 min and 1 and 2 h of incubation at 37°C. Incubation was stopped at each time point by removal of the medium; medium was washed twice with ice-cold internalization medium. The cells were then treated with 1 mL of 50 mM glycine (pH 2.8) and 0.1 M NaCl twice for 5 min each, and the acid wash was collected after each incubation and placed in counting tubes along with a further 1 mL of PBS/1% BSA rinse to comprise the membrane-bound fraction. The cells were then lysed with 1 M NaOH, which was removed to counting tubes after 15 min, along with 2 × 1 mL of PBS wash (the internalized fraction). After counting, we calculated the specifically bound mean values and represented them as a percentage of total activity added.
Biodistribution Studies
Biodistribution studies were performed in a level-2 containment facility according to Advisory Committee for Dangerous Pathogens guidelines and in accordance with British Home Office regulations governing animal experimentation. CCK-2R–positive xenografts were induced in immunodeficient CD1 nude mice by subcutaneous injection of 5–7.5 million AR42J rat pancreatic cancer calls into the flank or shoulder of the animal. Animals were used 7–10 d later when tumors had grown to around 5 mm in diameter. A total of 100–200 μL of 111In-labeled peptide was injected intravenously into the tail of the AR42J tumor–bearing mice (n = 3–6 per condition). After 4 h, the mice were anesthetized, and a 0.5-mL blood sample was obtained by cardiac puncture. The tumor, pancreas, kidneys, heart, stomach, spleen, intestine, liver, lung, and muscle were also sampled; each sample was weighed and counted with a γ-counter (LKB Compugamma), and standards were prepared from a sample of the injected material. The percentage of injected dose per gram of tissue was calculated for each tissue type. Statistical comparisons were performed using a 2-tailed, unpaired t test.
Imaging
Static Imaging
AR42J tumor–bearing female CD1 nude mice were injected intravenously in the tail with 10–15 MBq (0.5–1.5 μg) of 111In-MGD5 or APH070. At a 4-h time point, the animals were anesthetized using halothane and imaged using a NanoSPECT/CT animal scanner (Bioscan). Whole-body SPECT images were obtained in 24 projections over 20 min using a 4-head scanner with 4 × 9 (1.4 mm) pinhole collimators in helical scanning mode; CT images were acquired with a 45-kVP x-ray source in 180 projections over 6 min. Images were reconstructed in a 256 × 256 matrix using proprietary Bioscan InVivoScope software and fused using PMOD (Mediso).
Dynamic Imaging
Two AR42J tumor–bearing female CD1 nude mice were injected intravenously in the tail with 40–50 MBq (4.7–6 μg) of either 111In-MGD5 or APH070. Twenty-four helical SPECT scans were acquired in 16 projections for 5 min each over the upper abdomen. Images were reconstructed in a 256 × 256 matrix using InVivoScope. Three-dimensional regions of interest of approximately 1 cm3 were drawn around the tumor and an area of muscle on the contralateral shoulder, and time–activity curves were generated. Washout of radioactivity from the regions of interest was calculated by dividing the counts in the area during the final acquisition period (115–120 min) with those acquired in the first period (0–5 min).
RESULTS
Stability Studies
The stability of 111In-MGD5 was tested in mouse plasma and compared with that in the PBS standard (pH 7.4). At 5 h 95% and at 24 h approximately 90% of 111In-MGD5 remained intact in the plasma (compared with 95% and 62%, respectively, for the monomeric compound) (10).
Receptor Binding Studies
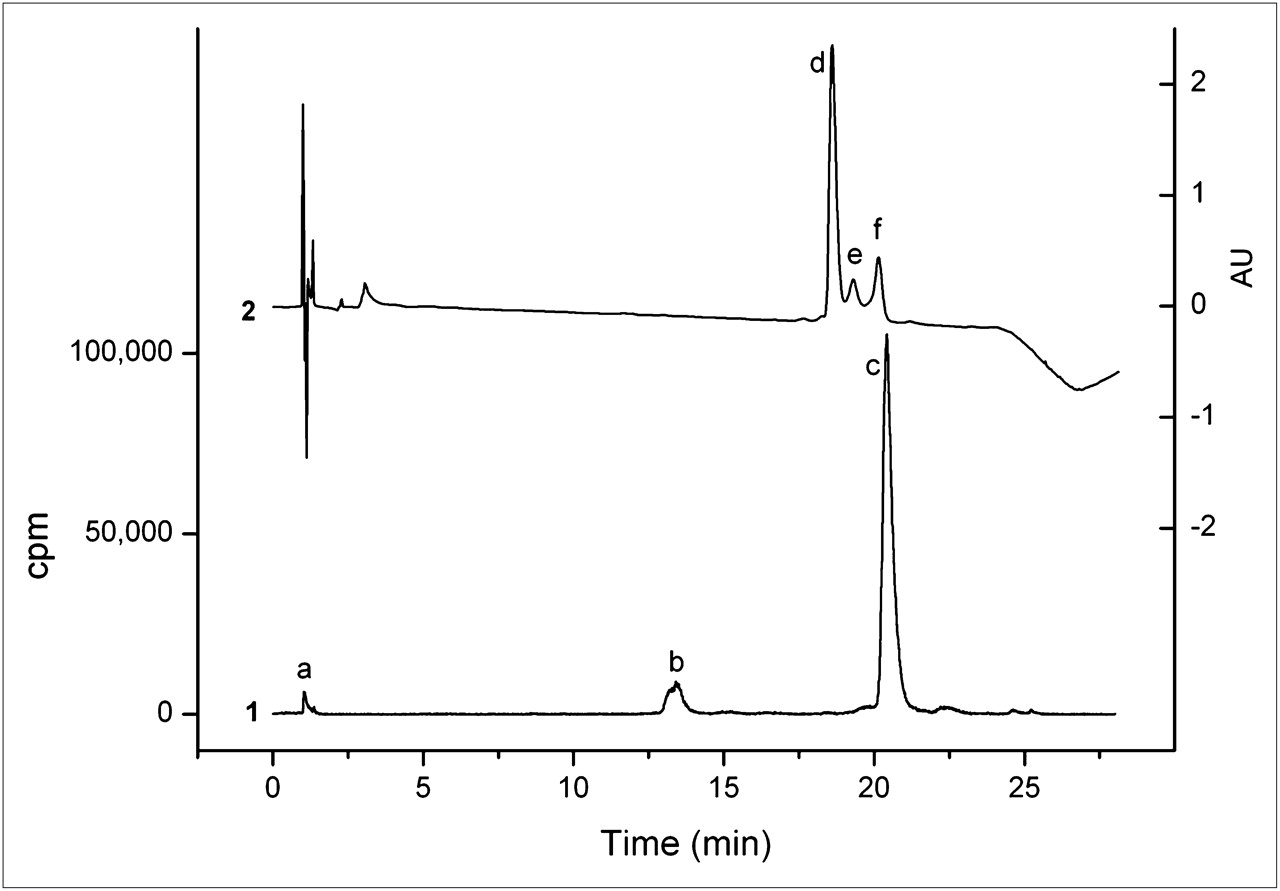
MGD5 was compared with APH070 in competition binding assays on AR42J cell membranes using 111In-APH070 as the radioligand. To obtain the highest possible specific activity for these studies, the radioligand was purified using a reversed-phase HPLC ion pairing method, with TEAA as the ion pairing agent. Using a 0.01 M TEAA (pH 5.5)/acetonitrile gradient (method 3), we separated unlabeled APH070 from 111In-APH070 (Fig. 2). In the ultraviolet chromatogram, 3 peaks were visible, with retention times of 18.6, 19.3, and 20.1 min, respectively. The 20.1-min ultraviolet peak (f) corresponded to the radioactive 111In-APH070 peak (c) and was isolated by fraction collection. The peak eluting at 18.6 min (d) coeluted with unlabeled APH070. The peak eluting at 19.3 min (e) was unknown, but did not appear in the radiochromatogram and could arise from nonradioactive divalent metal-DOTA complexes.
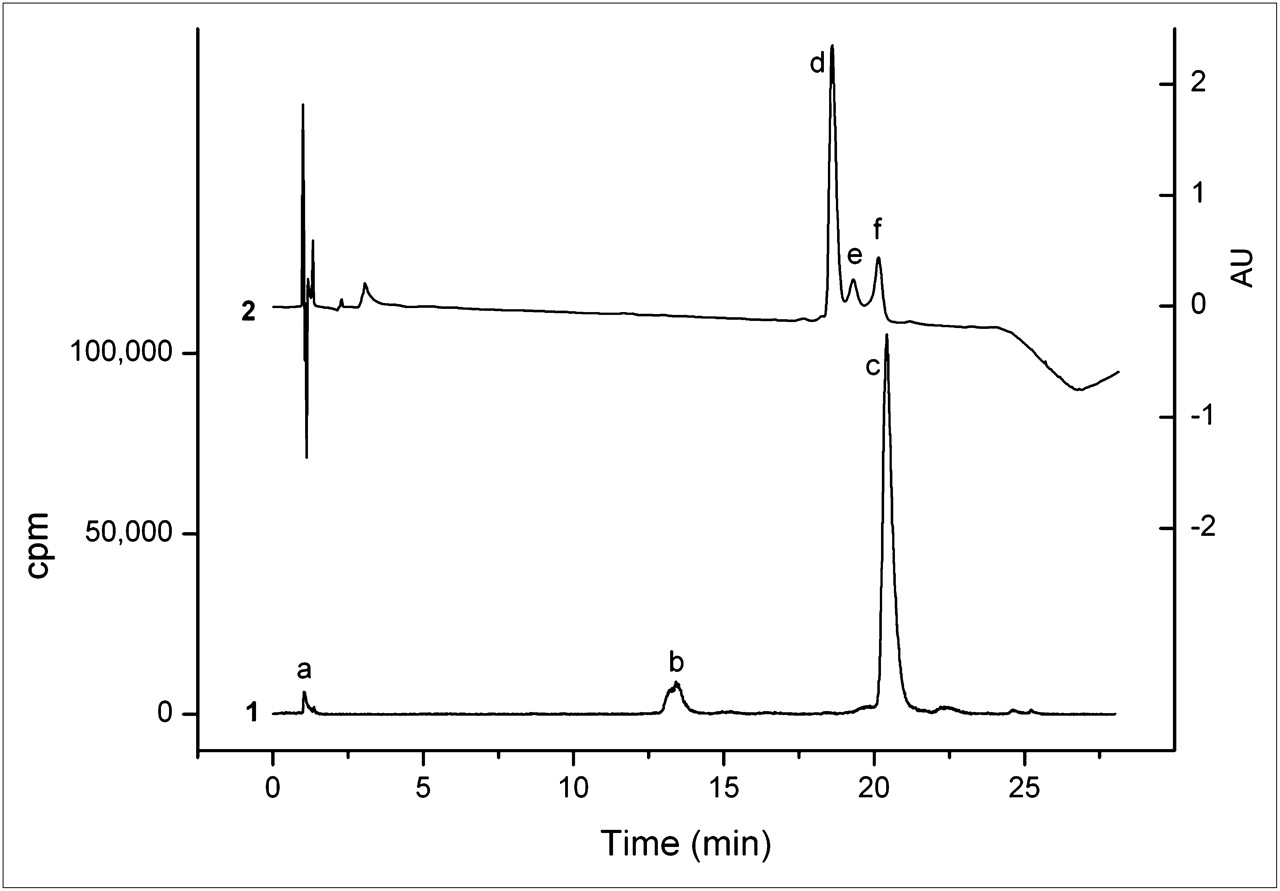
Reversed-phase HPLC analysis of typical 111In-APH070 reaction mixture coinjected with 5 μg of unlabeled APH070 showing both radio- (1) and ultraviolet chromatograms with measurement at 220 nm (2). Peak a is 111In-EDTA, b is 111In-APH070(O) (in which methionine side chain is oxidized to sulphoxide), c is 111In-APH070 (which corresponds to f in ultraviolet trace); unlabeled APH070 elutes earlier (d). Peak e is unknown.
111In-APH070 purified in this way was used as the radioligand in competition binding assays on AR42J cell membranes (Fig. 3). These assays showed that the affinity of MGD5 for the CCK-2R was higher than that of APH070 (IC50 of 1.04 ± 0.16 and 5.59 ± 1.46, respectively; n = 3). Saturation binding assays on 111In-MGD5 and 111In-APH070 were performed in intact AR42J cells. The Kd of indium-APH070 in this system was 2.9 ± 0.6 (average of 5 independent assays ± SE), and the Kd of indium-MGD5 was 0.69 ± 0.14 (average of 3 independent assays ± SE). Figure 4 shows an example of the results obtained using 111In-MGD5.
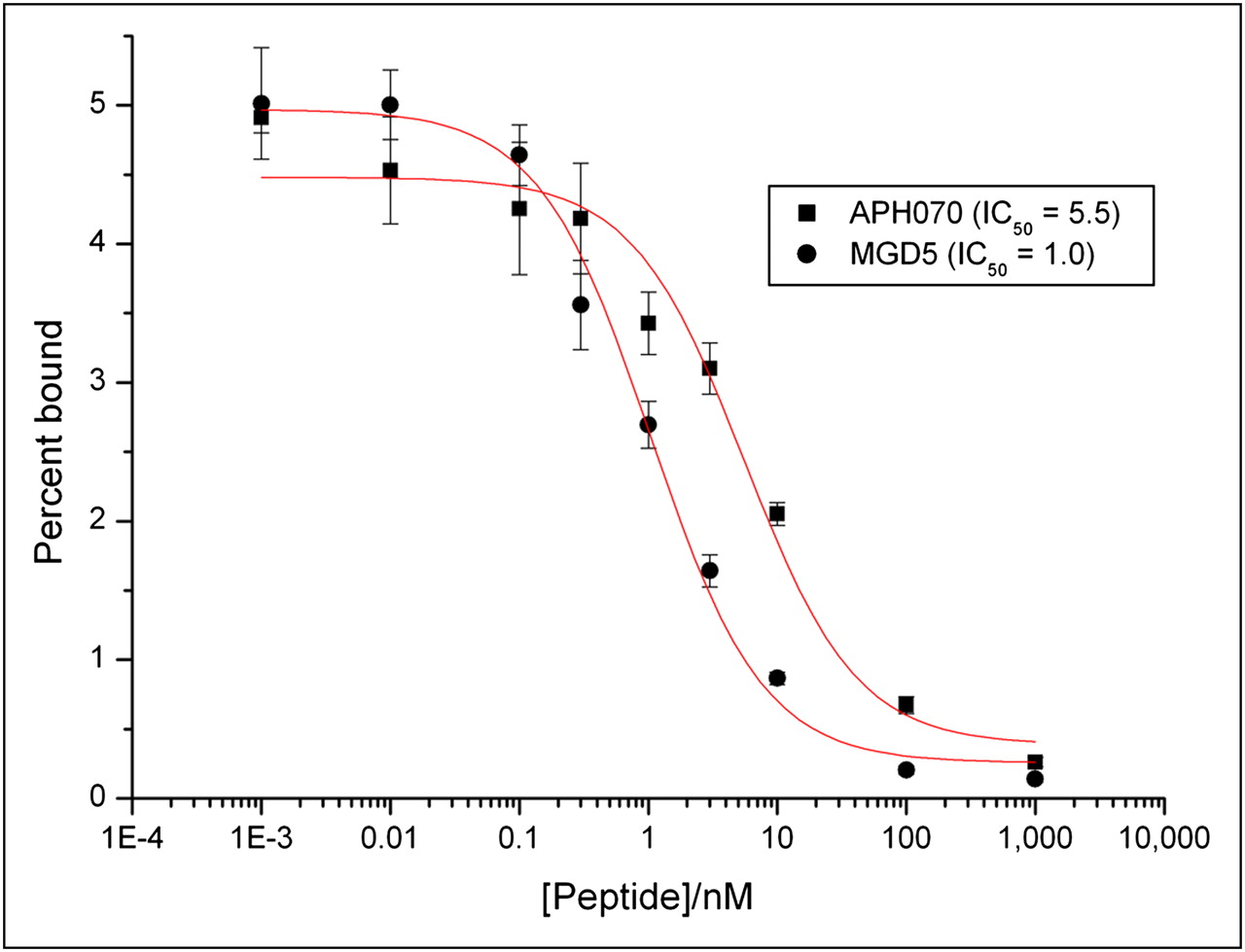
Competition binding assay of APH070 and MGD5 on AR42J cell membranes using 111In-APH070 as radioligand.

Saturation binding assay of 111In-MGD5 with Scatchard analysis.
Internalization Assays
Internalization assays performed in AR42J cells showed that after 120 min, 31.1% of 111In-MGD5 added was internalized as opposed to 16.7% of 111In-APH070 (Fig. 5). The rate of internalization of 111In-MGD5 was 0.24%/min, nearly double that of the rate of internalization of 111In-APH070 (0.14%/min).

Internalization of 111In-APH070 and 111In-MGD5 into AR42J cells over time (n = 2 independent assays ± SE).
Biodistribution Studies
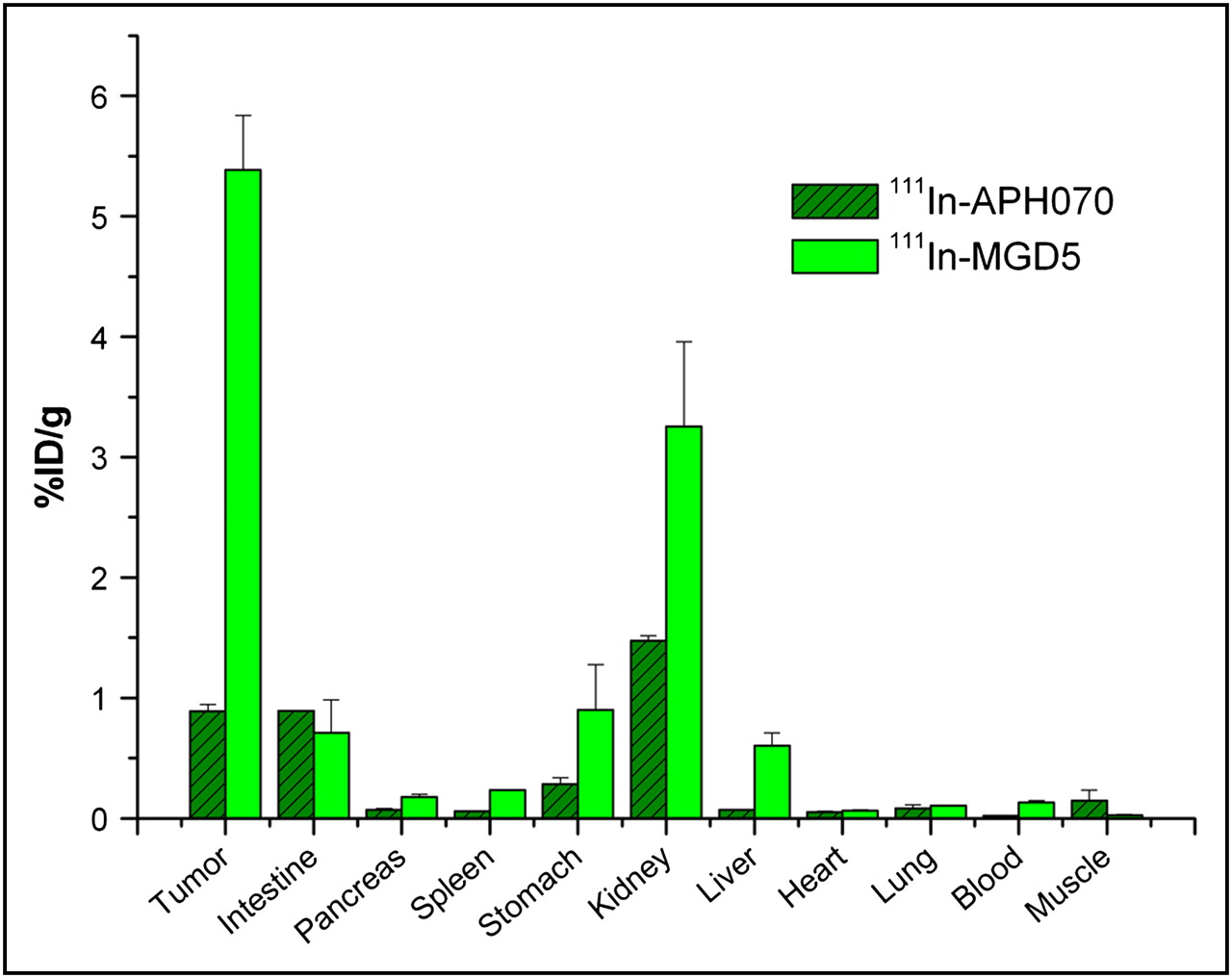
The biodistribution of 111In-MGD5 was studied at 4 h after injection in AR42J tumor–bearing CD1 nude mice and compared with that of 111In-APH070 (Fig. 6). This result shows that the tumor uptake of 111In-MGD5, compared with that of 111In-APH070, is significantly increased (6-fold) (P = 0.01). Kidney uptake also increased but not as much as the tumor uptake. Thus, the calculated tumor-to-kidney ratio of the divalent peptide compound is 1.66, which is a significant improvement on that of the monomer (0.6).
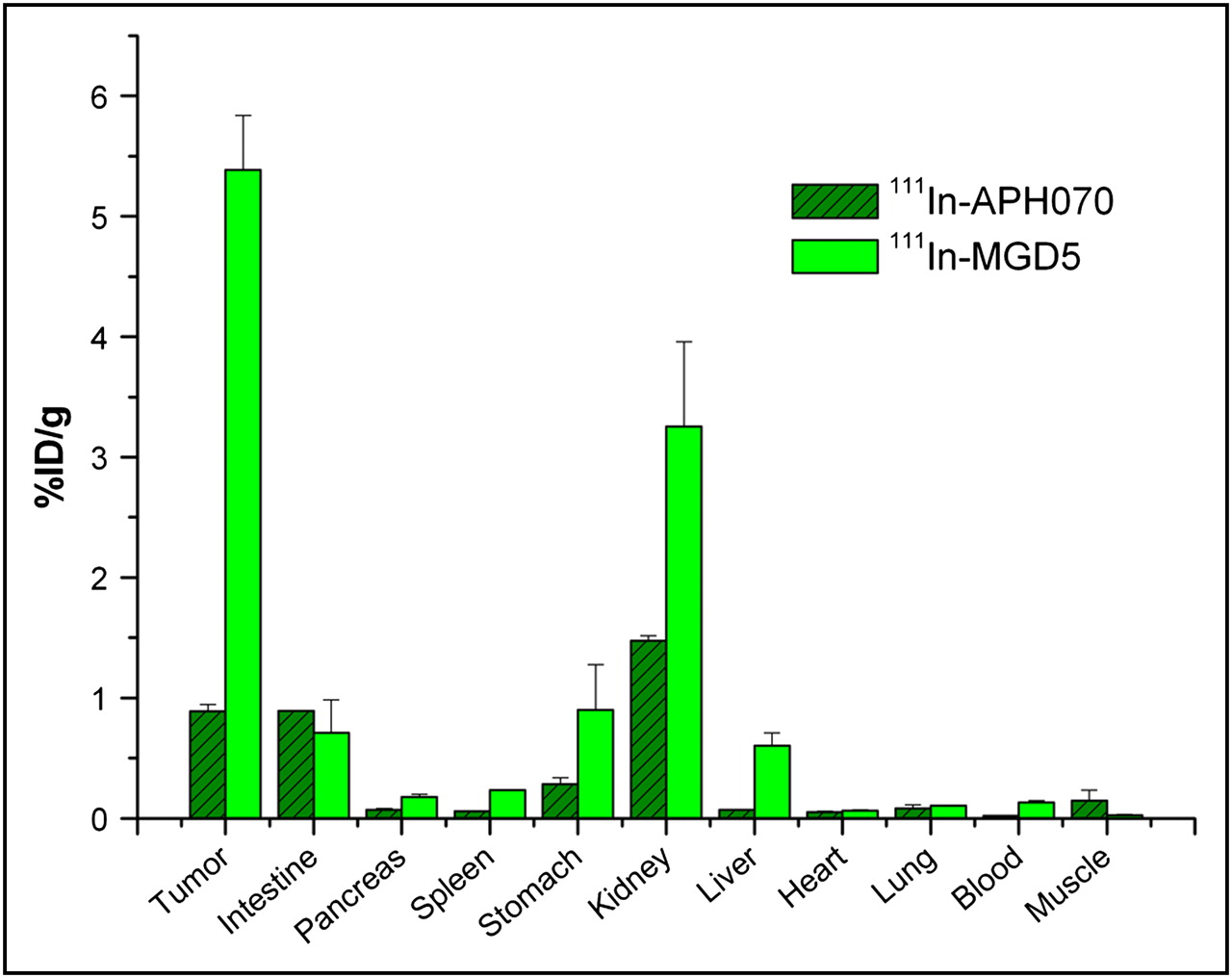
Biodistribution of 111In-MGD5 (n = 3 and n = 4) and 111In-APH070 (n = 4 and n = 5) in AR42J tumor–bearing CD1 nude mice at 4 h (mean of 2 separate experiments ± SE). Tumor uptake of 111In-MGD5 is significantly greater than that of 111In-APH070 (P = 0.01).
CCK-2R blocking experiments were performed by coinjecting 111In-MGD5 with 100 μg of the unlabeled compound. Tumor uptake was reduced 10-fold in the blocked animals, indicating specific uptake (P = 0.008, Table 1).
Biodistribution of 111In-MGD5 With and Without Coinjection of 100 μg of Unlabeled MGD5 in AR42J Tumor–Bearing CD1 Nude Mice at 4 Hours
Imaging
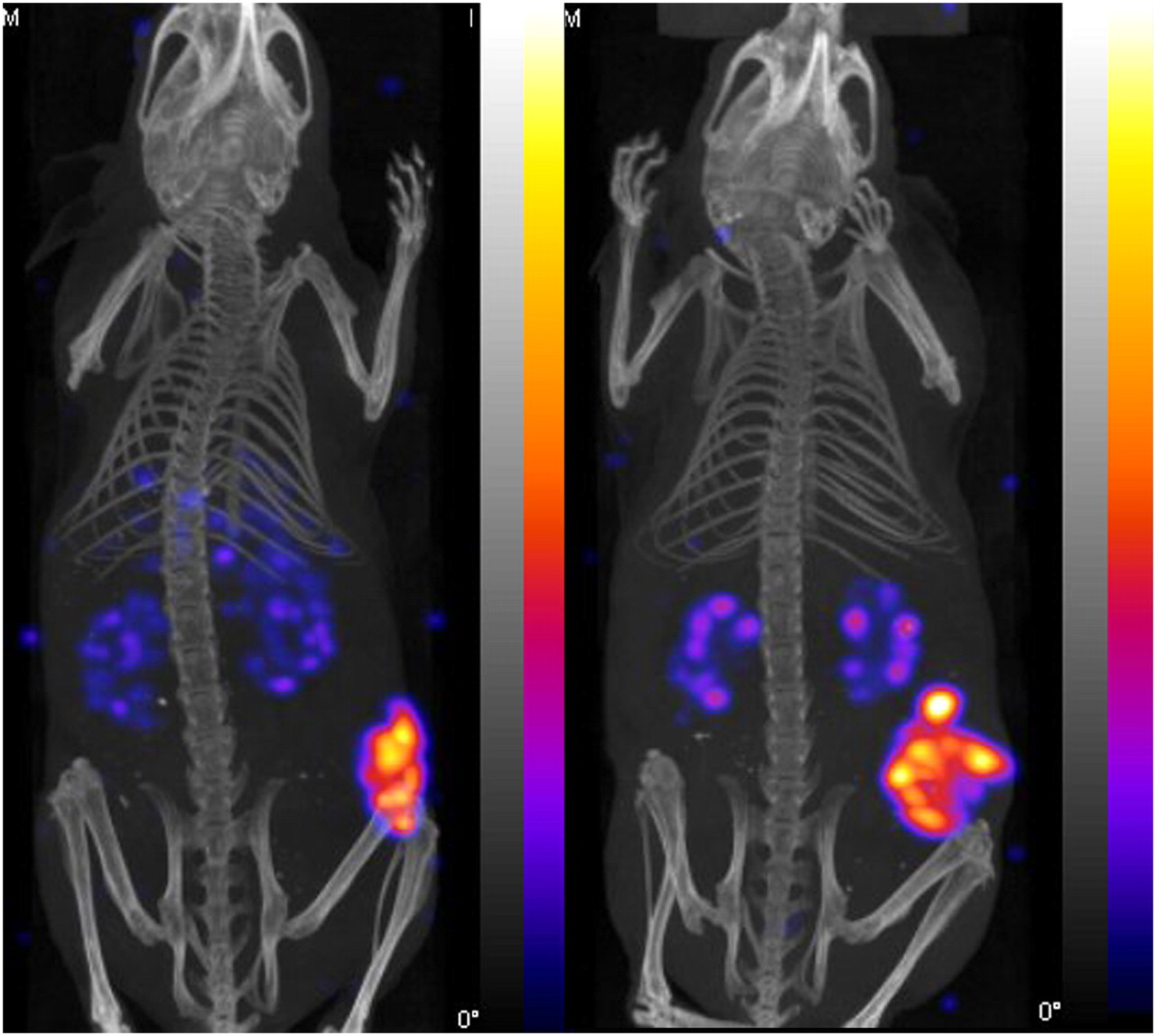
Static imaging was performed on AR42J tumor–bearing CD1 nude mice at 4 h after injection of either 111In-MGD5 or 111In-APH070. Representative examples of the images obtained are shown in Figure 7. The results obtained mirror those seen in the biodistribution study. Significant uptake is visible only in the tumor and kidney, with the tumor uptake on the 111In-MGD5 being clearly greater than that of 111In-APH070 while the kidney uptake was less than that in the tumor. To try and measure the kinetics of uptake and washout of 111In-MGD5 and 111In-APH070, dynamic SPECT was undertaken in just 1 animal injected with each tracer. Uptake was rapid, with maximum uptake occurring within the first 5-min imaging period. Thereafter, there was a gradual washout of both tracers from tumor and background tissues. However, APH070 washed out considerably more quickly than did MGD5. After 2 h, 89% of the initial level of uptake of the monomer was washed out, whereas the corresponding washout for the divalent peptide was only 58%. The rates of washout from background tissues were 99.7% and 99.5%, respectively.
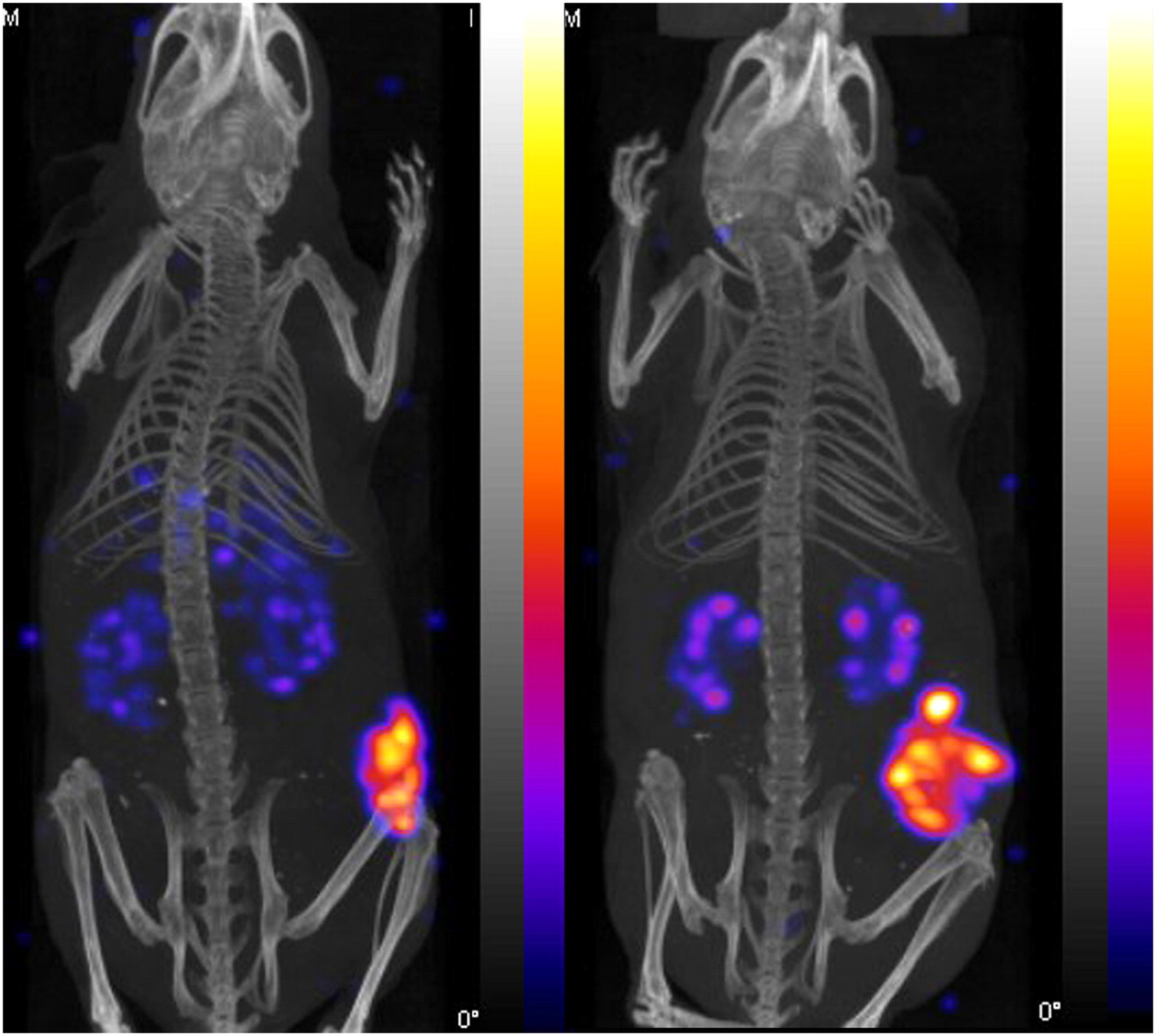
Representative NanoSPECT/CT images showing biodistribution of 111In-APH070 (right) and 111In-MGD5 (left) at 4 h after administration. Images are thresholded to indicate similar levels of tumor uptake, but kidney uptake is relatively lower in MGD5 image. Tumor-to-kidney ratios (measured postmortem) are 1.1 for APH070 and 2.7 for MGD5.
DISCUSSION
It has been known for some time that multimerization of small molecules results in an increase in affinity to their complementary binding site (11). This phenomenon has previously been applied to radiopharmaceutical development by synthesis and radiolabeling of small oligomers of 2–8 peptide subunits (15). The most extensive exploration has been undertaken with RGD-containing radiotracers that bind to the αvβ3 integrin, a protein upregulated in angiogenesis and on some malignant cells (12,16,17). Thus, Janssen et al. (18) showed that the binding affinity of oligomers of RGD peptides increased in direct proportion to the number of subunits incorporated. This increase in binding affinity translated into an improvement in tumor targeting in vivo by 18F-labeled oligomers (19). Subsequently, several other dimeric and tetrameric αvβ3 binding peptides have shown similar promise. The ability of other dimeric peptides to target several tumor-associated molecular targets including α-melanocyte–stimulating hormone receptor (20), the neurotensin receptor NTR1 (21), and bombesin receptor (22) has also been studied; these studies all showed an improvement in receptor binding or tumor targeting.
To our knowledge, this is the first report of a similar approach to targeting the gastrin CCK-2R. CCK-2R is a molecular target of considerable clinical potential, but our ability to harness this potential has been limited by the high renal uptake or low tumor uptake of most radioligands. Dimerization represents 1 approach to overcoming these limitations. We have previously synthesized a small library of divalent CCK-2R binding peptides, with variation in either the receptor-binding sequence or the chemistry of the dimerization. Preliminary biodistribution studies with these compounds identified a maleimide-linked compound, MGD5, as the most promising (14). In the studies described herein, we have compared the performance of this compound with a previously well-characterized monomeric peptide, APH070 (10). We have shown that this divalent peptide has a higher binding affinity than does the monomer, that it internalizes more rapidly and to a greater extent, and that it targets tumors more effectively in vivo. The fact that the interaction of the divalent peptide with the tumor is mediated by a specific receptor interaction is confirmed because the coadministration of a blocking dose of unlabeled peptide reduces the tumor uptake of the radioligand to only 10% of the unblocked value.
The mechanism of this improvement is likely to be due to an increase in the apparent ligand concentration in the vicinity of the receptor, resulting in an increase in the rate of association or a decrease in the rate of dissociation (23). The latter seems most likely because preliminary dynamic imaging studies indicate that the rate of washout of MGD5 from the tumor is considerably slower than that of the monomer, whereas the rate of washout from receptor negative tissues is similar. Actual simultaneous bivalent binding of the peptide to the receptor can be discounted because the distance between adjacent receptors is certain to be much greater than that separating the 2 binding sites in the peptide. It could be argued that the improvement could also be partially explained by pharmacokinetic changes induced by the approximate doubling in molecular weight. These pharmacokinetic changes require further exploration through more detailed dynamic imaging studies. In our previous detailed analysis of the pharmacokinetics of APH070 over a 3-d period, we showed that the greatest rate of washout of the peptide occurred in the first few hours after injection. However, the radioactivity continued to wash out throughout the 3 d, albeit at a slow rate, so that the tumor uptake at 3 d was only 25% of that seen at 1 h (10). The results of this preliminary study of the pharmacokinetics of the divalent peptide MGD5 indicate that the slower rate of washout should be maintained over the days after injection, although this remains to be confirmed by further study. This slower rate of washout would have significant implications on the therapeutic application of this peptide, because the integrated area under the time–activity curve would be expected to be significantly greater that that of the monomeric compound.
CONCLUSION
These results confirm those obtained with other radiolabeled peptide systems, namely that dimerization of the receptor binding site results in an increase in binding affinity and an increase in tumor uptake both in vitro and in vivo. A likely consequence would be an improvement in the targeting efficiency of the peptide in patients with tumors expressing the CCK-2R, but confirmation by clinical trials is needed.
Acknowledgments
We gratefully acknowledge financial support from Cancer Research U.K. and Aphton Inc.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication April 2, 2009.
- Accepted for publication August 28, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}