


The integrin αvβ3 is currently being evaluated as a molecular target for antiangiogenic therapies. Several targeting strategies and probes are being studied to enable noninvasive imaging of αvβ3 expression in vivo. In the September issue of The Journal of Nuclear Medicine, Line et al. investigated peptide-polymer conjugates to exploit the “enhanced permeability and retention” (EPR) effect and high-affinity binding for high-level targeting of αvβ3 integrins (1).
PEPTIDES, PROTEINS, AND EPR
Antibodies are increasingly being used in radioimmunoimaging and therapy. In addition, an impressive collection of smaller antibody fragments and constructs is currently being evaluated to overcome the drawback of applying intact antibodies (2,3). On the other hand, peptides with a molecular weight in the range of ∼1–2 kDa have been presented as valuable tracers that offer considerable advantages: minimum-sized amino acid sequences with high affinity to their binding sites; no immunogenicity; fast accumulation at the target and fast blood clearance via the kidneys; a defined chemical labeling position, with structures that tolerate even large, bulky chelators or complexes; the availability of chemical tools to adjust kinetics and to improve in vivo stability; and cost-effective, large-scale production under good-manufacturing-practice conditions (4,5). Hence, radiolabeled peptides have been introduced into the diagnostic (7–9) and therapeutic (10,11) fields as powerful radiopharmaceuticals—a development that generally corroborates the peptide concept.
In addition to expressing high-affinity binding sites for peptides or antibodies, tumors are known to exhibit enhanced vascular permeability, similarly to or even more than inflammatory tissues, (12–14), resulting in a facilitated permeability and entry of macromolecules into the interstitium. It is interesting that the proteinaceous vascular permeability factor, one of several vascular endothelial permeability mediators, was later found to be similar to vascular endothelial growth factor (15). Together with inadequate lymphatic drainage of solid tumors, macromolecular plasma components remain in the tumor for a long time, up to days (Fig. 1) (13,16). According to the EPR effect, biocompatible macromolecules accumulate in much higher concentrations in tumor tissue (>6-fold) than in normal tissues and organs or even than in plasma. The EPR effect was found to be effective for molecules with a molecular size of >45 kDa (13,16,17).
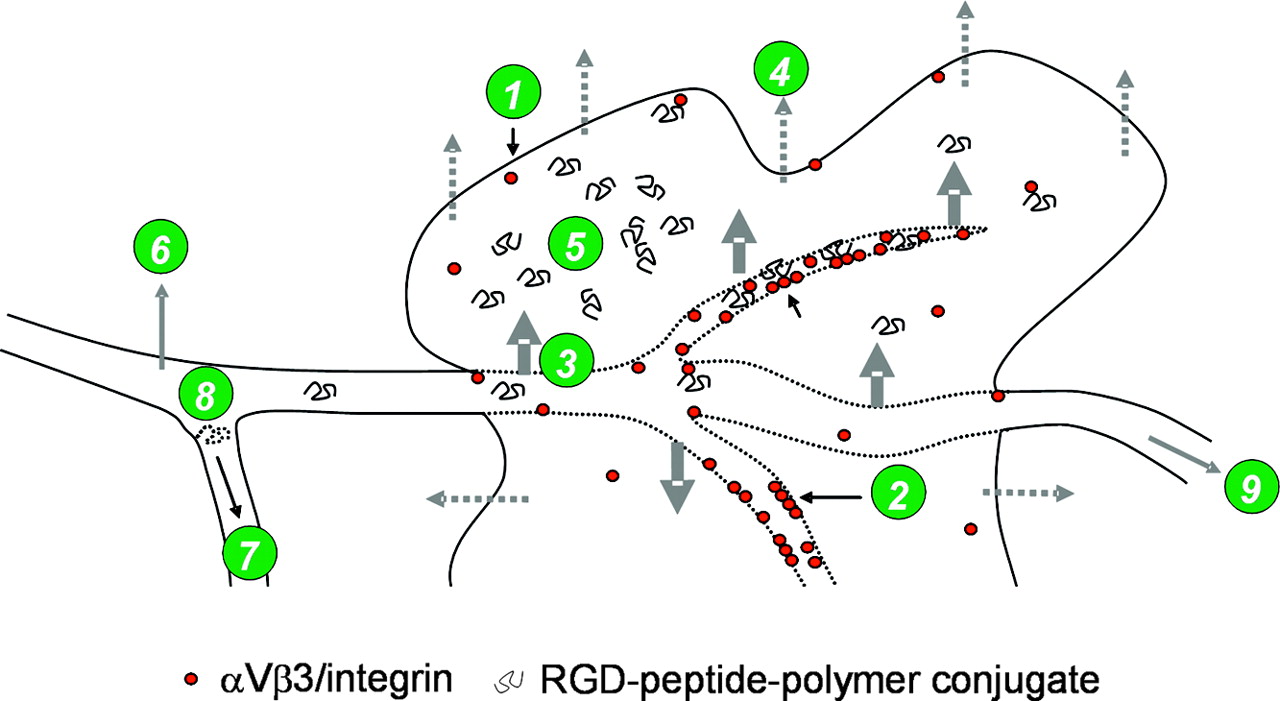
Factors influencing uptake of RGD-peptide-polymer conjugates: expression of αvβ3 integrins on tumor cells and vasculature (1), formation of clusters of activated αvβ3 integrins (2), enhanced permeability (3), impaired lymphatic drainage result in EPR (4), nonspecific tumor accumulation of peptide-polymer conjugates by EPR (5), nonspecific uptake in nontumor tissue (6), blood clearance (kinetics) (7), catabolism (8), and whole-body-retention and excretion kinetics (9).
Consequently, a combination of both methodologies—that is, the use of a macromolecule as an EPR-sensitive vehicle conjugated with a specific high-affinity-binding peptide—seems to be an appropriate strategy to improve peptide-receptor targeting. However, intuitively, most interested readers would suggest that more than one peptide be coupled to this vehicle. In fact, on the basis of examples in nature, this approach seems to be even better although theoretically and mechanistically much more complicated. All classes of antibodies have multiple equivalent binding sites, and viruses use polyvalent interactions to bind to their host cells, to mention only 2 important examples.
Several studies have already shown that the affinity of small molecules to their binding site can be dramatically increased by formation of multimeric units (18–23). Linear or branched polymers, dendrimers, and starlike structures have already been evaluated to enhance antigen immunogenicity, binding affinity, cytotoxic potency, or selectivity. Two different mechanistic models can be used to explain increased binding affinity for multimeric systems, at least qualitatively: First, when small multimeric structures are used and simultaneous binding to the corresponding receptors cannot occur, an increase in receptor-ligand binding is determined by an apparent increase in ligand concentration. Second, in cases of polyvalent binding, simultaneous interactions between multiple ligands and receptors occur, resulting in an overall increased affinity (avidity) of the entire (receptor-ligand) interaction.
Whether a multimeric construct can bind according to the first or the second kind of interaction depends on several characteristics of the construct: its overall size, the distance between 2 ligands, the flexibility of the used linker or backbone, and the receptor density. When short linkers are used with low-molecular-weight multimers (dimers, tetramers, octamers), even high receptor densities or the formation of receptor clusters will hardly lead to polyvalent binding. Longer linkers may result in polyvalent binding after initial monovalent interaction, when receptor clustering occurs or is initiated by ligand binding. Large, flexible linkers seem to have the highest probability of polyvalent interaction of the entire construct, resulting in a decrease of the dissociation rate constant and thus long-lasting retention in the receptor-expressing tissue.
Thus, high-level targeting with radiopharmaceuticals designed by conjugation of multiple peptides to a high-molecular-weight polymer or protein consists of 3 independent concepts: molecular targeting by peptide-receptor interaction, the possibility of polyvalent binding (“polyvalence concept”), and increased tracer delivery and retention by the EPR effect.
The entire concept is not new and has already been used for tumor delivery of RGD peptides coupled to a chitin backbone (24–26). The potency of monomeric RGD peptides and RGD peptides coupled to poly(carboxyethylmethacylamide) (poly(CEMA)) to inhibit spontaneous metastasis after subcutaneous inoculation of B16-BL6 melanoma cells was investigated. Similar to the results obtained with polypeptides containing repetitive RGD sequences (poly(RGDS)), the polymeric integrin ligands inhibited experimental and spontaneous metastasis more efficiently than did the corresponding monomeric peptides. Furthermore, the effect of 2 monomeric peptides, YIGSR and RGD, and 2 polymeric compounds, poly(CEMA-RGDS) and 6-O-carboxymethyl-chitin-RGDS (CM-chitin-RGDS), on invasiveness and survival in mice after inoculations of peritoneal-seeding OCUM-2MD3 (α2β1+ und α3β1+) human scirrhous gastric carcinoma cells was investigated and compared. All peptides and especially the polymers significantly inhibited the invasiveness of OCUM-2MD3 cells and resulted in improved survival time (27).
In a similar strategy, a combination of polyvalent high-affinity-peptide binding and EPR for targeting αvβ3-integrin expression during tumor angiogenesis was investigated by Line et al. and reported in the September issue (1). In that study, uptake of a macromolecular methacrylamide polymer conjugated with RGD4C peptides (28) and labeled via the [99mTc(CO)3]+ approach was investigated in PC3 and DU145 prostate tumor xenografts and compared with the corresponding RGE4C polymers with no affinity to αvβ3 and monomeric 99mTc-labeled RGD4C and RGE4C peptides (29,30).
Noteworthy, this study not only enlarged the repertoire of radiopharmaceuticals suggested for imaging of angiogenesis by various groups but filled a systematic and developmental gap in the design of αvβ3-integrin–directed radiotracers. Thus, for the first time, an entire series of tracers, ranging from peptidomimetics over small peptides to peptide dimers, tetramers, octamers, polymers, proteins, and even larger structures, such as liposomes and nanoparticles, is available to address the same molecular target (31). When compared in suitable test systems and models, this series of tracers will allow valid assessment of the contribution of individual concepts for tracer delivery and enrichment and will help to benchmark further developments.
To comprehend why such a series of compounds with different sizes and variable degrees of high-affinity-binding units per molecule has been designed especially for molecular targeting of integrins, one needs to look somewhat more deeply into the molecular structure and function of the target addressed.
αVβ3-INTEGRIN AND TRACER DESIGN
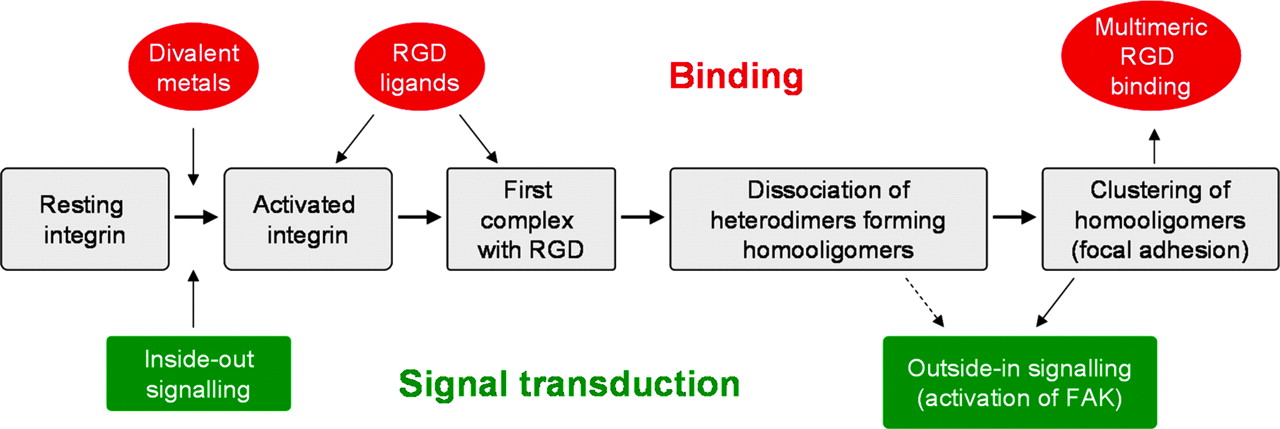
Integrins are bidirectional heterodimeric receptors consisting of one α- and one β-subunit. They mediate cell adhesion to the extracellular matrix or between cells (32). Activation of ligand binding can be modulated from inside the cell (inside-out signaling) or by binding of divalent metals at the extracellular part of the integrins (Fig. 2). Subsequent docking of high-affinity ligands induces signal transduction and activation of intracellular focal adhesion kinase (outside-in signaling). Several members of the integrin family (αvβ3, αvβ5, αvβ6, αIIbβ3, α1β5) recognize the tripeptide sequence -Arg-Gly-Asp- (RGD) (33), and certain classes of integrins (e.g., αv integrins and α5β1) were shown to be involved in cancer metastasis and angiogenesis (34). Hence, peptides containing the RGD sequence have long been evaluated as recognition elements for treatment of cancer or as inhibitors of angiogenesis in mice and humans, and ligands with superactivity and selectivity for the cancer integrins were developed. Via systematically designed conformational restrictions, cyclic pentapeptides, such as cyclo(-RGDfV-) (35), were discovered. This peptide exhibits high affinity (2 nmol/L) for αvβ3 binding to vitronectin but low affinity (>2,000 nmol/L) for αIIbβ3 (expressed on platelets) binding to fibronectin (36). Cyclo(-RGDyV-) was shown to bind with similar affinity and selectivity to αvβ3, and consequently, the iodinated peptides [*I]-3-iodo-d-Tyr4-cyclo(-RGDyV-) and [*I]-3-iodo-Tyr5-cyclo(-RGDfY-) were evaluated as the first imaging agents for αvβ3 integrins (37). Because Val5 of cyclo(-RGDfV-) can be replaced by a variety of other amino acids without affecting high-affinity binding to αvβ3 (38), suitable modified analogs, such as cyclo(-RGDfK-) and cyclo(-RGDfE-), have been used for radiolabeling with a variety of isotopes (8,39–42) and for creating strategies to optimize the in vivo behavior (43–45). The most promising ligand developed so far, [18F]galacto-RGD (8,39,41), is currently being evaluated in patients. The first results demonstrated that [18F]galacto-RGD PET enables noninvasive quantitative assessment of the αvβ3 expression pattern on tumor and endothelial cells in patients with malignant tumors (8).

Subsequent steps of integrin activation and multimeric RGD binding. FAK = focal adhesion kinase.
Apart from having high affinity to αvβ3, cyclic pentapeptides also exhibit high metabolic stability in vivo, another important prerequisite for successful in vivo application. The therapeutic potential of the N-methylated analog cyclo(-RGDfNMeVal-), named cilengitide (46), is now being assessed for the treatment of gliomas in a clinical phase II trial (47). Meanwhile, orally available nonpeptidic drug candidates have been developed (48,49). Peptidomimetics have also been evaluated as PET tracers (31,50–53).
To investigate the effect of ligand dimers and oligomers on binding characteristics to integrins and to improve imaging, a group has designed oligomeric constructs by tethering together multiple cyclo(-RGDfE-) units via linkers to form a multimeric structure (53–57). Subsequently, other groups have followed similar strategies using different isotopes (58,59).
This multimer strategy was chosen to match ligand design with the molecular structure of αvβ3 integrin, and especially with hypothesized structural changes after functional ligand binding (Fig. 2). Ligand binding to heterodimeric integrins is known to provoke altered interactions of the transmembrane helices of the α- and β-subunits (60–62). Studies on the αIIbβ3 integrin have shown that the α,β-heterodimeric integrin dissociates after ligand binding to form β-homotrimers and α-homodimers (63,64). Thus, ligand binding initiates a structural reorganization. Once a β-homotrimer is formed, the ligand binding sites are provided in a multimeric form in the focal adhesion (65), which finally results in signal transduction via binding of talin (66).
We assume that such a mechanism is also valid for αvβ3. Formation of these focal adhesion complexes by clustering of integrins thus provides a high focal density of activated binding sites capable of forming a strong adhesion with, or of binding to, multimeric ligands. It is interesting to note that cell spreading and the formation of focal adhesions have been shown to require a certain maximal distance of about 65 nm of single RGD units (67). This indicates that the functional integrity of these integrin clusters necessitates corresponding ligand clusters or at least a related high-focus-ligand concentration. One can easily calculate that the local concentration of a ligand tethered via a spacer to a second binding unit already interacting with a receptor is approximately 2.6 μmol/L, assuming the distance between the 2 ligands is 65 nm. Obviously, these concentrations directly correlate or increase with the number of binding units per multimer. Thus, through use of ligand multimers instead of single binding units, high-focus concentrations can be realized even at the no-carrier-added tracer level.
Multimers were developed to address the question of multimeric integrin binding by “polypotent” ligands and trying to initiate and to target integrin clusters with no-carrier-added radiopharmaceuticals. The αvβ3-selective peptides cyclo(-RGDfK-) or cyclo(-RGDfE-) were linked to polyethylene glycol–amino acid and other spacers. Monomeric units were bridged by lysine or a lysine tree to form dimeric, tetrameric, and octameric RGD oligomers in a well-controlled, defined, and characterized manner. Labeling was performed by oxime ligation, by using, for example, 4-[18F]fluorobenzaldehyde (53–56). Comparison of the inhibitory concentration of 50% of cyclo(-RGDfK-)- and cyclo(-RGDfE-)-containing monomers, dimers, tetramers, and octamers for vitronectin binding to αvβ3 revealed a significantly increasing affinity in the series monomer < dimer < tetramer < octamer (53,54,56). In contrast, the affinity of reference and control peptides carrying only one cyclo(-RGDfK-) (or cyclo(-RGDfE-) peptide, but otherwise cyclo(-RADfK-) or cyclo(-RADfE-) sequences, respectively, was lower or just similar to that of the corresponding monomers. Together, these experiments clearly demonstrated the multimer effect in vitro with similar molecular structures and thus independent of differences in charge, size, or shape. These data were confirmed in vivo in mice bearing M21 melanoma tumor (55,56). Both tumor uptake and tumor-to-organ ratios increased in the series monomer < dimer < tetramer, leading to significantly improved imaging with the 18F-labeled RGD tetramer.
With a molecular weight of ∼1–10 kDa, none of these tracers will show an EPR effect. Thus, the study of Line et al. in the September issue can be regarded as a further step toward con-structs with even higher numbers of binding units per molecule (1). One polymer consists of approximately 15 peptide units. The estimated molecular weight per polymer is ∼30 kDa. Consequently, we can expect a multimer effect and a moderate EPR effect for the peptide-polymer conjugate. Although a quantitative assessment of both effects is not possible, the increase in tumor uptake in the series (nonaffine) RGE4C monomer < (nonaffine) RGE4C polymer < RGD4C monomer < RGD4C polymer (∼1:4:12:16, respectively, for PC3 and DU145 tumor-bearing mice) seems to reflect a major contribution of high-affinity binding and a lower, but existing, contribution of EPR to tumor uptake at 24 h after injection. Further studies comparing the in vivo behavior of the used (RGD4C)15 polymer and (RGD4C)1-(RGE4C)14 polymer, or comparing the in vivo behavior of the (RGD4C)15 polymer and the (RGD4C)1 polymer in lower doses, would allow for a separation and analyses of the contribution of both effects. In addition, competition studies would be valuable to demonstrate the level of specificity. Nevertheless, the study of Line et al. demonstrated that tumor uptake may benefit from tracers exploiting the EPR effect. Furthermore, we would expect that one of the aforementioned experiments would reveal a strong contribution of the multimer effect on uptake.
TRACER FOR IMAGING AND THERAPY
What is the benefit of using EPR for peptide-receptor imaging? Primarily, uptake by EPR is nonspecific and thus will not reflect a process addressed by the peptides conjugated to the vehicle, at least not early after injection. Furthermore, EPR is a slow process, and significant uptake often needs several hours. Assuming that specific binding will occur in a suitable time after delivery by EPR (which has to be shown), such a peptide-polymer conjugate could be used for imaging—for example, receptor mapping or quantification. However, as shown in Figure 1, a variety of processes can influence net tumor uptake, most of which often differ individually. Consequently, to exploit EPR and to overcome these uncertainties, imaging with the aim to receive target-specific information would have to be performed at late times after injection. Thus, compared with the peptide-polymer conjugates, low-molecular-weight multimers exhibit striking advantages for imaging. The first human studies with 18F-labeled tetrameric cyclo(-RGDfE-) revealed excellent and specific uptake, fast clearance, low background activity, and high-contrast imaging 1 h after injection (unpublished data; Fig. 3).

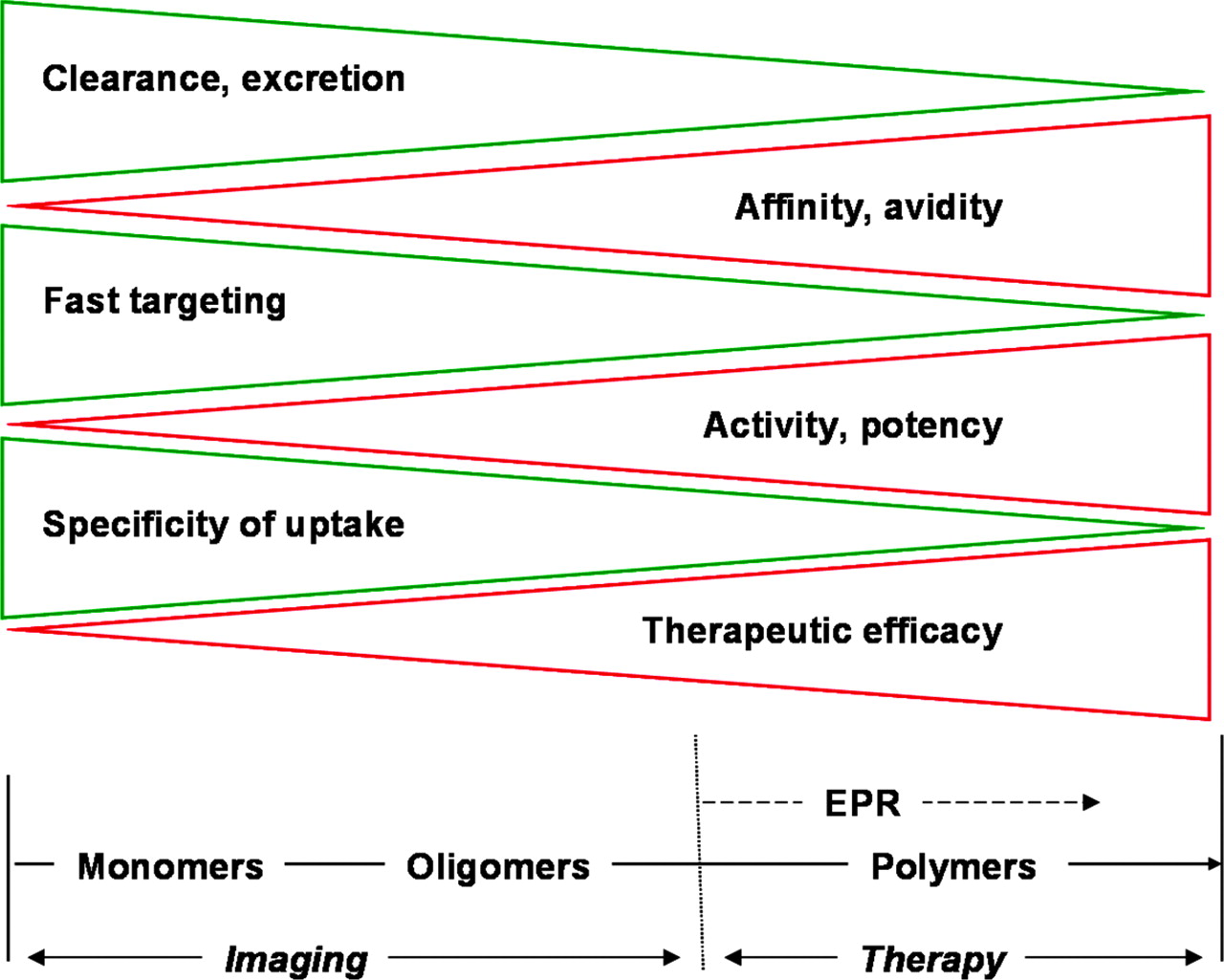
Selected characteristics of monovalent and polyvalent ligands and consequences for molecular targeting.
Although not ideal for peptide-receptor imaging, peptide-polymer conjugates may generally represent interesting and promising tracers for strategies aiming to deliver a payload to tumors (68–70). As already successfully demonstrated with αvβ3-targeted nanoparticles and liposomes for imaging of angiogenesis with MRI (71–73), targeting with polyvalent vehicles may offer a suitable alternative for an integrin-targeted EPR-enhanced PRRT radiotherapy. Here, EPR-mediated radiotherapy of the entire tumor would act in parallel with an RGD/integrin-mediated dose enhancement for the neovasculature and single tumor cells expressing αvβ3, and both the polyvalent binding and EPR would help to retain the activity over a long period inside the tumor.
In conclusion, polyvalent αvβ3-integrin antagonists, such as peptide multimers or peptide-polymer conjugates, are extremely promising ligands for the molecular targeting of integrins involved in angiogenesis. On the basis of their high specificity of uptake, peptide oligomers are outstanding tracers for imaging, whereas peptide-polymer conjugates may represent interesting constructs for high-level targeting and, thus, radiotherapeutic approaches.
Footnotes
Received Aug. 2, 2005; revision accepted Sep. 16, 2005.
For correspondence or reprints contact: Hans-Jürgen Wester, PhD, Department of Nuclear Medicine, Technische Universität München, Ismaninger Strasse 22, 81675 Munich, Germany.
E-mail: h.j.wester{at}lrz.tum.de
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Radionuclide Tumor Targeting Using ADAPT Scaffold Proteins: Aspects of Label Positioning and Residualizing Properties of the Label
- ADAPT, a Novel Scaffold Protein-Based Probe for Radionuclide Imaging of Molecular Targets That Are Expressed in Disseminated Cancers
- Tumor Targeting Using Affibody Molecules: Interplay of Affinity, Target Expression Level, and Binding Site Composition
- Imaging of Insulinlike Growth Factor Type 1 Receptor in Prostate Cancer Xenografts Using the Affibody Molecule 111In-DOTA-ZIGF1R:4551
- Targeting of CCK-2 Receptor-Expressing Tumors Using a Radiolabeled Divalent Gastrin Peptide
- Identification and Characterization of a Peptide with Affinity to Head and Neck Cancer
- Phase I Trial of the Positron-Emitting Arg-Gly-Asp (RGD) Peptide Radioligand 18F-AH111585 in Breast Cancer Patients
- Identification and Evaluation of a New Tumor Cell-Binding Peptide, FROP-1