


Abstract
Both bombesin (BBN) analogs and cyclic RGD peptides have been suitably radiolabeled for prostate cancer imaging. However, the limited expression of gastrin-releasing peptide receptor (GRPR) and integrin αvβ3 as well as unfavorable in vivo kinetics limited further applications of these imaging agents. We hypothesize that a peptide ligand recognizing both GRPR and integrin will be advantageous because of its dual-receptor–targeting ability. Methods: A BBN-RGD heterodimer was synthesized from bombesin(7–14) and c(RGDyK) through a glutamate linker and then labeled with 18F via the N-succinimidyl-4-18F-fluorobenzoate (18F-SFB) prosthetic group. The receptor-binding characteristics and tumor-targeting efficacy of 18F-FB-BBN-RGD were tested in vitro and in vivo. Results: FB-BBN-RGD had comparable integrin αvβ3-binding affinity with c(RGDyK) and comparable GRPR-binding affinity with BBN(7–14). 18F-FB-BBN-RGD had significantly higher tumor uptake compared with monomeric RGD and monomeric BBN peptide tracer analogs at all time points examined. The PC-3 tumor uptake of 18F-FB-BBN-RGD was inhibited only partially in the presence of an excess amount of unlabeled BBN(7–14) or c(RGDyK) but was blocked completely in the presence of both BBN(7–14) and c(RGDyK). Compared with 18F-FB-BBN and 18F-FB-RGD, 18F-FB-BBN-RGD also had improved pharmacokinetics, resulting in a significantly higher imaging quality. Conclusion: Dual integrin αvβ3 and GRPR recognition showed significantly improved tumor-targeting efficacy and pharmacokinetics compared with 18F-labeled RGD and BBN analogs. The same heterodimeric ligand design may also be applicable to other receptor system combinations and other imaging modalities.
Prostate cancer remains one of the leading causes of cancer-related deaths in the United States and Europe (1). As life expectancy increases, so will the incidence of this disease, creating what will become an epidemic male health problem. Overexpression of gastrin-releasing peptide receptor (GRPR) has been discovered primarily in androgen-independent human prostate tissues and, thus, provides a potential target for prostate cancer diagnosis and therapy (1–4). Various approaches have been explored for the imaging of GRPR expression in vivo. Bombesin (BBN), which was originally isolated from the skin of a frog, is an analog of the gastrin-releasing peptide (GRP). The truncated sequence BBN(7–14) was considered to be sufficient for the specific binding interaction with GRPR and metabolically stable for in vivo application. Several BBN peptides have been labeled with various radioisotopes for diagnosis and treatment of GRPR-positive prostate lesions (5–9). We have been particularly interested in developing an 18F-labeled imaging agent for PET due to the favorable properties of this radioisotope (18F) and the high sensitivity of PET. 18F-Labeled BBN peptides were successfully used for detecting GRPR-positive prostate cancer in vivo (5). However, 18F-labeled tracers derived from monomeric BBN had a relatively low tumor accumulation and retention as well as unfavorable hepatobiliary excretion (5). Therefore, modifications are necessary to obtain a better tumor-targeting effect and imaging quality.
It is well documented that most solid tumors are angiogenesis dependent and that integrin is a key player. In particular, integrin αvβ3 was found to be necessary for the formation, survival, and maturation of new blood vessels (10–12). Synthetic peptides containing the arginine-glycine-aspartate (RGD) sequence motif are active modulators of cell adhesion and can bind specifically to integrin αvβ3. Recently, we and others found that excellent tumor integrin-targeting efficacy and favorable in vivo kinetics were obtained for radiolabeled mutimeric RGD peptides due to the polyvalency effect (13–19). However, RGD peptide–based probes—including multimeric RGD peptides with high affinity for integrin αvβ3—had only moderate uptake in prostate cancer models, presumably because of the insufficient expression of this receptor in prostate cancer tumors.
As androgen-independent prostate cancer expresses both GRPR and integrin αvβ3, we hypothesize that a peptide ligand recognizing both receptors would be advantageous over a single receptor-binding probe. Thus, we have designed a BBN-RGD conjugate, in which cyclic RGD peptide c(RGDyK) was connected with BBN(7–14) through a glutamate linker. The dual GRPR- and integrin αvβ3-targeting ability of BBN-RGD heterodimer is illustrated in Figure 1. The tumor-targeting efficacy and in vivo kinetics of 18F-FB-BBN-RGD were compared with 18F-FB-BBN and 18F-FB-RGD counterparts.

(A) Strategy for enhancing effective binding of heterodimeric imaging agent. Dissociation of 18F-FB-BBN-RGD from GRPR may lead to rapid recomplexation of the same ligand with integrin αvβ3 (arrows). (B) Chemical structure of 18F-FB-BBN-RGD.
MATERIALS AND METHODS
All chemicals obtained commercially were of analytic grade and used without further purification. No-carrier-added 18F-F− was obtained from an in-house PETtrace cyclotron (GE Healthcare). Reversed-phase extraction C18 Sep-Pak cartridges were obtained from Waters and were pretreated with ethanol and water before use. The syringe filter and polyethersulfone membranes (pore size, 0.22 μm; diameter, 13 mm) were obtained from Nalge Nunc International. 125I-Echistatin, labeled by the lactoperoxidase method to a specific activity of 74 TBq/mmol (2,000 Ci/mmol) and 125I-[Tyr4]BBN (74 TBq/mmol (2,000 Ci/mmol)) were purchased from GE Healthcare. The peptides Aca-BBN(7–14) and c(RGDyK) were synthesized by Peptides International. Analytic as well as semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC) were performed on a Dionex 680 chromatography system with a UVD 170U absorbance detector and model 105S single-channel radiation detector (Carroll & Ramsey Associates). The recorded data were processed using Chromeleon version 6.50 software. Isolation of peptides and 18F-labeled peptides was performed using a Vydac protein and peptide column (218TP510; 5 μm, 250 × 10 mm). The flow was set at 5 mL/min using a gradient system starting from 95% solvent A (0.1% trifluoroacetic acid [TFA] in water) and 5% solvent B (0.1% TFA in acetonitrile [ACN]) (0–2 min) and ramped to 35% solvent A and 65% solvent B at 32 min. The analytic HPLC was performed using the same gradient system, but with a Vydac column (218TP54, 5 μm, 250 × 4.6 mm) and a flow of 1 mL/min. The ultraviolet (UV) absorbance was monitored at 218 nm and the identification of the peptides was confirmed based on the UV spectrum acquired using a photodiode array detector.
Preparation of NH2-Glu-BBN(7–14)-c(RGDyK) (BBN-RGD)
The Boc-protected glutamic acid activated ester Boc-E(OSu)2 was prepared as previously reported (20). To a solution of Boc-E(OSu)2 (4.4 mg, 10 μmol) in 2 mL anhydrous N,N-dimethylformamide (DMF), 0.8 eq. Aca-BBN(7–14) (8.4 mg, 8 μmol) was added. The pH of the resulting mixture was adjusted to 8.5–9.0 with diisopropylethyl amine (DIPEA). After stirring at room temperature for 2 h, 1.2 eq. c(RGDyK) (7.6 mg, 12 μmol) was added. The desired product Boc-BBN-RGD was isolated by preparative HPLC. The Boc- group was then removed by anhydrous TFA, and the crude product was again purified by HPLC. A total of 6.9 mg BBN-RGD was obtained as white powder in 48.4% overall yield. Analytic HPLC (retention time [Rt] = 16.8 min) and mass spectrometry (MALDI-TOF-MS [matrix-assisted laser desorption/ionization time-of-flight mass spectrometry]: m/z 1,783.03 for [MH]+ (C81H123N24O20S, calculated molecular weight [MW] 1,783.90)) confirmed the identity of the purified product.
Preparation of FB-NH-Glu-BBN(7–14)-c(RGDyK) (FB-BBN-RGD)
N-Succinimidyl-4-fluorobenzoate (SFB) (4 mg, 16.8 μmol) and BBN-RGD (2 mg, 1.12 μmol) were mixed in 0.05 M borate buffer (pH 8.5) at room temperature. After 2 h, the desired product FB-BBN-RGD was isolated by semipreparative HPLC in 62% yield. Analytic HPLC (Rt = 18.1 min) and mass spectrometry (MALDI-TOF-MS: m/z 1,905.90 for [MH]+ (C88H126FN24O21S, calculated [MW] 1,905.92)) analyses confirmed the product identification.
Radiochemistry
N-Succinimidyl-4-18F-fluorobenzoate (18F-SFB) was synthesized according to our previously reported procedure (21). Recently, we adapted the procedure into a commercially available synthesis module (GE TRACERlab FXFN). The purified 18F-SFB was rotary evaporated to dryness, reconstituted in dimethyl sulfoxide (DMSO, 200 μL), and added to a DMSO solution of peptides (0.1 μmol of BBN, RGD, or BBN-RGD) with DIPEA (20 μL). The peptide mixture was incubated at 60°C for 30 min. After dilution with 700 μL of 1% TFA, the mixture was purified by semipreparative HPLC. The desired fractions were combined and rotary evaporated to remove the solvent. The 18F-labeled peptides were then formulated in normal saline and passed through a 0.22-μm Millipore filter into a sterile multidose vial for in vitro and in vivo experiments.
Octanol/Water Partition Coefficient
Approximately 111 kBq of 18F-FB-BBN, 18F-FB-RGD, or 18F-FB-BBN-RGD in 500 μL of PBS (pH 7.4) were added to 500 μL of octanol in an Eppendorf microcentrifuge tube. The mixture was vigorously vortexed for 1 min at room temperature. After centrifugation at 12,500 rpm for 5 min in an Eppendorf microcentrifuge (model 5415R; Brinkman), 200-μL aliquots of both layers were measured using a γ-counter (Packard Instruments). The experiment was performed in triplicates.
Cell Lines and Animal Models
Animal procedures were performed according to a protocol approved by the Stanford University Institutional Animal Care and Use Committee. The PC-3 and DU-145 human prostate carcinoma cell lines were purchased from American Type Culture Collection. PC-3 cells were grown in F-12K nutrient mixture (Kaighn's modification) (Invitrogen Corp.), and DU-145 cells were grown in minimum essential medium (Eagle) mixture supplemented with 10% (v/v) fetal bovine serum (Invitrogen) at 37°C with 5% CO2. The PC-3 and DU-145 tumor models were generated by subcutaneous injection of 5 × 106 tumor cells into the front flank of male athymic nude mice (Harlan). The mice were subjected to microPET studies when the tumor volume reached 100–300 mm3 (3–4 wk after inoculation).
In Vitro Cell-Binding Assay
In vitro integrin αvβ3-binding affinities and specificities of RGD, BBN-RGD, and FB-BBN-RGD were assessed via displacement cell-binding assays using 125I-echistatin as the radioligand. Experiments were performed on U87MG human glioblastoma cells by a previously described method (20). In vitro GRPR-binding affinities and specificities of BBN, BBN-RGD, and FB-BBN-RGD were assessed via displacement cell-binding assays using 125I-[Tyr4]BBN as the radioligand. Experiments were performed on PC-3 human prostate carcinoma cells by a previously described method (22). The best-fit 50% inhibitory concentration (IC50) values were calculated by fitting the data with nonlinear regression using Graph-Pad Prism (GraphPad Software, Inc.). Experiments were performed with triplicate samples.
Cell Uptake and Efflux Studies
Uptake and efflux of 18F-FB-BBN, 18F-FB-RGD, and 18F-FB-BBN-RGD into PC-3 cells were examined according to the following protocol. In the cell uptake experiment, PC-3 cells were seeded into 12-well plates at a density of 5 × 105 cells per well for overnight incubation. Cells were rinsed 3 times with phosphate-buffered saline (PBS), followed by the addition of 18F-FB-RGD, 18F-FB-BBN, or 18F-BBN-RGD to the cultured wells in triplicate (∼2 μCi/well). After incubation at 37°C for 5, 15, 30, 60, and 120 min, cells were rinsed 3 times with PBS and lysed with NaOH-sodium dodecyl sulfate (SDS) (0.2 M NaOH, 1% SDS). The cell lysate was collected in measurement tubes for counting. The cell uptake was normalized in terms of added radioactivity. In the cell efflux experiment, PC-3 cells were seeded into 12-well plates at a density of 5 × 105 cells per well for overnight incubation. Cells were rinsed 3 times with PBS and then the appropriate 18F-labeled peptide tracer was added. The cells were incubated at 37°C for 2 h, washed with PBS, and then reincubated with serum-free medium. The cells were washed at different time points (0, 15, 30, 60, 120, 180 min) with PBS and lysed with NaOH-SDS (0.2 M NaOH, 1% SDS). The cell lysate was collected in measurement tubes for counting. Efflux values at different time points were calculated by subtracting retention from 0-min retention and normalized by dividing the total counts at 0 min.
microPET Studies
PET scans and image analysis were performed using a microPET R4 rodent model scanner (Siemens Medical Solutions) as reported previously (19,20). Tumor-bearing mice were each tail-vein injected with ∼3.7 MBq (100 μCi) of 18F-FB-RGD, 18F-FB-BBN, or 18F-FB-BBN-RGD under isoflurane anesthesia. Five-minute static PET images were then acquired at 0.5, 1, and 2 h after injection. The images were reconstructed by a 2-dimensional ordered-subsets expectation maximum (OSEM) algorithm. No attenuation or scatter correction was applied. For the receptor-blocking experiment, c(RGDyK) (10 mg/kg), Aca-BBN(7–14) (15 mg/kg), or RGD+BBN (10 mg/kg RGD and 15 mg/kg BBN) were coinjected with 3.7 MBq of 18F-FB-BBN-RGD to PC-3 tumor mice. The 5-min static PET scans was then acquired at 1 h after injection. For each microPET scan, regions of interest (ROIs) were drawn over the tumor, normal tissue, and major organs by using vendor software (ASI Pro 5.2.4.0) on decay-corrected whole-body coronal images. The average radioactivity concentration (accumulation) within a tumor or an organ was obtained from mean pixel values within the multiple ROI volume, which were converted to counts/mL/min using a conversion factor. Assuming a tissue density of 1 g/mL, the ROIs were converted to counts/g/min and then divided by the administered activity to obtain an imaging ROI–derived percentage injected dose per gram of tissue (%ID/g).
Metabolic Stability of 18F-FB-BBN-RGD
A PC-3 tumor mouse was injected intravenously with 3.7 MBq of 18F-FB-BBN-RGD. At 1 h after injection, the mouse was sacrificed, the blood, urine, liver, kidneys, and tumor were collected, and metabolite analysis was performed as reported previously (23). In brief, the blood sample was immediately centrifuged for 5 min at 13,200 rpm. Other tissues were homogenized and then centrifuged for 5 min at 13,200 rpm. Each supernatant was passed through a C18 Sep-Pak cartridge. The urine sample was diluted directly with 1 mL of PBS and passed through a C18 Sep-Pak cartridge. The cartridges were each washed with 2 mL of water and eluted with 2 mL of ACN containing 0.1% TFA. The ACN eluent was concentrated and injected onto the analytic HPLC system. The eluent was collected with a fraction collector (0.5 min/fraction), and the radioactivity of each fraction was measured with a γ-counter.
Statistical Analysis
Quantitative data are expressed as mean ± SD. Means were compared using 1-way ANOVA and the Student t test. P values of < 0.05 were considered statistically significant.
RESULTS
Chemistry and Radiochemistry
The synthesis of BBN-RGD heterodimer was performed through an active ester method by coupling Boc-Glu(OSu)2 with BBN and RGD peptides sequentially. After TFA deprotection, BBN-RGD was obtained as a fluffy white powder with a yield of 48.4%. Four products were generated from the coupling reaction [Boc-Glu(BBN)2, Boc-Glu(RGD)2, Boc-Glu-BBN-RGD (RGD on the side-chain δ-position), and Boc-Glu-RGD-BBN (BBN on the side-chain δ-position)]. The Boc-Glu(BBN)2 and Boc-Glu(RGD)2 impurities can be efficiently removed. However, we observed no difference in HPLC retention time between Boc-Glu-BBN-RGD and Boc-Glu-RGD-BBN. Therefore, as shown in Figure 1, the final product is a mixture of 2 closely related compounds. During the following text of this article, we will not clarify these 2 peptides and simply refer to them as BBN-RGD peptide. The total synthesis time for 18F-SFB was about 100 min, and the decay-corrected yield was 67% ± 11% (n = 10) using the modified GE synthetic module (TRACERlab FXFN). The decay-corrected radiochemical yield of 18F-FB-BBN-RGD based on 18F-SFB was 12.0% ± 0.7% (n = 4). The radiochemical purity of 18F-FB-BBN-RGD was >99% according to analytic HPLC. The specific radioactivities of 18F-FB-BBN, 18F-FB-RGD, and 18F-FB-BBN-RGD were estimated to be ∼100 TBq/mmol on the basis of the labeling agent 18F-SFB, as the unlabeled peptides were efficiently separated from the product. The octanol/water partition coefficient (logP) for 18F-FB-BBN-RGD was −0.92 ± 0.04 (18F-FB-RGD, −1.75 ± 0.03; 18F-FB-BBN, 1.49 ± 0.02), indicating that this tracer is more hydrophilic than 18F-FB-BBN but less hydrophilic than 18F-FB-RGD.
In Vitro Cell Receptor-Binding Assay
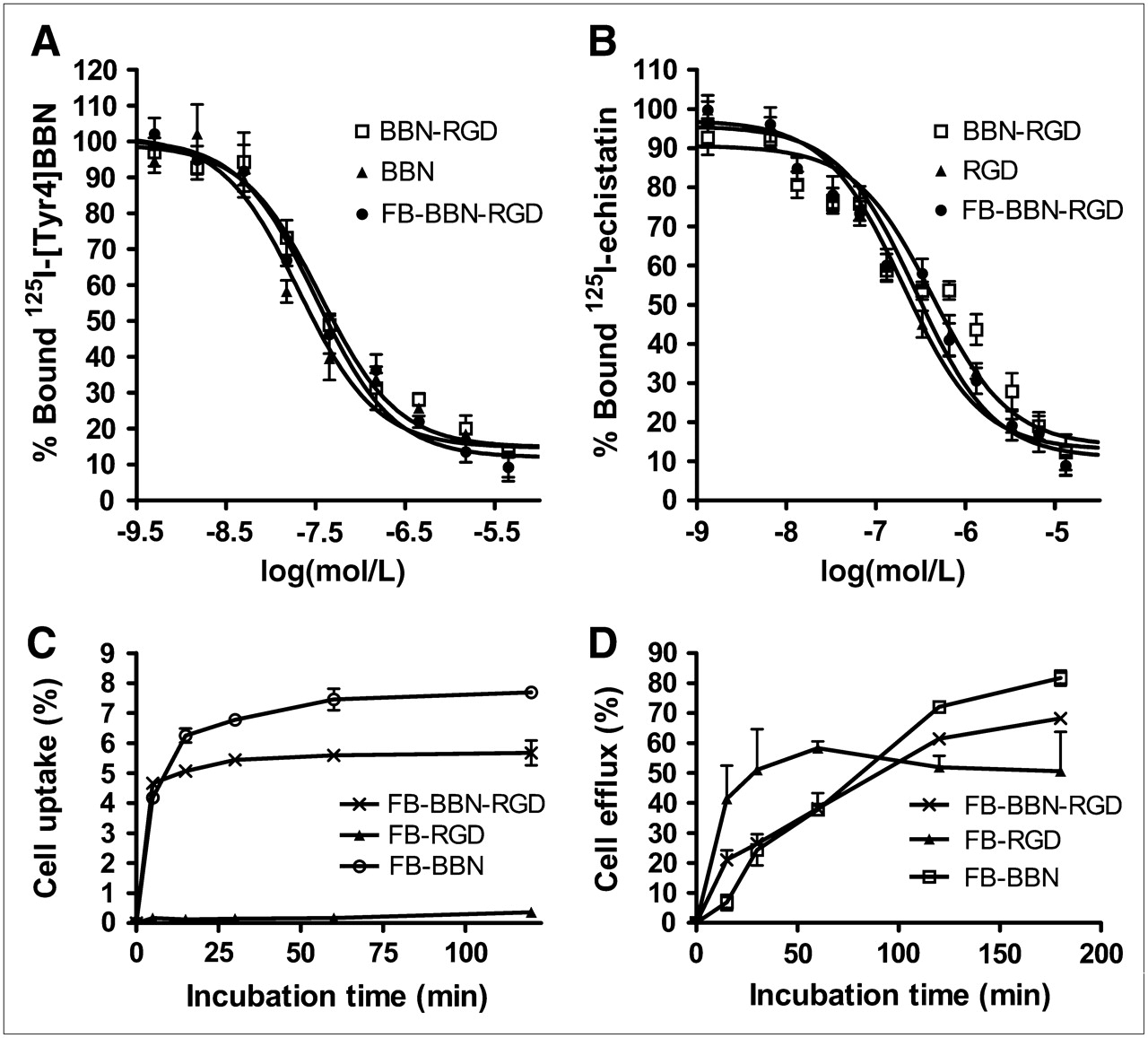
The binding affinities of Aca-BBN(7–14), BBN-RGD, and FB-BBN-RGD for GRPR were evaluated for PC-3 cells. Results of the cell-binding assay were plotted in sigmoid curves for the displacement of 125I-[Tyr4]BBN from PC-3 cells as a function of increasing concentration of BBN analogs. The IC50 values were determined to be 20.7 ± 3.2 nM for BBN monomer, 35.7 ± 4.4 nM for heterodimer BBN-RGD, and 32.0 ± 1.9 nM for FB-BBN-RGD on 105 PC-3 cells (Fig. 2A). The integrin αvβ3 receptor–binding affinity of RGD, BBN-RGD, and FB-BBN-RGD was determined by performing competitive displacement studies with 125I-echistatin. All peptides inhibited the binding of 125I-echistatin (integrin αvβ3–specific) to U87MG cells in a concentration-dependent manner. The IC50 values for RGD, BBN-RGD, and FB-BBN-RGD were 202 ± 28, 428 ± 57, and 282 ± 34 nM, respectively (n = 3) (Fig. 2B). The comparable IC50 values from these 2 sets of experiments suggest that the BBN-RGD peptide possesses comparable GRPR and integrin αvβ3 receptor–binding affinities as the corresponding monomer.

(A) Inhibition of 125I-[Tyr4]-BBN (GRPR-specific) binding to GRPR on PC-3 cells by BBN, BBN-RGD, and FB-BBN-RGD (n = 3, mean ± SD). (B) Inhibition of 125I-echistatin binding to integrin αvβ3 on U87MG cells by RGD BBN-RGD and FB-BBN-RGD (n = 3, mean ± SD). (C) Cell uptake assay of 18F-FB-BBN-RGD, 18F-FB-BBN, and 18F-FB-RGD on PC-3 tumor cells (n = 3, mean ± SD). (D) Cell efflux assay of 18F-FB-BBN-RGD, 18F-FB-BBN, and 18F-FB-RGD on PC-3 tumor cells (n = 3, mean ± SD).
Cell Uptake and Efflux Studies
Due to the relatively low receptor-binding affinity of the monomeric RGD peptides and moderate integrin receptor density of PC-3 cells, 18F-FB-RGD had relatively low cell uptake (<0.5%). On the other hand, PC-3 cells express a high level of GRPR, 18F-FB-BBN binding to GRPR facilitates effective internalization of this radioligand, and the uptake of 18F-FB-BBN is thus rapid and high, reaching about 7% within 30 min of incubation, and plateaus afterward. The cell uptake behavior of 18F-FB-BBN-RGD is similar to that of 18F-FB-BBN but the uptake value is slightly lower (Fig. 2C). All 3 tracers showed substantial efflux when the labeled cells were cultured in serum-free medium devoid of radioactivity (Fig. 2D). Note that the cell uptake protocol used in this study does not distinguish between cell-surface bound and internalized activity.
microPET of Tumor-Bearing Mice
Static microPET scans were performed on a PC-3 xenograft model (n = 3, both GRPR- and integrin αvβ3-positive) (24–27), and selected coronal images at different time points after injection of 18F-FB-BBN-RGD, 18F-FB-BBN, or 18F-FB-RGD are shown in Figure 3. The tumor was clearly visible with high contrast to contralateral background for 18F-FB-BBN-RGD. Quantitation of tumor and major organ activity accumulation in microPET scans was realized by measuring ROIs encompassing the entire organ on the coronal images. The averaged time–activity curves of these 3 tracers for the PC-3 tumor, liver, kidneys, and muscle are shown in Figure 4. 18F-FB-BBN and 18F-FB-RGD indicated moderate initial tumor uptake in this dual-receptor–positive tumor model; however, the unfavorable hepatobiliary excretion (high liver, bile, and intestinal activity accumulation) limited further applications of these 2 radiotracers, especially in an attempt to detect lesions in the lower abdomen. The PC-3 tumor uptake of 18F-FB-BBN-RGD was calculated to be 5.00 ± 0.28, 3.57 ± 0.27, and 2.79 ± 0.28 %ID/g at 30, 60, and 120 min after injection—significantly higher than those for 18F-FB-BBN (0.63 ± 0.16, 0.44 ± 0.10, and 0.29 ± 0.08 %ID/g at 30, 60, and 120 min after injection, P < 0.001), 18F-FB-RGD (2.21 ± 0.32, 1.30 ± 0.10, and 0.89 ± 0.08 %ID/g at 30, 60, and 120 min after injection P < 0.01), and even the sum of the uptake for these 2 monomeric tracers (dotted line in Fig. 4A, P < 0.01). 18F-FB-BBN-RGD also showed substantially lower liver and significantly decreased renal uptake compared with 18F-FB-BBN (slightly increased kidney uptake compared with 18F-FB-RGD). Because of the enhanced tumor-targeting efficacy and improved in vivo pharmacokinetics, 18F-FB-BBN-RGD had higher tumor-to-organ ratios than 18F-FB-BBN and 18F-FB-RGD as shown in Figure 4E. We also tested this tracer in an integrin αvβ3–positive but low GRPR-expressing DU-145 tumor model (Supplemental Fig. S1; supplemental material is available online only at http://jnm.snmjournals.org) (24,27,28). No significant difference in normal organs and tissues was found between these 2 tumor models. Because of the low GRPR expression, 18F-FB-BBN was unable to detect the DU-145 tumor. However, both 18F-FB-BBN-RGD and 18F-FB-RGD were able to visualize this tumor model as it is integrin αvβ3–positive. The DU-145 tumor uptake for 18F-FB-BBN-RGD was calculated to be 2.18 ± 0.24, 1.49 ± 0.17, and 1.02 ± 0.23 %ID/g at 30, 60, and 120 min after injection.

Decay-corrected whole-body coronal microPET images of athymic male nude mice bearing PC-3 tumor at 30, 60, and 120 min after injection of 18F-FB-BBN-RGD, 18F-FB-BBN, or 18F-FB-RGD (3.7 MBq [100 μCi]). Images shown are 3-min static scans of a single mouse, which is representative of the 3 mice tested in each group. Tumors are indicated by arrowheads.
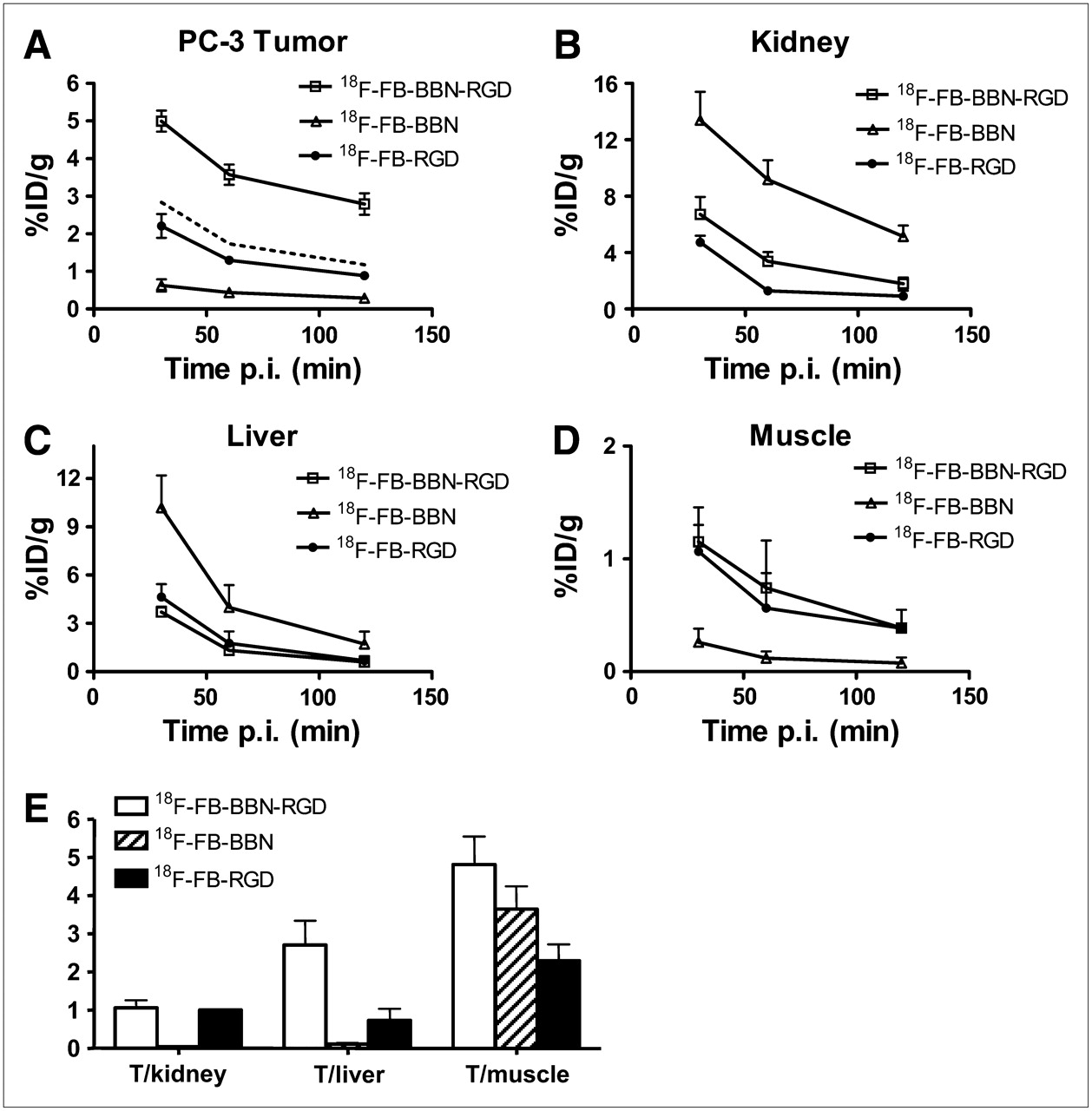
(A–D) Comparison between uptake of 18F-FB-BBN-RGD, 18F-FB-BBN, and 18F-FB-RGD in PC-3 tumor (A), kidneys (B), liver (C), and muscle (D). Dotted line in A represents the addition of 18F-FB-BBN and 18F-FB-RGD. (E) Comparison of tumor (T) with muscle, kidney, and liver ratio of 18F-FB-BBN-RGD, 18F-FB-BBN, and 18F-FB-RGD at 1 h after injection (p.i.) for athymic male nude mice bearing PC-3 tumor (n = 3, mean ± SD).
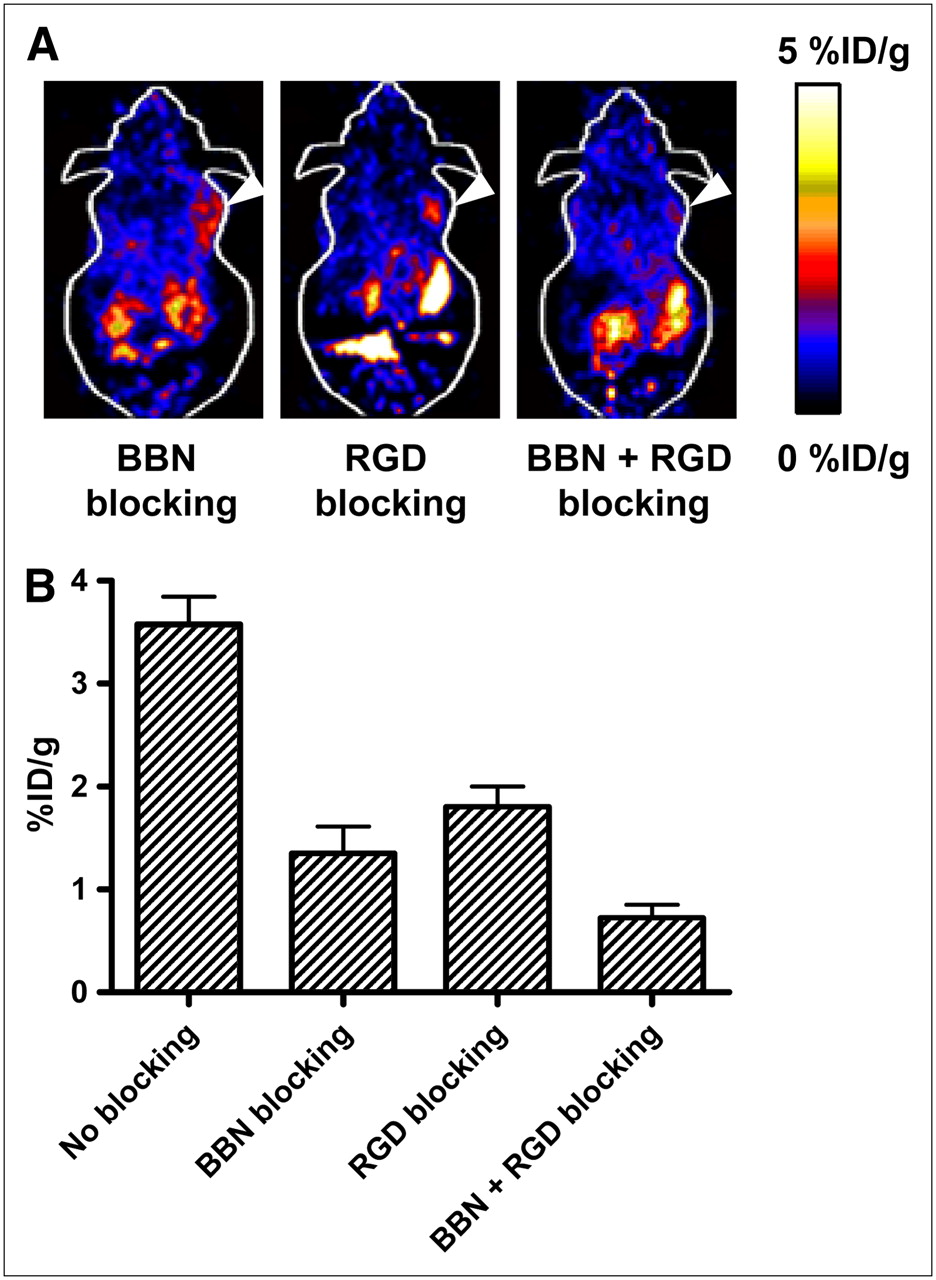
The receptor specificity of 18F-FB-BBN-RGD in vivo was confirmed by several blocking experiments (Fig. 5). Representative coronal images of PC-3 tumor mice after injection of 18F-FB-BBN-RGD in the presence of c(RGDyK) (10 mg/kg), Aca-BBN(7–14) peptide (15 mg/kg), or RGD + BBN (10 mg/kg for RGD and 15 mg/kg for BBN) are illustrated in Figure 5A. Uptake in the tumor at 1 h after injection (3.57 ± 0.27 %ID/g) was inhibited only partially by either RGD (1.80 ± 0.19 %ID/g) or BBN peptide alone (1.35 ± 0.26 %ID/g). When both RGD and BBN were coadministered with 18F-FB-BBN-RGD, the tumor uptake was reduced further to the background level (0.87 ± 0.27 %ID/g at 1 h after injection).

(A) Decay-corrected whole-body coronal microPET images of a PC-3 tumor-bearing mouse at 1 h after injection of 18F-FB-BBN-RGD and a blocking dose of c(RGDyK) (10 mg/kg of mouse body weight), BBN peptide (15 mg/kg mouse body weight), or RGD + BBN peptides (10 mg/kg for RGD and 15 mg/kg for BBN). Images shown are 3-min static scans of a single mouse, which is representative of the 3 mice tested in each group. Tumors are indicated by arrowheads. (B) Comparison between uptake of 18F-FB-BBN-RGD in PC-3 tumor with or without preinjection of blocking dose of peptides (c(RGDyK) (10 mg/kg of mouse body weight), BBN peptide (15 mg/kg mouse body weight), or RGD + BBN peptides (10 mg/kg for RGD and 15 mg/kg for BBN)). ROIs are shown as %ID/g ± SD (n = 3).
Metabolism of 18F-FB-BBN-RGD
The metabolic stability of 18F-FB-BBN-RGD was determined in mouse blood, urine, liver, kidneys, and tumor homogenates at 60 min after injection. The extraction efficiencies were 91.4% for blood, 73.6% for liver, 95.2% for kidneys, and 94.6% for PC-3 tumor, respectively. The elution efficiencies of the soluble fractions were 93.2% for blood, 68.6% for liver, 89.1% for kidneys, and 90.0% for PC-3 tumor. HPLC analysis results of the ACN-eluted fractions were shown in Figure 6. The average fraction of intact tracer was from 19.2% to 34.6% (Table 1). Although we did not identify the composition of the metabolites, we found that all metabolites came off the HPLC column earlier than those for the parent compound. No defluoridation of 18F-FB-BBN-RGD was observed, as no visible bone uptake was found on any of the microPET scans. Overall, 18F-FB-BBN-RGD exhibited comparable metabolic stability with 18F-FB-BBN (5).
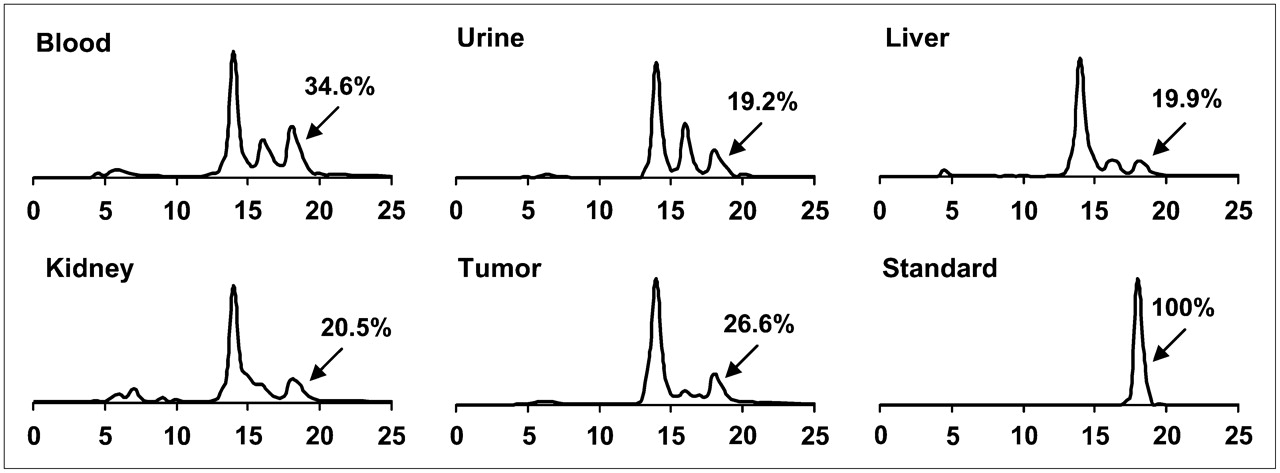
Metabolic stability of 18F-FB-BBN-RGD in mouse blood and urine samples and in liver, kidney, and PC-3 tumor homogenates at 1 h after injection. The HPLC profile of pure 18F-FB-BBN-RGD (standard) is also shown.
Extraction Efficiency, Elution Efficiency, and HPLC Analysis of Soluble Fractions of Tissue Homogenates at 1 Hour After Injection of 18F-FB-BBN-RGD
DISCUSSION
There has been an exponential growth in the development of radiolabeled peptides for diagnostic and therapeutic applications in the last decade. Peptidic radiopharmaceuticals have many favorable properties—including fast clearance, rapid tissue penetration, and low antigenicity—and can be produced easily and inexpensively (13,29). 18F with almost 100% positron efficiency and relatively short physical half-life (t1/2 = 109.7 min) is ideally suited for peptide labeling and PET. For example, 18F-labeled RGD monomeric peptides have been synthesized for the imaging of integrin αvβ3–positive tumors (30,31). 18F-Labeled BBN peptides were also reported to be successful for GRPR-targeted imaging (5). However, the tumor uptake and contrast of these peptide tracers were suboptimal owing, in part, to the relatively low binding affinity of this unmodified monomeric peptide and the imperfect pharmacokinetics.
As PC-3 tumor cells express both GRPR and integrin αvβ3 (24–27), we wanted to test whether the dual GRPR- and integrin αvβ3-targeting approach would allow us to develop significantly improved imaging probes over those that recognize only 1 receptor type. In this study, we synthesized and radiolabeled 18F-FB-BBN-RGD (BBN peptide motif for GRPR targeting and RGD peptide motif for integrin αvβ3 targeting). Our receptor-binding assay data demonstrated that BBN-RGD peptide is similar to Aca-BBN(7–14) for GRPR binding and is similar to c(RGDyK) for integin αvβ3 binding. 18F-FB-RGD showed the lowest cell uptake, as integrin αvβ3 binding does not tend to internalize the tracer. However, in vivo uptake of the 18F-FB-BBN derivative is lower than that for the RGD derivative, which might be partially attributed to their difference in pharmacokinetics. The cell uptake of 18F-FB-BBN-RGD is slightly lower than that of 18F-FB-BBN at early time points but is significantly higher than that of 18F-FB-RGD in PC-3 cells. 18F-FB-BBN-RGD tends to have a slower washout than 18F-FB-BBN, which might be the result of enhanced effective binding due to dual targeting. However, additional experiments are needed to confirm this statement.
The imaging quality of 18F-FB-BBN-RGD was tested in the PC-3 xenograft model. Compared with 18F-FB-BBN and 18F-FB-RGD, the PC-3 tumor uptake of 18F-FB-BBN-RGD was much higher than the sum of the monomeric tracers at all time points examined (Fig. 4A). The pharmacokinetics were also improved for 18F-FB-BBN-RGD compared with that of 18F-FB-BBN and 18F-FB-RGD in the liver and kidneys, which may be attributed to the differences in molecular size, charge, and hydrophilicity of these 3 compounds. 18F-FB-BBN-RGD also had the highest tumor-to-nontumor ratios when compared with 18F-FB-BBN and 18F-FB-RGD. Overall, a synergistic effect has been observed for 18F-FB-BBN-RGD, and significantly higher tumor uptake and contrast have been obtained in the PC-3 tumor model. In the blocking experiment, neither nonradioactive BBN peptide nor nonradioactive RGD peptide could totally inhibit the uptake of 18F-FB-BBN-RGD in PC-3 tumor, as the tracer could still bind to the unblocked receptors. The BBN and RGD double blocking could further reduce the tumor uptake, which strongly supports the dual-receptor specificity of 18F-FB-BBN-RGD in vivo. For 18F-FB-BBN-RGD, the RGD blocking resulted in a slightly higher tumor uptake than BBN blocking, which may be due to the fact that the PC-3 cells express a high level of GRPR but express a medium level of integrin αvβ3. Moreover, the advantage of this heterodimer tracer is also obvious when only 1 receptor type is overexpressed. For example, DU-145 tumor expresses a moderate level of integrin αvβ3 but expresses a low level of GRPR (24,27,28), 18F-FB-BBN that binds to GRPR but not to integrin αvβ3 is unable to provide enough tumor uptake and tumor-to-background contrast. 18F-FB-BBN-RGD, on the other hand, had a tumor uptake similar to that of 18F-FB-RGD but had a significantly lower background (Supplemental Fig. S1). We did not test the heterodimer probe in a GRPR-positive but integrin-negative tumor model. If such a model exists, we expect that the heterodimer will also allow tumor detection with comparable tumor contrast to 18F-FB-BBN.
In the metabolic stability study, the metabolites of 18F-FB-BBN-RGD may be determined primarily by the FB unit and BBN sequence as cyclic RGD-containing pentapeptide is highly stable in vivo. In this regard, a BBN derivative with better proteolytic stability may be advantageous for further improving the tumor-targeting efficacy. By analyzing the structure of the BBN-RGD heterodimer (Fig. 1), the peptide is expected to bind to either GRPR or integrin αvβ3, as the glutamate linker is too short to allow simultaneous GRPR and integrin αvβ3 binding. Thus, the total number of binding sites for this ligand is the sum of GRPR and integrin αvβ3, higher than that for RGD peptide and for BBN analogs. The advantage of BBN-RGD over BBN or RGD is not only the increased number of receptors for signal amplification but also the binding kinetics would contribute to the in vivo behavior of 18F-FB-BBN-RGD. Assuming that the ligand binds to GRPR through the BBN moiety, the remaining RGD moiety will then be in close vicinity of integrin αvβ3. The dissociation of BBN-RGD from GRPR will lead to rapid recomplexation of the same ligand with integrin αvβ3. If the heterodimer is initially bound to integrin instead, the dissociation of the RGD motif from integrin will reorient the BBN-RGD to bind to GRPR, resulting in an apparent low off-rate of the ligand binding. Both the increased number of binding sites and the apparent low off-rate of the dual-receptor–targeting ligand are expected to have enhanced tumor uptake and retention as compared with those single-receptor–recognizing ligands. The added molecular size and change of overall molecular charge and hydrophilicity will also have certain effects on the in vivo kinetics of the resulting probes. The synthesis of heterodimers with a scrambled sequence that have the same molecular weight but no receptor binding may be needed in the future to study the pharmacokinetic effects.
For integrin binding, we and others have shown that multimeric RGD peptides are superior to the monomeric counterparts in terms of receptor-binding affinity in vitro and tumor-targeting efficacy in vivo, presumably due to the so-called “multivalency effect” (13,19,21). It is probably worthwhile to study whether BBN analogs linked with dimeric or oligomeric RGD through the glutamate linker will be better than the BBN-RGD heterodimer described here using monomeric RGD peptide. Note that the heterodimer connected with glutamate is actually a mixture of Glu-BBN-RGD (where RGD is on the side-chain δ-position) and Glu-RGD-BBN (where BBN is on the side-chain δ-position), which are not separable by HPLC. It is desirable to synthesize similar BBN-RGD heterodimers through a symmetric linker or a side-chain–protected glutamic acid, which will result in only 1 possible structure. Moreover, the radiolabeling yield for the heterodimeric BBN-RGD peptide is significantly lower than that for the monomeric BBN or RGD peptide using 18F-SFB as the synthon. We also observed a low 18F-labeling yield for RGD homodimer and homotetramer in which 18F-SFB was reacted with the glutamate amine group, presumably due to the steric hindrance and relatively low reactivity of the glutamate α-amino group. A mini-PEG (polyethylene glycol) spacer has been successfully used to reduce the spatial hindrance and improve the labeling yield for the multimeric RGD peptides (21,23). A similar strategy may also be applied here. Furthermore, other 18F-labeling strategies (such as click chemistry, reductive amination, Michael addition for thiol-specific coupling, and oxime formation) (32–35) may be used to simplify the labeling procedure and improve the labeling yield.
CONCLUSION
We have successfully developed a heterodimeric peptide that binds to both GRPR and integrin αvβ3. Dual-receptor binding results in more than an additive effect in vivo in terms of tumor uptake and retention in an androgen-receptor–negative PC-3 tumor model. The same peptide labeled with 18F for PET described in this study may also be labeled with other tags for different imaging modalities and may also be coupled with chemo- and radiotherapeutics. The synergistic effect observed for GRPR and integrin targeting may also be applicable to other peptide systems designed for dual- or multiple-receptor targeting. To our knowledge, this is the first example of a PET probe that targets 2 different types of receptors on the same cell surface.
Acknowledgments
We thank Drs. Gang Niu and Hui Wang for their excellent technical support and also thank the cyclotron teams at Stanford University for 18F-F− production. This work was supported by the National Cancer Institute (grants R01 CA119053, R21 CA121842, R21 CA102123, P50 CA114747, U54 CA119367, and R24 CA93862) and the Department of Defense (grants W81XWH-07-1-0374, W81XWH-04-1-0697, W81XWH-06-1-0665, W81XWH-06-1-0042, and DAMD17-03-1-0143).
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication October 9, 2007.
- Accepted for publication November 20, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Dual-Targeted Molecular Imaging of Cancer
- Reply: Dual-Integrin {alpha}v{beta}3- and Gastrin-Releasing Peptide Receptor-Targeting PET Radiotracer (68Ga-BBN-RGD)
- Approaches to Multireceptor Targeting: Hybrid Radioligands, Radioligand Cocktails, and Sequential Radioligand Applications
- Clinical Translation of a Dual Integrin {alpha}v{beta}3- and Gastrin-Releasing Peptide Receptor-Targeting PET Radiotracer, 68Ga-BBN-RGD
- Bombesin-Targeted PET of Prostate Cancer
- In Vivo Cancer Dual-Targeting and Dual-Modality Imaging with Functionalized Quantum Dots
- Dosimetry and First Clinical Evaluation of the New 18F-Radiolabeled Bombesin Analogue BAY 864367 in Patients with Prostate Cancer
- 18F-Labeled Bombesin Analog for Specific and Effective Targeting of Prostate Tumors Expressing Gastrin-Releasing Peptide Receptors
- Small-Animal PET of Tumors with 64Cu-Labeled RGD-Bombesin Heterodimer
- Integrins in prostate cancer progression