


Abstract
Two bombesin analogs, Demobesin 4 and Demobesin 1, were characterized in vitro as gastrin-releasing peptide (GRP) receptor agonist and antagonist, respectively, and were compared as 99mTc-labeled ligands for their in vitro and in vivo tumor-targeting properties. Methods: N4-[Pro1,Tyr4,Nle14]Bombesin (Demobesin 4) and N4-[d-Phe6,Leu-NHEt13,des-Met14]bombesin(6–14) (Demobesin 1) were characterized in vitro for their binding properties with GRP receptor autoradiography using GRP receptor–transfected HEK293 cells, PC3 cells, and human prostate cancer specimens. Their ability to modulate calcium mobilization in PC3 and transfected HEK293 cells was analyzed as well as their ability to trigger internalization of the GRP receptor in transfected HEK293 cells, as determined qualitatively by immunofluorescence microscopy and quantitatively by enzyme-linked immunosorbent assay (ELISA). Further, their internalization properties as 99mTc-labeled radioligands were tested in vitro in both cell lines. Finally, their biodistribution was analyzed in PC3 tumor–bearing mice. Results: A comparable binding affinity with the 50% inhibitory concentration (IC50) in the nanomolar range was measured for Demobesin 4 and Demobesin 1 in all tested tissues. Demobesin 4 behaved as an agonist by strongly stimulating calcium mobilization and by triggering GRP receptor internalization. Demobesin 1 was ineffective in stimulating calcium mobilization and in triggering GRP receptor internalization. However, in these assays, it behaved as a competitive antagonist as it reversed completely the agonist-induced effects in both systems. 99mTc-Labeled Demobesin 1 was only weakly taken up by PC3 cells or GRP receptor–transfected HEK293 cells (10% and 5%, respectively, of total added radioactivity) compared with 99mTc-labeled Demobesin 4 (45% of total added radioactivity in both cell lines). Remarkably, the biodistribution study revealed a much more pronounced uptake at 1, 4, and 24 h after injection of 99mTc-labeled Demobesin 1 in vivo into PC3 tumors than 99mTc-labeled Demobesin 4. In vivo competition experiments demonstrated a specific uptake in PC3 tumors and in physiologic GRP receptor–expressing tissues. The tumor-to-kidney ratios were 0.7 for Demobesin 4 and 5.2 for Demobesin 1 at 4 h. Conclusion: This comparative in vitro/in vivo study with Demobesin 1 and Demobesin 4 indicates that GRP receptor antagonists may be superior targeting agents to GRP receptor agonists, suggesting a change of paradigm in the field of bombesin radiopharmaceuticals.
Bombesin receptors—in particular, the gastrin-releasing peptide (GRP) receptor subtype—have been shown to be massively overexpressed in several human tumors, including breast cancer, prostate cancer, small cell lung cancer, ovarian cancers, endometrial cancers, and gastrointestinal stromal tumors (1–6). These receptors represent a molecular target for radiolabeled bombesin analogs as diagnostic or radiotherapeutic applications in these tumors, in analogy to somatostatin receptors in neuroendocrine tumors (7). In recent years, several bombesin analogs suitable for targeting have been synthesized and characterized. After the first proof-of-concept study by Van de Wiele et al. (8) reporting the targeting of breast and prostate cancers with a bombesin(7–14) conjugate labeled with 99mTc (RP527), several other bombesin radioligands were developed and characterized both in vitro and in vivo. These include, among others, analogs synthesized by Volkert's group (9,10), the bombesin analogs Demobesin 1 and Demobesin 4 developed by Nock et al. (11,12), a pan-bombesin analog reported by Zhang et al. (13), and the 177Lu-AMBA developed by Bracco (14,15). Up to now the consensus has been to develop compounds with good radioligand internalization properties, as a high in vivo accumulation of radioligands into the tumors appeared to be required for optimal visualization and radionuclide therapy in vivo (16). It is well known from molecular–pharmacologic investigations that efficient internalization is usually provided predominantly by agonists (16–18). Interestingly, however, we have recently been able to show that high-affinity somatostatin receptor antagonists that poorly internalize into tumor cells can perform equally or even better in terms of in vivo uptake into tumor in animal tumor models than the corresponding agonists, which massively internalize (19). This observation was made both for sst2- as well as for sst3-selective somatostatin analogs, suggesting that such a change of paradigm may be valid for more than just one particular G protein–coupled receptor, as these radioligands bind to distinct receptors (19).
In the present study, we have compared a potent radiolabeled bombesin agonist with a comparably potent radiolabeled bombesin antagonist and investigated in the same assays, under identical conditions and using the same batch of cells, their in vitro and in vivo characteristics as tumor-targeting agents. The aim was to investigate whether the change of paradigm in peptide receptor tumor targeting toward antagonists, as described recently for somatostatin (19), could also be extended to the bombesin receptor system. Because of the large spectrum of GRP receptor–expressing tumors (1–7), the bombesin receptor system may be considerably more important as a clinically relevant target than the somatostatin receptor system.
MATERIALS AND METHODS
Reagents and Peptides
The mouse monoclonal hemagglutinin (HA) epitope antibody was purchased from Covance. The secondary antibodies Alexa Fluor 488 goat anti-mouse IgG (H+L) was from Molecular Probes, Inc., and the horseradish peroxidase–conjugated goat anti-mouse IgG was from Bio-Rad Laboratories, Inc. Peptides were purchased from Bachem. [d-Phe6,Leu-NHEt13,des-Met14]Bombesin(6–14) and [Pro1,Tyr4,Nle14]bombesin were coupled with the tetraamine precursor and radiolabeled with 99mTc, to obtain [99mTc]Demobesin 1 (11) and [99mTc]Demobesin 4 (12), respectively.
Cell Lines
Human embryonic kidney 293 (HEK293) cells, stably expressing the HA epitope–tagged human GRP receptor (HEK-GRPR), were generated by transfection of HEK293 cells with the 3xHA-GRPR pcDNA3.1+ plasmid (UMR cDNA Resource Center) using the calcium phosphate precipitation method (20). Stable transfectants were selected in the presence of 750 μg/mL G418. HEK-GRPR cells were cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle medium with GlutaMAX-I (DMEM) containing 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 750 μg/mL G418 (GIBCO). These HEK-GRPR cells have been validated for correct expression and functionality of the HA-tagged GRP receptor by different methods: (a) in vitro receptor autoradiography of HEK-GRPR cell pellets showed high binding affinity for the established bombesin radioligands 125I-[Tyr4]bombesin and 125I-[d-Tyr6,β-Ala11,Phe13,Nle14]bombesin(6–14) (21), comparable to the binding affinities obtained in other GRP receptor–expressing tissues; (b) calcium mobilization and internalization assays with HEK-GRPR cells identified the expected rank order of potency for a series of well-characterized bombesin analogs; (c) Western blot analysis of lysates of HEK-GRPR cells produced a band with the correct molecular weight corresponding to the HA-tagged GRP receptor; (d) the mouse monoclonal HA epitope antibody recognized the HA-tagged GRP receptor in enzyme-linked immunosorbent assay (ELISA) and immunofluorescence microscopy assays. Human prostate cancer cells (PC3 cells) were obtained from the DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH; DSMZ no: ACC465) or from the American Type Culture Collection (LGC Promochem). Cells were cultured at 37°C and 5% CO2 in Ham's F-12K medium containing 2 mM l-glutamine and supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin or in DMEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Binding-Affinity Measurements
The GRP receptor–binding affinity of the various compounds was determined by in vitro receptor autoradiography on cryostat sections of either well-characterized prostate carcinomas or on sections from HEK-GRPR or PC3 cell pellets as described previously (1,21,22). The radioligands used were 125I-[Tyr4]bombesin, known to preferentially label GRP receptors (23), and 125I-[d-Tyr6,β-Ala11,Phe13,Nle14]bombesin(6–14) as the universal bombesin receptor ligand (23).
Immunofluorescence Microscopy
Immunofluorescence microscopy–based internalization assays with HEK-GRPR cells were performed as described by Cescato et al. (18). Briefly, cells were treated either with bombesin, Demobesin 1, or Demobesin 4 at concentrations ranging from 100 pM to 1 μM, or—to evaluate potential antagonism—with 10 nM bombesin in the presence of a 1,000-fold excess of Demobesin 1 or Demobesin 4 for 30 min at 37°C and 5% CO2 in growth medium and then processed for immunofluorescence microscopy using the mouse monoclonal HA epitope antibody at a dilution of 1:1,000 as the first antibody and Alexa Fluor 488 goat anti-mouse IgG (H+L) at a dilution of 1:600 as the secondary antibody. The cells were imaged as described previously (18).
Quantitative Assay for Receptor Internalization
Receptor internalization was quantitated in HEK-GRPR cells using an ELISA as described previously for other peptide receptors (18,24). HEK-GRPR cells grown on poly-d-lysine–coated 24-well plates were incubated for 30 min at 37°C either without or with ligands added. Incubations were terminated by placing the plates on ice, and cells were subsequently processed for ELISA. The GRP receptor remaining at the cell surface after ligand treatment was calculated as the absorbance measured in treated cells expressed as a percentage of the absorbance in untreated cells as described previously for other peptide receptors (18,24).
Intracellular Calcium Mobilization
Intracellular calcium mobilization was measured in PC3 and HEK-GRPR cells using the Fluo-4NW Calcium Assay kit (Molecular Probes, Inc.) as described by Magrys et al. and Michel et al. (25,26). In brief, PC3 cells (10,000 cells per well) or in HEK-GRPR cells (25,000 cells per well) were seeded in 96-well plates and cultured for 2 or 1 d, respectively, at 37°C and 5% CO2. The cells were then washed with assay buffer (1× Hank's balanced salt solution and 20 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) [HEPES]) containing 2.5 mM probenecid and then loaded with 100 μL per well Fluo-4 dye in assay buffer containing 2.5 mM probenecid for 30 min at 37°C and 5% CO2 and then for a further 30 min at room temperature. To measure the intracellular calcium mobilization after stimulation, the cells were transferred to a SpectraMax M2e (Molecular Devices). Intracellular calcium mobilization was recorded at room temperature for 60 s in a kinetic monitoring fluorescence emission at 520 nm (with λex = 485 nm) in the presence of the peptides to be tested. Maximum fluorescence (Fmax) was measured after the addition of ionomycin. Baseline (control) measurements were taken for untreated cells. Data are shown as % Fmax obtained with ionomycin as reported previously (25,26). All experiments were repeated at least 3 times in triplicate.
Radioligand Internalization
Internalization of [99mTc]Demobesin 1 or [99mTc]Demobesin 4 was performed in PC3 and HEK-GRPR cells. For internalization experiments, cells were seeded in 35-mm-diameter dishes (Greiner Labortechnik) and cultured for 48 h (1.0–1.5 × 106 cells per well). The cells were then rinsed twice with ice-cold internalization medium comprising DMEM supplemented with 1% (v/v) FBS (11,12,27). After addition of fresh medium (1.2 mL), approximately 300,000 cpm of [99mTc]Demobesin 1 or [99mTc]Demobesin 4 (in 150 μL phosphate-buffered saline [PBS]/0.5% bovine serum albumin buffer, corresponding to 200 fmol total peptide) were added, and the experiment was performed as described previously (27).
Biodistribution
All protocols were approved by national authorities and were consistent with European guidelines for animal welfare. The biodistribution of [99mTc]Demobesin 1 and [99mTc]Demobesin 4 was compared directly in SCID (severely compromised immunodeficient) mice from the same colony and age (National Center for Scientific Research Demokritos). After brief acclimatization, animals were injected subcutaneously with an ∼150-μL suspension of 1.5–2 × 107 PC3 cells in PBS, freshly harvested from the same batch. Two weeks later, well-palpable tumors (70–150 mg) formed at the inoculation site in animals kept under aseptic conditions, and the biodistribution study was conducted according to a published protocol (11,12).
RESULTS
Table 1 summarizes the affinity binding data for bombesin, Demobesin 1, and Demobesin 4. The 50% inhibitory concentrations (IC50) in the low nanomolar range are comparable for the 3 compounds. The tissues used for binding experiments were human prostate cancers, one of the main target tissues for bombesin receptors targeting in vivo, HEK293 cells transfected with the HA-tagged GRP receptor, and PC3 cells endogenously expressing the GRP receptor. IC50 values are comparable for the various peptides in all tissues.
In Vitro GRP Receptor Binding, Signaling, and Internalization Properties of Bombesin Analogs
The compounds were evaluated for their effect on signaling by using a calcium mobilization assay in PC3 and HEK-GRPR cells. Although bombesin and Demobesin 4 behaved as agonists, Demobesin 1 behaved as an antagonist (Table 1). As seen in Figure 1, 100 nM bombesin alone can stimulate calcium mobilization, an effect that is antagonized when 10 μM of the bombesin receptor antagonist [d-Phe6, Leu-NHEt13, des-Met14]bombesin(6–14) (ANTAG) is added at the same time. Demobesin 4 also behaved as an agonist and, when given together with 100 nM bombesin, was unable to inhibit the bombesin-stimulated calcium mobilization, indicating the absence of antagonist properties for this compound. Conversely, Demobesin 1 has no effect when given alone at 1,000 nM; however, if given at 10 μM concentration together with 100 nM bombesin, it was able to completely inhibit the bombesin-induced calcium mobilization, indicating full antagonistic properties. The results were comparable in PC3 and HEK-GRPR cells (Fig. 1).
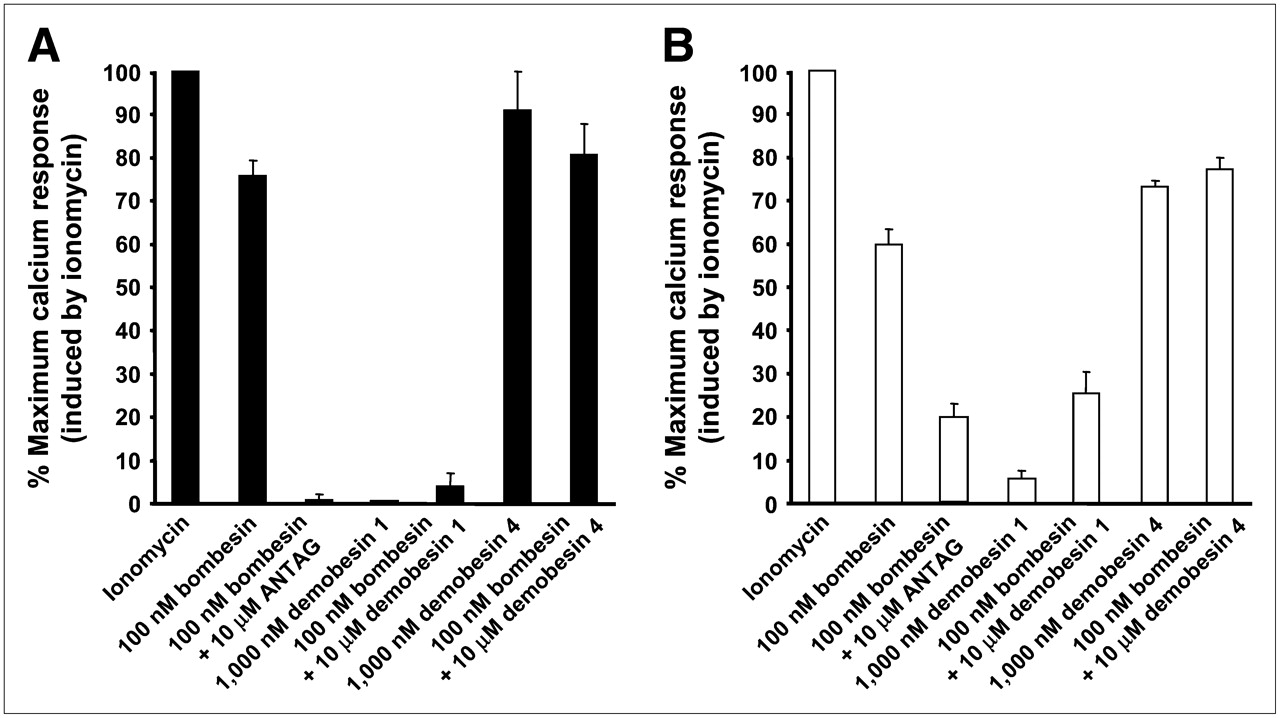
Intracellular calcium mobilization induced in PC3 (A) and HEK-GRPR (B) cells by various bombesin analogs. Cells were loaded with Fluo-4 dye as described and analyzed for calcium mobilization in response either to 100 nM bombesin, 1,000 nM Demobesin 4, or Demobesin 1 or to 100 nM bombesin in the presence of 10 μM Demobesin 1 or Demobesin 4 or the antagonist (d-Phe6,Leu-NHEt13,des-Met14)bombesin(6–14) (ANTAG). Maximum calcium response was obtained by treating the cells with ionomycin (100% value). Results are shown as percentage of maximum calcium response induced by ionomycin.
The agonistic and antagonistic properties of the bombesin analogs were also observed in immunofluorescence-based internalization assays with HEK-GRPR cells. Figure 2 illustrate that 10 nM bombesin can trigger a massive GRP receptor internalization in HEK-GRPR cells, compared with the condition without peptide. Demobesin 4 at 10 or 1,000 nM also induces a massive internalization of GRP receptors. Conversely, Demobesin 1 does not trigger the internalization of GRP receptors at 1,000 nM; however, when given at a concentration of 1,000 nM together with 10 nM bombesin, it is able to completely prevent the bombesin-induced receptor internalization. Conversely, Demobesin 4, at the same concentration of 1,000 nM, is unable to inhibit the bombesin-induced receptor internalization. Furthermore, we used an ELISA to quantify the GRP receptor internalization in HEK-GRPR cells, as illustrated in Figure 3. Whereas Demobesin 4 triggers receptor internalization in the picomolar range (half-maximally effective concentration [EC50] = 0.20 ± 0.11 nM), Demobesin 1 is unable to induce a significant GRP receptor internalization up to 10 μM concentration (Fig. 3A and Table 1). However, it is able to completely antagonize the Demobesin 4-induced internalization (Fig. 3B).

GRP receptor internalization in HEK-GRPR cells shown by immunofluorescence microscopy. HEK-GRPR cells were treated for 30 min either with vehicle (no peptide), 10 nM bombesin, 10 nM or 1,000 nM Demobesin 4, or with 10 nM bombesin in the presence of 1,000 nM Demobesin 1 or 1,000 nM Demobesin 1 alone. GRP receptor internalization is induced by bombesin and Demobesin 4 but not by Demobesin 1. However, the bombesin-induced internalization is completely abolished by Demobesin 1.
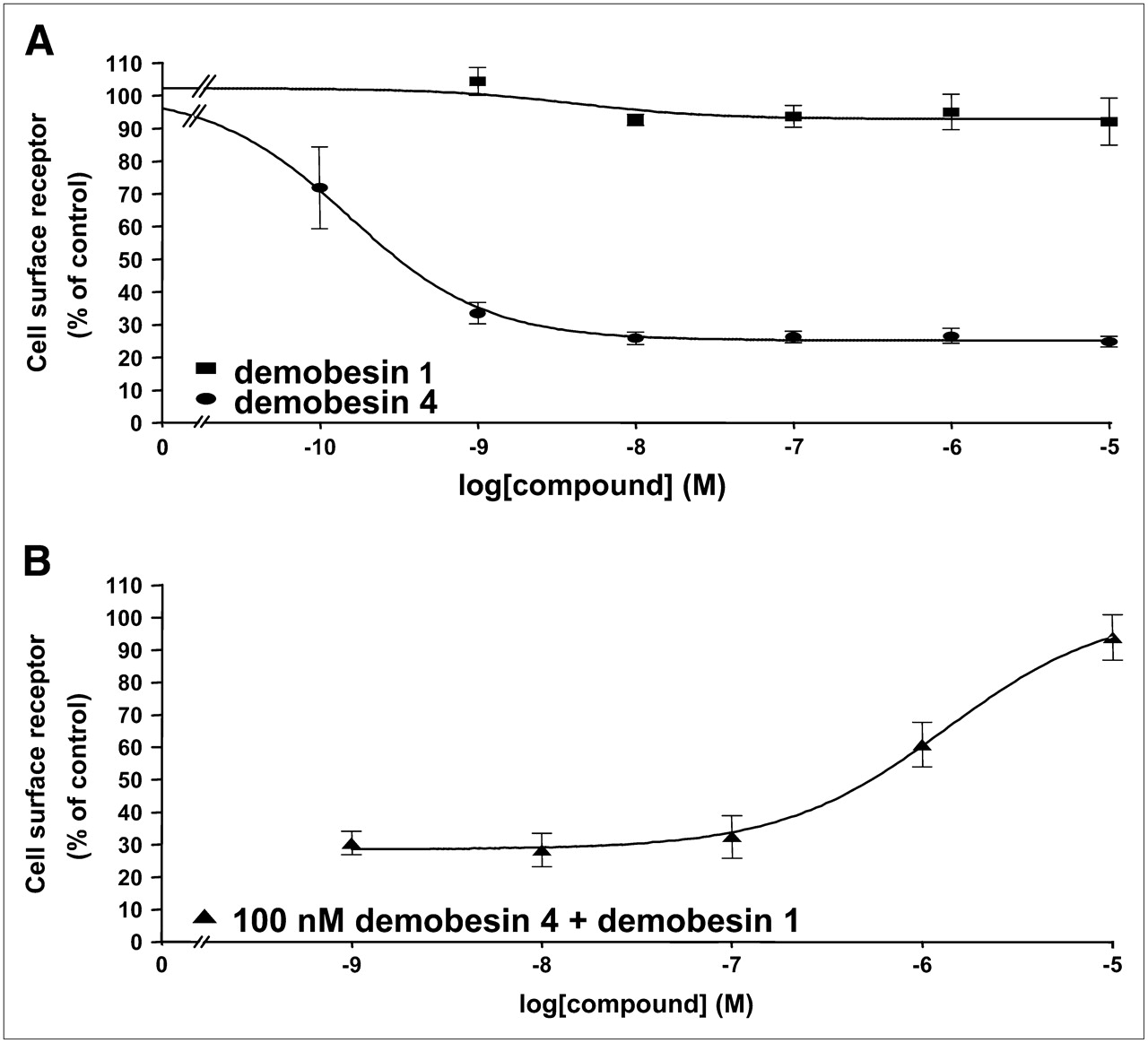
(A) Quantitation of Demobesin 4 and Demobesin 1-induced GRP receptor internalization by ELISA. HEK-GRPR cells were preincubated with mouse monoclonal HA epitope antibody (1:1,000) at room temperature for 2 h. Cells were treated in concentrations ranging from 0.1 nM to 10 μM with Demobesin 4 ( ) or Demobesin 1 (▪) for 30 min at 37°C and 5% CO2, fixed with paraformaldehyde, and then processed for ELISA. Demobesin 4 in picomolar range triggers GRP receptor internalization, whereas Demobesin 1 is inactive up to 10 μM. (B) Dose dependence of antagonist inhibition of GRP receptor internalization. HEK-GRPR cells were treated with increasing concentrations of Demobesin 1 in the presence of 100 nM Demobesin 4 (▴). Cell-surface receptor was measured by ELISA and is expressed as a percentage of untreated cells as described. Demobesin 1 antagonizes the agonistic effect of Demobesin 4.
) or Demobesin 1 (▪) for 30 min at 37°C and 5% CO2, fixed with paraformaldehyde, and then processed for ELISA. Demobesin 4 in picomolar range triggers GRP receptor internalization, whereas Demobesin 1 is inactive up to 10 μM. (B) Dose dependence of antagonist inhibition of GRP receptor internalization. HEK-GRPR cells were treated with increasing concentrations of Demobesin 1 in the presence of 100 nM Demobesin 4 (▴). Cell-surface receptor was measured by ELISA and is expressed as a percentage of untreated cells as described. Demobesin 1 antagonizes the agonistic effect of Demobesin 4.
The internalization properties of the radiolabeled ligands, [99mTc]Demobesin 1 and [99mTc]Demobesin 4, were studied in PC3 and HEK-GRPR cells. In Figure 4 the percentage of internalized radioligand is plotted versus time and reveals a massive difference in internalization between [99mTc]Demobesin 1 and [99mTc]Demobesin 4 in both cell lines. The agonist already achieves a specific internalization of ∼45% within 2 h of incubation in both PC3 and HEK-GRPR cells, whereas the value for the antagonist lies well below 10% in PC3 cells and 5% in HEK-GRPR cells. Of interest is the difference in the radioactivity fraction specifically bound to the cell membranes observed between the 2 agents. As characteristically shown by the corresponding curves with PC3 cells in Figure 4, the antagonist, [99mTc]Demobesin 1, reaches a plateau value of ∼25% at 30 min of incubation, whereas for the agonist, [99mTc]Demobesin 4, the percentage of membrane-bound radioactivity remains stable and never exceeds 10% during the period of the assay. Similar differences are seen with HEK-GRPR cells. Furthermore, internalization assays in PC-3 cells at 37°C revealed an identical behavior between [99mTc/99gTc]Demobesin 1 (isolated by high-performance liquid chromatography as a single-chemical species, applied at a 0.13 nM concentration) and [99mTc]Demobesin 1 (applied as a fraction of the labeling solution in the presence of a high excess of unlabeled Demobesin 1 in a total peptide concentration of 0.13 nM). This finding strongly suggests that incorporation of the radiometal does not alter the internalization behavior of the peptide conjugate.

Internalization of [99mTc]Demobesin 1 in PC3 (A) or HEK-GRPR (C) cells was found significantly lower as compared with massive internalization of [99mTc]Demobesin 4 in PC3 (B) or HEK-GRPR (D). Conversely, a higher percentage of [99mTc]Demobesin 1 remained bound to cell membrane of PC3 or HEK-GRPR cells in comparison with [99mTc]Demobesin 4. Cell-associated radioactivity in presence of 1 μM [Tyr4]bombesin (total nonspecific) remained <1% for both radiopeptides. ▪, % internalized radioactivity; □, % membrane-bound radioactivity; ⋄, % total nonspecific radioactivity.
In Tables 2 and 3, the in vivo biodistribution of the antagonist [99mTc]Demobesin 1 is reported in SCID nude mice bearing GRP receptor–expressing PC3 tumor xenografts and is compared with that of the agonist [99mTc]Demobesin 4. Both agents were capable of rapidly targeting the pancreas as well as the PC3 tumor in a high percentage. Uptake in these GRP receptor–rich tissues was found to be significantly reduced (P < 0.001) in the animals that received excess [Tyr4]bombesin along with the radioligand (in vivo blockade), indicating a GRP receptor–mediated process. However, the specific uptake significantly differed between the 2 agents in these GRP receptor–expressing tissues. Indeed, despite its poor in vitro internalization properties, [99mTc]Demobesin 1 consistently showed a significantly higher PC3 tumor accumulation at all time intervals compared with the massively internalizing [99mTc]Demobesin 4, which showed 2- to 4-fold lower tumor values at 1, 4, and 24 h after injection. Similarly, a 2- to 3-fold higher pancreas uptake was achieved by [99mTc]Demobesin 1 at 1 and 4 h after injection compared with [99mTc]Demobesin 4. Interestingly, the pancreas uptake of [99mTc]Demobesin 1 was rapidly declining to 1.29 %ID/g (percentage injected dose per gram) at 24 h after injection, whereas the agonist [99mTc]Demobesin 4, despite its lower initial pancreas uptake, showed prolonged retention in this organ, with 14.4 %ID/g still found in the pancreas at 24 h after injection. Blood and background clearance was fast for both agents, with the radioactivity excreted into the urine predominantly via the kidneys and the urinary system. [99mTc]Demobesin 1 showed a higher percentage of hepatobiliary excretion with higher liver and bowel values compared with [99mTc]Demobesin 4. It is interesting to note that intestinal uptake of [99mTc]Demobesin 4 could be significantly blocked (P < 0.001) at 4 h after injection by coinjection of excess [Tyr4]bombesin, implying a GRP receptor–mediated mechanism in the bowel wall (12,28). Blockade of intestinal uptake was “masked” in the case of [99mTc]Demobesin 1 as a result of its higher hepatobiliary excretion. The superior PC3 tumor values of [99mTc]Demobesin 1 compared with [99mTc]Demobesin 4 resulted in significantly higher tumor-to-background ratios for the antagonist, especially as far as the kidneys are concerned. In particular, a tumor-to-kidney ratio > 5 at 4 and 24 h with the antagonist represents a considerable improvement over the agonist. Only the tumor-to-liver ratio was found to be in favor of the more-hydrophilic radioligand [99mTc]Demobesin 4.
Biodistribution of [99mTc]Demobesin 1 in Human PC3 Xenograft–Bearing SCID Mice at 1, 4, and 24 Hours After Injection
Biodistribution of [99mTc]Demobesin 4 in Human PC3 Xenograft–Bearing SCID Mice at 1, 4, and 24 Hours After Injection
DISCUSSION
Demobesin 1 was developed a few years ago by us as a potent bombesin radioligand suitable for targeting of GRP receptor–expressing tissues (11). The well-established and commercially available bombesin antagonist [d-Phe6,Leu-NHEt13,des-Met14]bombesin(6–14) was chosen because of its potency and stability (29); a tetraamine 99mTc-binding unit was then covalently attached to the d-Phe6 through a benzylaminodiglycolic acid spacer. It was not known, at that time, whether the modified structure of the chelator-linked peptide would retain the antagonist property of the original peptide or whether it would switch to an agonist. However, the fact that moderate in vitro internalization of the radioligand was observed and that an excellent uptake in vivo into animal tumor models was found suggested that the compound was worth developing. Here, we demonstrate—to our knowledge, for the first time—that Demobesin 1 not only has the characteristics of an excellent bombesin antagonist, but also that its radiolabeled version can be compared advantageously to a similarly potent radiolabeled bombesin receptor agonist as a potential targeting agent for GRP receptor–positive tissues.
A comparison between the agonist Demobesin 4 and the antagonist Demobesin 1 has been performed at several levels in this study. In binding assays, the IC50 values obtained in the receptor autoradiography experiments were in the low nanomolar range and were found to be almost identical for the agonist and the antagonist, indicating that both compounds are comparable in terms of their binding to the GRP receptors. This binding similarity can be considered an excellent prerequisite for the thorough comparison of both peptides in this study. However, functionally, both compounds were found to differ in several instances. Whereas Demobesin 4 stimulated calcium mobilization, Demobesin 1 had no effect; whereas Demobesin 4 did not influence the bombesin-induced calcium mobilization, Demobesin 1 completely abolished it. Similarly, Demobesin 4 triggered a very strong internalization of the GRP receptor proteins, whereas Demobesin 1 had no effect. However, Demobesin 1 abolished completely the agonist-triggered internalization, whereas Demobesin 4 had no visible effect. These 2 different functional assays, defining 2 completely different receptor-mediated cellular pathways—such as signaling and internalization—represent strong in vitro evidence for Demobesin 4 as a GRP receptor agonist and Demobesin 1 as antagonist.
A further comparison was the analysis of the behavior of Demobesin 1 and Demobesin 4 when they are used as radioligands in internalization assays. Whereas the agonist [99mTc]Demobesin 4 massively and rapidly internalizes, the antagonist [99mTc]Demobesin 1, in comparison, internalizes poorly. This difference was observed with PC3 cells and, to an even greater extent, with HEK-GRPR cells. These observations go in the same direction as the in vitro studies measuring the internalization of the GRP receptor after Demobesin 1 or Demobesin 4 application. However, although modest, the internalization of [99mTc]Demobesin 1 is more than one would expect from the immunofluorescence microscopy–based and ELISA-based internalization study. Indeed, in the latter 2 assays, there is no sign of GRP receptor internalization, even after large doses of the peptide. At present, it is unclear whether the weak internalization of [99mTc]Demobesin 1 reflects a weak, but real, internalization process of the antagonist that may not be detected with the less-sensitive immunofluorescence test or whether it is due to another, yet unidentified, phenomenon.
Considering the poor receptor-mediated internalization of Demobesin 1 in the 3 different types of internalization assays, it is remarkable to observe such a high in vivo uptake of this radioligand in GRP receptor–positive tissues. This uptake not only is highly specific, as shown by the successful in vivo competition experiments but also is organ-selective, being restricted to GRP receptor–expressing tissues, such as the pancreas and the PC3 tumor. Even more remarkable, the [99mTc]Demobesin 1 tumor uptake is considerably better than the uptake of the agonist [99mTc]Demobesin 4, itself characterized in vitro by strong internalization properties. The uptake of [99mTc]Demobesin 1 in the PC3 tumor is 4-fold at 4 h, and 2-fold at 24 h, as compared with [99mTc]Demobesin 4. This is a considerable improvement for an in vivo targeting agent—in particular, when we consider that the uptake in the kidney, the major excreting organ for the injected radioactivity, remains comparable with both ligands. The consequence of that is a much improved tumor-to-kidney ratio for [99mTc]Demobesin 1. Furthermore, it is worth noting that the washout of the radioactive antagonist from the PC3 tumor is much longer than the washout from physiologic targets such as the pancreas. This is not the case for the radioactive agonist. This factor further increases the efficiency and tumor selectivity of antagonist labeling and, consequently, favors the use of radiolabeled bombesin antagonists as radiotherapeutic agents.
On the basis of the present in vitro and in vivo data, it is clear that the excellent in vivo uptake of the antagonist Demobesin 1 cannot be due solely to the active internalization of the radioligand into the PC3 tumor. Other mechanisms must be hypothesized. It is well known for various receptors—such as 5-HT2A receptors, corticotropin-releasing factor receptors, or somatostatin receptors—that the respective radioactive ligand antagonists label more receptor sites in vitro than the corresponding radiolabeled agonists (19,30,31). The same may be true for bombesin receptors. However, other possible reasons for an efficient in vivo accumulation of radiolabeled antagonists may exist: Skillfully tailored antagonists may have a slow dissociation rate from the receptor (32,33); moreover, antagonists are often less degradable than agonists and may be less affected by membrane-bound enzymes.
The current bombesin radioligands developed for tumor targeting in the past decade have usually not been characterized functionally as bombesin receptor agonists or antagonists (8,11,13,14), mainly because one has relied primarily on the ability of the radioligand to internalize into the cells as key criterion for drug development. The present study should sensitize nuclear medicine researchers working in this field to include in their future reports a systematic evaluation of the agonistic/antagonistic properties of their novel bombesin analogs, as such knowledge may be of discriminative importance. Adequate functional tests may include, as illustrated in the present study, effects of the peptide ligands on receptor internalization as well as effects on a second-messenger signaling system.
The radiolabeled bombesin analogs proposed so far for targeted radionuclide therapy have been agonists tagged with short- and medium-range β-emitters. Short-range β-emitters are more cytotoxic by delivering most of their energy to the cell nucleus after radioligand internalization and are more effective in the case of micrometastases. However, medium-range β-emitters have been proven to be more efficient for destroying larger tumor masses by virtue of the “cross-fire effect” without requiring radioligand internalization. The longer penetration of the radioactivity destroys neighboring cells and reaches tumor areas that are difficult to access because of necrosis. Consequently, antagonists stably radiolabeled with “hard” β-emitters, such as [188Re]Demobesin 1, may successfully enter the arena of targeted radionuclide therapy of tumors in near future.
High tumor uptake, high tumor-to-kidney ratio, and long tumor washout of radiolabeled GRP receptor antagonists represent highly relevant features for potential radionuclide therapy of GRP receptor–expressing tumors and metastases. Other clinical advantages of GRP receptor antagonists are their reduced physiologic activity and radioactivity accumulation at physiologic GRP targets, implying fewer side effects than with bombesin agonists (34–36). Finally, because bombesin agonists stimulate tumor growth and angiogenesis (37,38), it is probable that the use of radiolabeled GRP receptor antagonists may prevent such tumor-proliferative side effects.
The present study generalizes previous data indicating that somatostatin receptor antagonists are preferable to somatostatin receptor agonists for in vivo tumor targeting (19) by extending these data to a further peptide receptor. Extending this change of paradigm to GRP receptors implies that, in the future, the strategy to develop successful bombesin analogs must move from the design of radiolabeled agonists to that of radiolabeled antagonists, relying for that on the seminal work by the groups of Jensen and Coy (39).
Acknowledgments
This project was financially supported in part by Biomedical Life Sciences, S.A.
Footnotes
-
↵* Contributed equally to this work.
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication July 9, 2007.
- Accepted for publication November 19, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Preclinical Investigation of [212Pb]Pb-DOTAM-GRPR1 for Peptide Receptor Radionuclide Therapy in a Prostate Tumor Model
- A Radiotracer for Molecular Imaging and Therapy of Gastrin-Releasing Peptide Receptor-Positive Prostate Cancer
- MITIGATE-NeoBOMB1, a Phase I/IIa Study to Evaluate Safety, Pharmacokinetics, and Preliminary Imaging of 68Ga-NeoBOMB1, a Gastrin-Releasing Peptide Receptor Antagonist, in GIST Patients
- Imaging the Distribution of Gastrin-Releasing Peptide Receptors in Cancer
- Development of Improved Tumor-Residualizing, GRPR-Targeted Agents: Preclinical Comparison of an Endolysosomal Trapping Approach in Agonistic and Antagonistic Constructs
- Development and Characterization of a Novel, High-Affinity, Specific, Radiolabeled Ligand for BRS-3 Receptors
- Proof of Therapeutic Efficacy of a 177Lu-Labeled Neurotensin Receptor 1 Antagonist in a Colon Carcinoma Xenograft Model
- 68Ga/177Lu-NeoBOMB1, a Novel Radiolabeled GRPR Antagonist for Theranostic Use in Oncology
- Highly Increased 125I-JR11 Antagonist Binding In Vitro Reveals Novel Indications for sst2 Targeting in Human Cancers
- Theranostic Perspectives in Prostate Cancer with the Gastrin-Releasing Peptide Receptor Antagonist NeoBOMB1: Preclinical and First Clinical Results
- Bombesin-Targeted PET of Prostate Cancer
- Comparative Evaluation of the Biodistribution Profiles of a Series of Nonpeptidic Neurotensin Receptor-1 Antagonists Reveals a Promising Candidate for Theranostic Applications
- In Vitro and In Vivo Application of Radiolabeled Gastrin-Releasing Peptide Receptor Ligands in Breast Cancer
- Preclinical Comparison of Al18F- and 68Ga-Labeled Gastrin-Releasing Peptide Receptor Antagonists for PET Imaging of Prostate Cancer
- N-Terminal Modifications Improve the Receptor Affinity and Pharmacokinetics of Radiolabeled Peptidic Gastrin-Releasing Peptide Receptor Antagonists: Examples of 68Ga- and 64Cu-Labeled Peptides for PET Imaging
- Targeting Neuropeptide Receptors for Cancer Imaging and Therapy: Perspectives with Bombesin, Neurotensin, and Neuropeptide-Y Receptors
- A High-Affinity, High-Stability Photoacoustic Agent for Imaging Gastrin-Releasing Peptide Receptor in Prostate Cancer
- In Vivo Imaging of Prostate Cancer Using [68Ga]-Labeled Bombesin Analog BAY86-7548
- PulmoBind, an Adrenomedullin-Based Molecular Lung Imaging Tool
- Bombesin Antagonist-Based Radioligands for Translational Nuclear Imaging of Gastrin-Releasing Peptide Receptor-Positive Tumors
- Evaluation of 177Lu-DOTA-sst2 Antagonist Versus 177Lu-DOTA-sst2 Agonist Binding in Human Cancers In Vitro
- First Clinical Evidence That Imaging with Somatostatin Receptor Antagonists Is Feasible
- In Vitro and In Vivo Evaluation of 64Cu-Labeled SarAr-Bombesin Analogs in Gastrin-Releasing Peptide Receptor-Expressing Prostate Cancer
- 18F-Labeled Bombesin Analog for Specific and Effective Targeting of Prostate Tumors Expressing Gastrin-Releasing Peptide Receptors
- Evaluation of a 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid-Conjugated Bombesin-Based Radioantagonist for the Labeling with Single-Photon Emission Computed Tomography, Positron Emission Tomography, and Therapeutic Radionuclides
- High expression of gastrin-releasing peptide receptors in the vascular bed of urinary tract cancers: promising candidates for vascular targeting applications
- Peptide-Based Probes for Cancer Imaging