


Abstract
Bombesin receptors are overexpressed on a variety of human tumors. In particular, the gastrin-releasing peptide receptor (GRPr) has been identified on prostate and breast cancers and on gastrointestinal stromal tumors. The current study aims at developing clinically translatable bombesin antagonist–based radioligands for SPECT and PET of GRPr-positive tumors. Methods: A potent bombesin antagonist (PEG4-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 [AR]) was synthesized; conjugated to the chelators DOTA, 6-carboxy-1,4,7,11-tetraazaundecane (N4), 1,4,7-triazacyclononane, 1-glutaric acid-4,7 acetic acid (NODAGA), and 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-TE2A); and radiolabeled with 111In, 99mTc, 68Ga, and 64Cu, respectively. The radioconjugates were evaluated in vitro and in vivo in PC-3 tumor–bearing nude mice. Antagonist potency was determined by Ca2+-flux measurements and immunofluorescence. Results: All the conjugates showed high binding affinity to GRPr (inhibitory concentration of 50% [IC50], 2.5–25 nmol/L). The immunofluorescence and Ca2+-flux assays confirmed the antagonist properties of the conjugates. Biodistribution revealed high and specific uptake in PC-3 tumor and in GRPr-positive tissues. Tumor uptake of 64Cu-CB-TE2A-AR (31.02 ± 3.35 percentage injected activity per gram [%IA/g]) was higher than 99mTc-N4-AR (24.98 ± 5.22 %IA/g), 111In-DOTA-AR (10.56 ± 0.70 %IA/g), and 68Ga-NODAGA-AR (7.11 ± 3.26 %IA/g) at 1 h after injection. Biodistribution at later time points showed high tumor-to-background ratios because of the fast washout of the radioligand from normal organs, compared with tumor. High tumor-to-background ratios were further illustrated by PET and SPECT images of PC-3 tumor–bearing nude mice acquired at 12 h after injection showing high tumor uptake, clear background, and negligible or no radioactivity in the abdomen. Conclusion: The chelators do influence the affinity, antagonistic potency, and pharmacokinetics of the conjugates. The promising preclinical results warrant clinical translation of these probes for SPECT and PET.
Peptide receptors are important targets for imaging and targeted radionuclide therapy of cancer (1–4). Receptors of the bombesin family are of particular interest because they are overexpressed on a variety of human cancers. In particular, the gastrin-releasing peptide receptor (GRPr) has been identified on prostate (5) and breast (6) cancers, on gastrointestinal stromal tumors (7), and on peritumoral vessels of ovarian cancer (8). Markwalder and Reubi (5) demonstrated GRPr expression on 30 of 30 invasive primary prostatic carcinomas with high density in most cases. Therefore, a variety of radiolabeled bombesin derivatives were developed and studied preclinically (9–11) and clinically (12,13). Despite some promising results, none of them seem to have provided a breakthrough in the clinic so far.
For therapeutic studies, bombesin(7-14) was conjugated to DOTA via a gly-4-aminobenzoic acid group and labeled with 177Lu. This compound showed promising results in preclinical models (14,15) and was studied in clinical settings (phase I) (16). Side effects during a first phase I dose escalation study included abdominal cramps, diarrhea, and nausea (16). We and others hypothesized that these side effects may be absent when using bombesin-based antagonists (17–20). In addition, agonists of the bombesin family were shown to have mitogenic properties (21).
For these reasons and because we have recently shown that radiolabeled somatostatin-based antagonists have superior pharmacokinetic properties in tumor models (22), we started a program toward developing antagonistic radiovectors targeting the GRPr (19,20).
In this work, we were particularly interested in performing a systematic study using the most important metallic radionuclides labeled to a potent bombesin antagonist '(PEG4-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 [AR]). Metallic radionuclides may have the potential advantage of allowing for kit formulations, which may be used by the addition of radionuclide solutions. We developed conjugates for labeling with 64Cu (4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo-[6.6.2]hexadecane [CB-TE2A]-AR) and 68Ga (1,4,7-triazacyclononane, 1-glutaric acid-4,7 acetic acid [NODAGA]-AR) for PET and with 111In (DOTA-AR) and 99mTc (6-carboxy-1,4,7,11-tetraazaundecane [N4]-AR) for SPECT (Fig. 1).
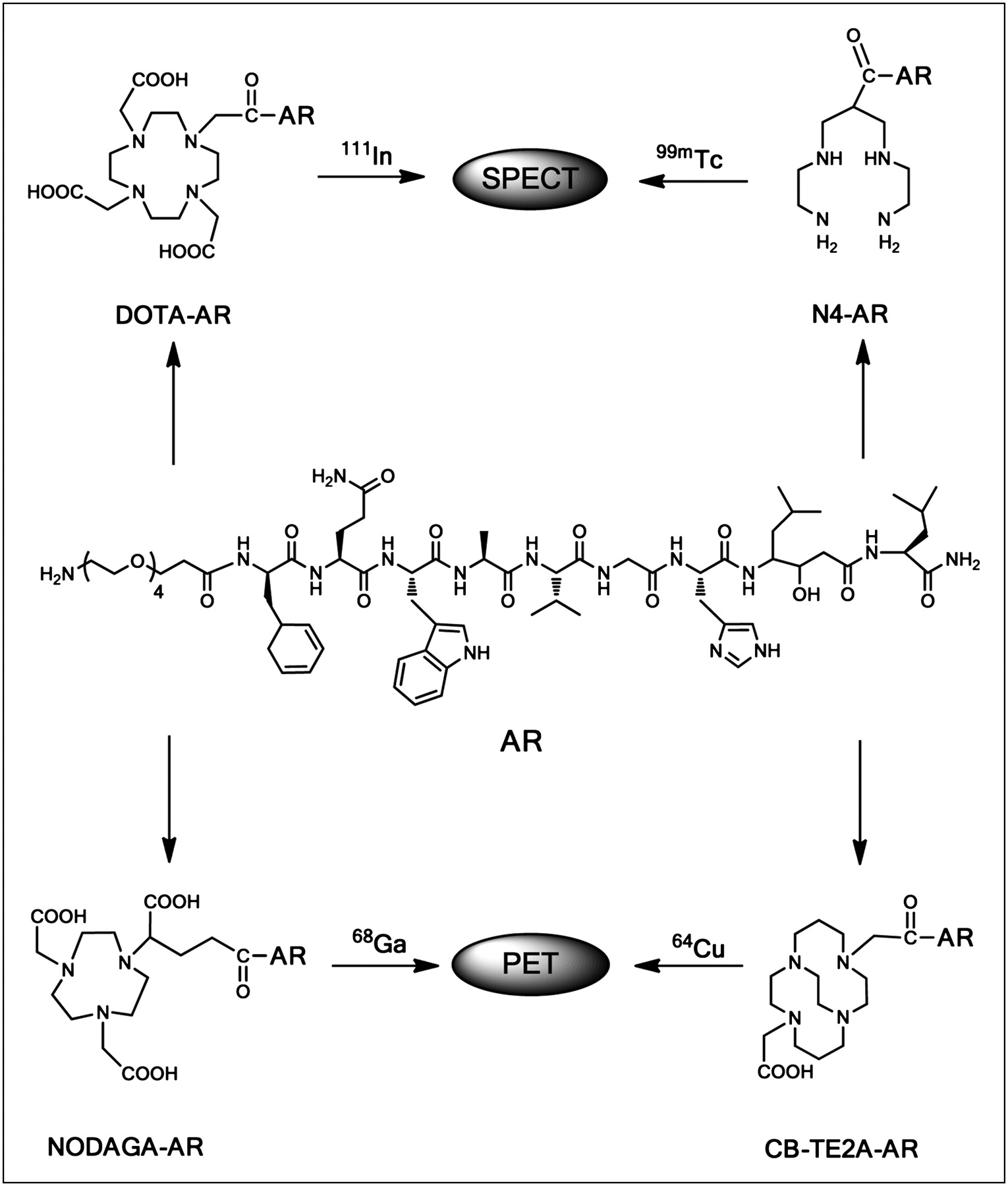
Structure of conjugates DOTA-AR, NODAGA-AR, CB-TE2A-AR, and N4-AR.
64Cu is widely used because it can be produced easily on a medical cyclotron (23). A variety of 64Cu-labeled bioactive molecules, such as folate derivatives (24), antibodies (25), and peptides (26), were developed for PET studies. Past experience indicated the high importance of the use of suitable bifunctional chelators for the stable in vivo complexation of 64Cu to avoid premature release of the radiometal (26). The generator-produced 68Ga is attracting great interest as a metallic positron emitter for PET. 68Ga-labeled peptides found their way into the clinic many years ago (27) and are used successfully. 99mTc is the workhorse of the nuclear medicine physician, and 111In is being used as an important SPECT label and as a surrogate of β-emitters such as 90Y and radiolanthanides.
The important outcome of this work is that antagonist potency and receptor affinity are conserved after modification with spacers and chelators, but both parameters are important determinants of overall performance. All radiovectors seem to be suitable for clinical translation, offering a broad range of molecular imaging possibilities. The results encouraged us to start early clinical use in primary prostate cancer patients using 64Cu-CB-TE2A-AR.
MATERIALS AND METHODS
The supplier information for all reagents, radionuclides, and generators and details of instruments used are provided in the supplemental data (available online only at http://jnm.snmjournals.org).
Cell Lines
Human embryonic kidney (HEK) 293 cells, stably expressing the hemagglutinine-epitope–tagged human GRPr (HEK-GRPr), were generated and cultured as previously described (17). The identity of the cells has been confirmed throughout the studies by determination of the receptor density and the growth pattern in vitro and in vivo. Human prostate cancer cells (PC-3) were obtained from American Type Culture Collection and cultured at 37°C and 5% CO2 either in Ham F12K or in Dulbecco modified Eagle medium containing 2 mM l-glutamine and supplemented with 10% (v/v) fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL). All culture reagents were from Invitrogen or BioConcept.
Synthesis and Radiolabeling of Chelator–Peptide Conjugates
The peptide–chelator conjugates were synthesized manually according to standard Fmoc chemistry (28) using Rink amide 4-methylbenzhydrylamine resin. The spacer and chelators were consecutively coupled to the peptide using 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate as an activating agent. The cleavage of the peptides and simultaneous deprotection of the side chain–protecting groups were performed using trifluoroacetic acid/thioanisole/triisopropylsilane/H2O (95/2/2/1). The crude conjugates were further purified by semipreparative high-performance liquid chromatography (HPLC). The analytic characterization data of the purified conjugates is provided in Supplemental Table 1.
Radiolabeling of Chelator–Peptide Conjugates
The radioligands were prepared as described in detail in the supplemental data. Briefly, 111In-DOTA-AR and 64Cu-CB-TE2A-AR were prepared by dissolving 10 μg of the peptides in acetate buffer followed by incubation with 111InCl3 (100–180 MBq) or 64CuCl2 (100–120 MBq) for 30 min at 95°C. Purified 68Ga(III) (250–300 MBq) and 67GaCl3 (100–180 MBq) were used for the labeling of NODAGA-AR-02 (10 μg) in NH4-acetate buffer (0.2 mol/L, pH 4.0), followed by incubation for 10 min at room temperature. For 99mTc-N4-AR preparation, N4-AR (1 mmol/L) was dissolved in a mixture (8:2 v/v) of acetic acid (50 mmol/L) and ethanol. A reaction mixture of phosphate buffer (0.5 mol/L, pH 11.5; 50 μL) and sodium citrate (0.1 mol/L, 5 μL), Na99mTcO4 generator eluate (650–750 MBq, 700 μL), N4-AR solution (20 nmol, 20 μL), and freshly prepared SnCl2 solution in ethanol (25 mg, 25 μL) was incubated at room temperature for 30 min. All radiolabeled peptides were analyzed with analytic HPLC. For biodistribution, the radioligands were diluted with 0.9% NaCl (0.1% bovine serum albumin).
Binding Affinity Measurements
The inhibitory concentration of 50% (IC50) values were determined by in vitro GRPr autoradiography on cryostat sections of well-characterized prostate carcinomas, as described previously (5). The radioligand used was 125I-Tyr4-bombesin, known to preferentially label GRPrs (29).
Cellular Uptake Kinetics
PC-3 cells were seeded into 6-well plates overnight (0.8–1.0·106 cells per well). On the day of the experiment the medium was removed and the cells were washed twice with fresh medium (Dulbecco modified Eagle medium, 1% fetal bovine serum, pH 7.4) and incubated for 1 h at 37°C. Approximately 3 kBq of the radioligand (0.25 pmol) were added to the medium, and the cells were incubated (in triplicates) for 0.5, 1, 2, and 4 h at 37°C, 5% CO2. A 1,000-fold excess of each blocking agent was used to determine nonspecific internalization. At each time point, the cells were treated exactly as described recently (11).
The Fate of GRPr-Bound Radiopeptides In Vitro
PC-3 cells were seeded into 6-well plates and treated as described in the “Cellular Uptake Kinetics” section. The plates were placed on ice for 30 min; an excess of blocking agent was added to selected wells to determine nonspecific binding. The radioligands (0.25 pmol, 3 kBq) were added to the medium and allowed to bind to the cells for 2 h at 4°C. After the incubation, the cells were washed twice with ice-cold phosphate-buffered saline, and 1 mL of fresh prewarmed culture medium was added to each well, followed by incubation for 10, 20, and 30 min and 1, 2, and 4 h (37°C, 5% CO2). At each time point, the plates were treated exactly as described recently (11).
Immunofluorescence Microscopy
Immunofluorescence microscopy–based internalization assays with HEK-GRPr–expressing cells were performed as previously described (17). The cells were treated either with 10 nmol of bombesin per liter or with 1 μmol of all conjugates per liter. Or, to evaluate potential antagonism, cells were treated with 10 nmol of bombesin per liter in the presence of a 100-fold excess of all conjugates for 30 min at 37°C, 5% CO2, in growth medium and then processed for immunofluorescence microscopy using the mouse monoclonal hemagglutinine-epitope antibody at a dilution of 1:1,000 as a first antibody (Covance) and Alexa Fluor 488 goat antimouse IgG (H+L) at a dilution of 1:600 as a secondary antibody (Molecular Probes). The cells were imaged using a Leica DM RB immunofluorescence microscope and an Olympus DP10 camera.
Calcium Mobilization Assay
Intracellular calcium mobilization was measured in PC-3 cells using the Fluo-4NW Calcium Assay kit (Molecular Probes) as described previously (17). Briefly, PC-3 cells were seeded (10,000 cells per well) in 96-well plates and cultured for 2 d at 37°C, 5% CO2. On the day of the experiment, the cells were washed with assay buffer (1× Hank's balanced salt solution, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)) containing 2.5 mmol of probenecid per liter. The cells were loaded with Fluo-4NW dye (100 μL/well) in assay buffer for 30 min at 37°C and 5% CO2 and for 30 min at room temperature. The dye-loaded cells were transferred to a SpectraMax M2e (Molecular Devices), and intracellular Ca2+ mobilization was recorded in a kinetic experiment for 60 s at room temperature monitoring fluorescence emission at 520 nm (λex = 485 nm, where λex is the excitation wavelength) in the presence of the compounds when applied at 1 μmol/L. Maximum fluorescence was measured after the addition of 25 μmol of ionomycin per liter (17). All experiments were repeated at least 3 times in triplicates.
Biodistribution in PC-3 Tumor–Bearing Nude Mice
All animal experiments were performed in compliance with the Swiss regulations (permit 789).
Female nude mice were implanted subcutaneously with 106 PC-3 cells, which were freshly expanded in sterilized phosphate-buffered saline (pH 7.4). Eleven days after inoculation, the tumors grew to a size of 4–7 mm. The mice were injected with 10 pmol of the radiotracers (0.15–0.40 MBq, 100 μL) via the tail vein. For the determination of nonspecific uptake in the tumor or receptor-positive organs, a group of 4 animals was preinjected (5 min) with 20 nmol of unlabeled peptide. Mice were sacrificed at 1, 4, and 24 h, and the organs of interest were collected, rinsed of excess blood, weighed, and counted in a γ-counter. The biodistribution studies of all radioconjugates were performed accordingly. The percentage injected activity per gram (%IA/g) was calculated for each tissue. For biodistribution studies of 68Ga-NODAGA-AR, mice were sacrificed at 1 h after injection.
SPECT/CT Using 111In-DOTA-AR and 99mTc-N4-AR
PC-3 tumor–bearing nude mice were injected with 5 MBq of 111In-DOTA-AR (150 pmol) and 15 MBq of 99mTc-N4-AR (200 pmol). The mice were anesthetized 12 h after injection, and images were acquired using a clinical SPECT/CT camera (Symbia T2; Siemens). Iteratively reconstructed SPECT images (4 subsets, 8 iterations) were fused with 3-dimensional reconstructed images from the CT study (two 1.25-mm slices, 130 kV, 48 mA).
PET/CT Using 68Ga-NODAGA-AR and 64Cu-CB-TE2A-AR
PC-3 tumor–bearing nude mice were sacrificed 1 h after injection of 2 MBq of 68Ga-NODAGA-AR (150 pmol) and 12 h after injection of 1.5 MBq of 64Cu-CB-TE2A-AR (150 pmol), and images were acquired using a clinical PET/CT scanner (Discovery STE; GE Healthcare). PET emission events were collected in 3-dimensional scanning mode (septa out) over 60 min. The acquired data were corrected for 68Ga decay and random events and reconstructed using the manufacturer's 3-dimensional ordered-subset expectation maximization algorithm. The images were fused with 3-dimensional reconstructed images from the CT study (sixteen 0.625-mm slices, 120 keV, 320 mA).
Statistical Analysis
Data are expressed as mean ± SD, calculated on Microsoft Excel. Prism 5 software (GraphPad Software) was used to determine statistical significance at the 95% confidence level, with a P value of less than 0.05 being considered significantly different.
RESULTS
Chemistry and Radiochemistry
The synthesis yields of the peptide–chelator conjugates ranged from 30% to 35%. All conjugates showed purities greater than 95%, as confirmed by HPLC; identities were confirmed by mass spectroscopy (Supplemental Table 1). 68Ga, 67Ga, and 99mTc were labeled at room temperature (30 min), with radiolabeling yields greater than 97% at specific activities of 50, 30, and 37 GBq μmol−1, respectively. 111In- and 64Cu-labeled conjugates were obtained by incubation at elevated temperature (95°C, 30 min), with labeling yields of 95% or greater at a maximum specific activity of 30 and 25 GBq μmol−1, respectively.
Binding Affinity
Compared with the reference peptide JMV594 (30) (IC50, 5.6 ± 1.8 nmol/L), DOTA-AR and NODAGA-AR still retained reasonable affinity to the GRPr (IC50, 18 ± 7 and 25 ± 6 nmol/L, respectively). The IC50 value of CB-TE2A-AR (IC50, 5.5 ± 1.3 nmol/L) was comparable to JMV594, whereas N4-AR showed the highest binding affinity (IC50, 2.5 ± 0.6 nmol/L).
Cellular Uptake Kinetics
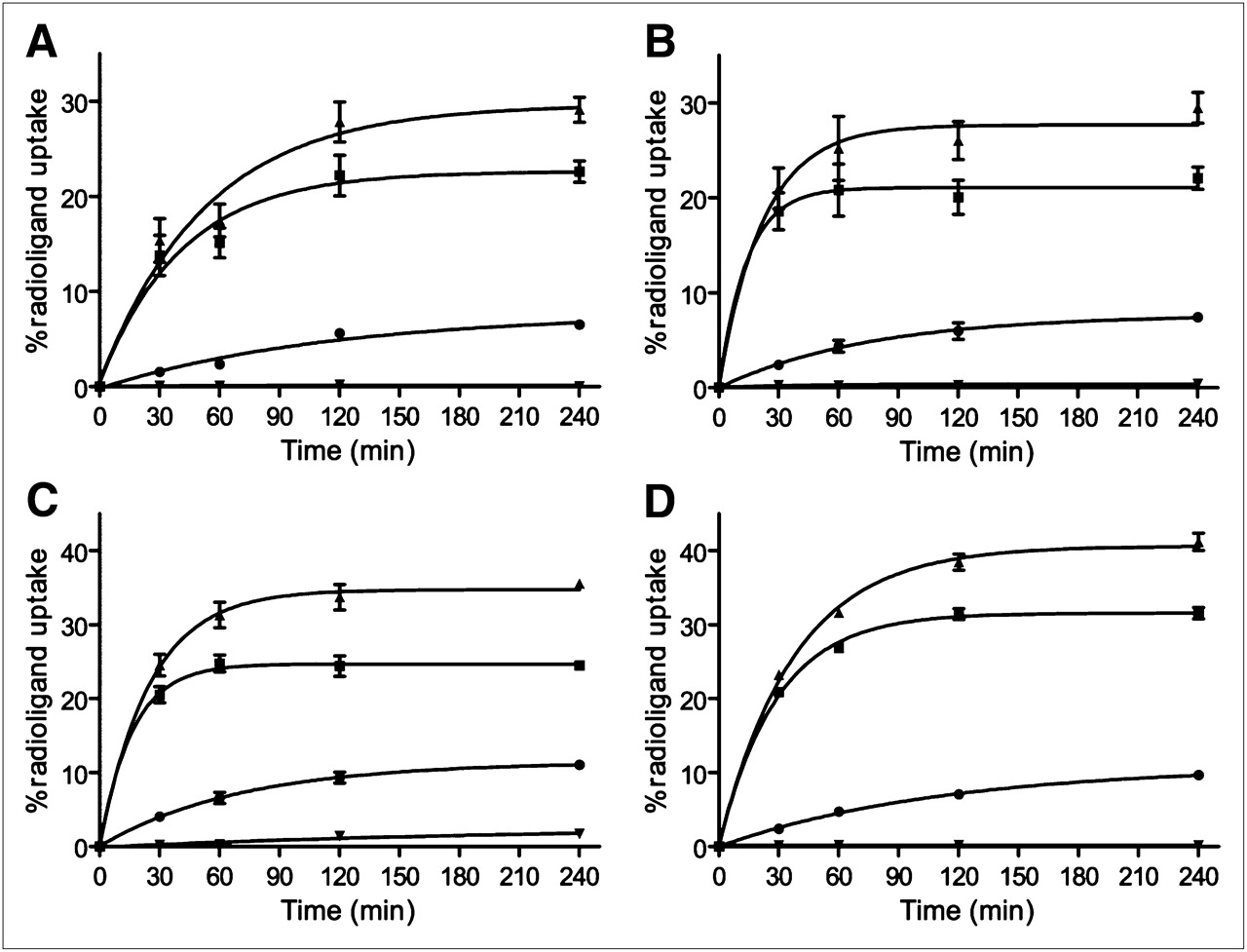
All conjugates showed specific and time-dependent cellular uptake at 37°C (Fig. 2). Blocking studies demonstrated that the uptake was receptor-mediated. 111In-DOTA-AR and 67Ga-NODAGA-AR showed rapid binding to the membrane receptors. Within 2 h of incubation, the specifically bound fraction leveled off at 20%. The specifically bound radiopeptide was 24.3% ± 1.4% for 64Cu-CB-TE2A-AR and 31.4% ± 0.7% for 99mTc-N4-AR. For all conjugates, the internalized fraction was less than 10% at 4 h.
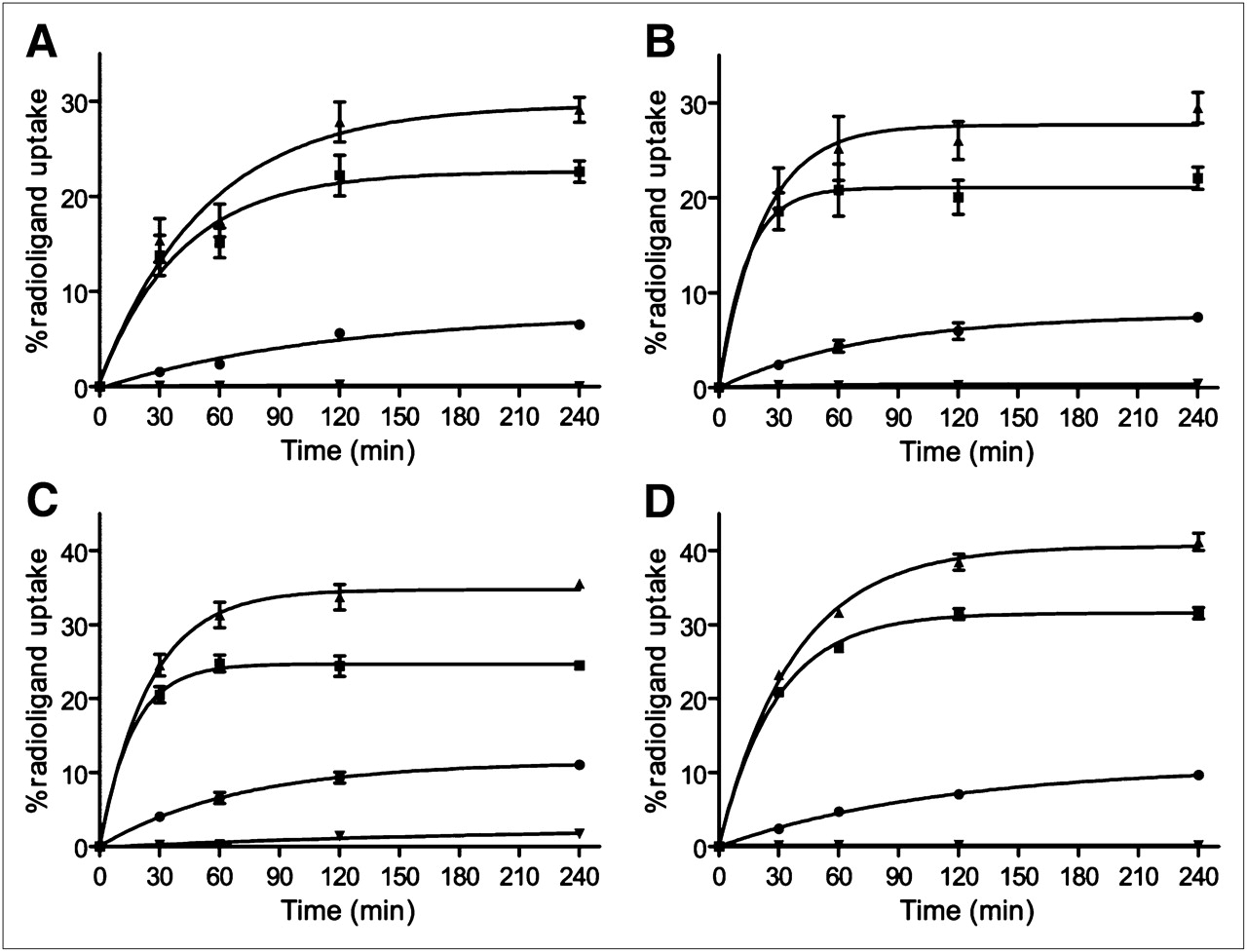
Cellular uptake profile of 111In-DOTA-AR (A), 67Ga-NODAGA-AR (B), 64Cu-CB-TE2A-AR (C), and 99mTc-N4-AR (D) as measured with PC-3 cells. Percentage of specific bound (■), specific internalized (•), and total specific cell uptake (▴) was determined with respect to total added activity. Nonspecific uptake (▾) was determined by pretreating cells with 1,000-fold excess of blocking agent (BIM26226) before adding radioligand. Values and SD are result of 2 independent experiments, with triplicates in each experiment.
The Fate of GRPr-Bound Radiopeptides In Vitro
The fate of the receptor-bound radiopeptides was studied by a temperature shift experiment; the 4 radiotracers showed similar behavior in that they presented a stable receptor–ligand interaction (Supplemental Fig. 1). 67Ga-NODAGA-AR showed the highest dissociated fraction (∼50%), whereas the 64Cu and 99mTc conjugates bound more strongly, with almost 60% of the peptide still bound to the receptor after 4 h at 37°C. The internalized fraction after 4 h at 37°C was less than 20% for all radiotracers.
Immunofluorescence Microscopy
The antagonistic properties of the conjugates were assessed by immunofluorescence-based internalization assay using HEK-GRPr–expressing cells. Figure 3A shows that 10 nmol of bombesin per liter were able to trigger receptor internalization. None of the 4 conjugates was able to stimulate GRPr internalization, even at a concentration of 1,000 nmol/L. However, at a concentration of 1,000 nmol/L together with 10 nmol of bombesin per liter, the peptides were able to prevent the bombesin-induced receptor internalization.

(A) Immunofluorescence microscopy–based internalization assay with HEK-GRPr cells, DOTA-AR (c, d), NODAGA-AR (e, f), CB-TE2A-AR (g, h), and N4-AR (i, j). Control experiment shows membrane-bound GRPr in absence of peptide (a); bombesin agonist (10 nmol/L) triggers massive GRPr internalization (b). Antagonist conjugates failed to induce GRPr internalization, even at concentration of 1 μmol/L (c, e, g, and i); antagonist conjugates at a concentration of 1 μmol/L efficiently blocked bombesin agonist–mediated GRPr internalization (d, f, h, and j). (B) Dose–response curves of bombesin analogs determined by calcium release assay as described in “Materials and Methods” section. All conjugates behave like antagonists, shifting dose–response curve of bombesin to higher molar range when applied at 1 μmol/L. Data are expressed as percentage of maximum calcium response induced by ionomycin.
Calcium Mobilization Assay
The Ca2+ mobilization assay allows determining quantitatively bombesin antagonist potency. Dose–response curves of the 4 conjugates in PC-3 cells are shown in Figure 3B. All 4 peptides behaved as antagonists, shifting the dose–response curve of Tyr4-bombesin (10 μmol/L) to a higher molar range. The CB-TE2A-AR and N4-AR showed a pronounced shift to the right, confirming the superior antagonist potency of these 2 peptides. Moreover, tested alone at 1 and 10 μmol/L, the peptides did not cause an intracellular Ca2+ flux (Fig. 3B).
Biodistribution
The comparative pharmacokinetics of the 4 radiopeptides are summarized in Table 1, and relevant tumor–to–normal-tissue ratios are summarized in Table 2. All 4 radiopeptides showed fast blood clearance (only 0.02 ± 0.001–0.27 ± 0.07 %IA/g left in blood at 4 h after injection). The tumor uptake was specific, as shown with blocking experiments, and ranged between 7.1 ± 3.3 and 31.0 ± 3.3 %IA/g at 1 h after injection for 4 radiopeptides. The tumor uptake depends on the chelate attached to the peptide and is higher for the 2 peptides that have higher binding affinity and higher antagonist potency. Tumor uptake was reduced by 80%–98.5% by preinjection of a 1,000-fold excess of cold peptide (Supplemental Table 2).
Biodistribution of 111In-DOTA-AR, 68Ga-NODAGA-AR, 64Cu-CB-TE2A-AR, and 99mTc-N4-AR in Human PC-3 Tumor–Bearing Nude Mice
Tumor-to-Tissue Ratios
64Cu-CB-TE2A-AR and 99mTc-N4-AR cleared somewhat more slowly, but at 4 h after injection there was little difference seen among the 4 radiopeptides. Tumor-to-blood ratios were high already at short time points and increased with time for all radiopeptides. An early high and specific uptake of the radiotracers was also found in the pancreas and the intestine, both known to express GRPr. The latter 2 organs show a much faster washout than the tumor.
A fast washout was also observed from the kidneys, in particular for 64Cu-CB-TE2A-AR, which showed 6.53 ± 0.52 %IA/g at 1 h and 0.41 ± 0.07 %IA/g at 24 h. Similarly, the kidney uptake of 99mTc-N4-AR dropped from 5.92% to 0.85 %IA/g in the same time interval. The 111In and 67/68Ga peptides had a low kidney uptake already at the 1-h time point (Table 1; Supplemental Fig. 2).
Imaging Studies
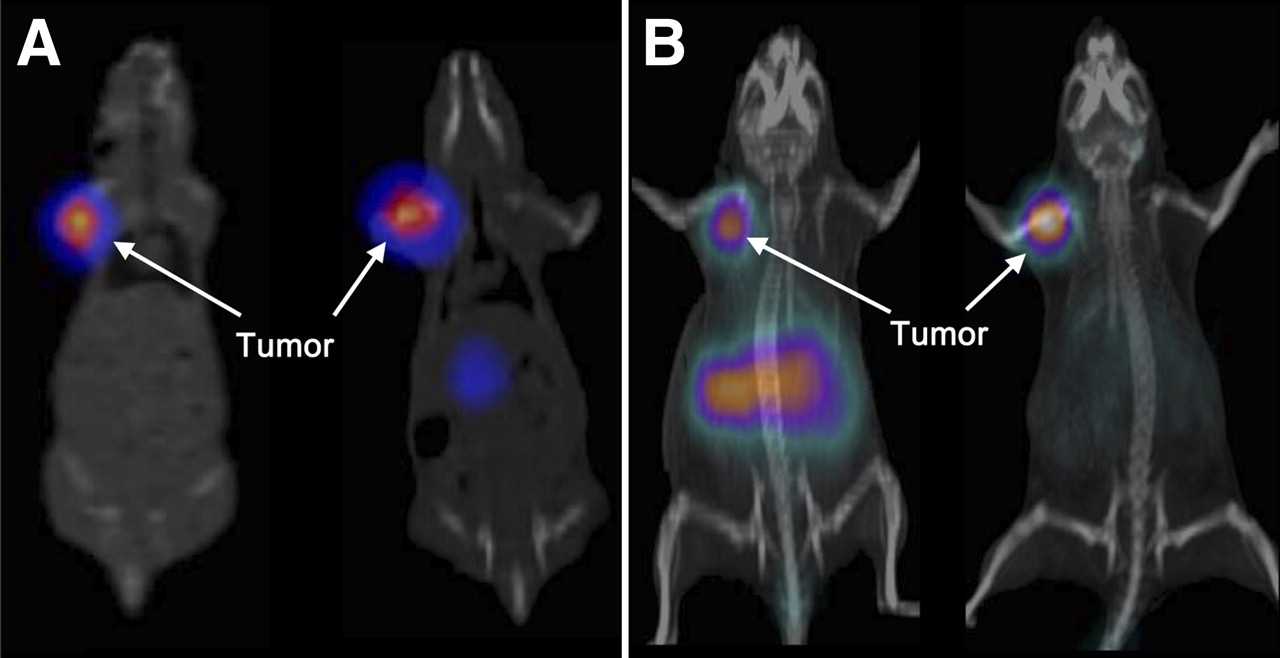
SPECT/CT images acquired at 12 h after injection clearly showed high tumor uptake and fast washout from other organs for both 111In-DOTA-AR and 99mTc-N4-AR (Fig. 4A). PET/CT images were acquired at 1 h after injection for 68Ga-NODAGA-AR and at 12 h for 64Cu-CB-TE2A-AR (Fig. 4B). 68Ga-NODAGA-AR exhibited high uptake in the abdomen, mainly because of pancreas uptake, and the image of 64Cu-CB-TE2A-AR at the late time point (12 h) resulted in an excellent tumor-to-background ratio due to washout of the radioligand from all organs except the tumor.
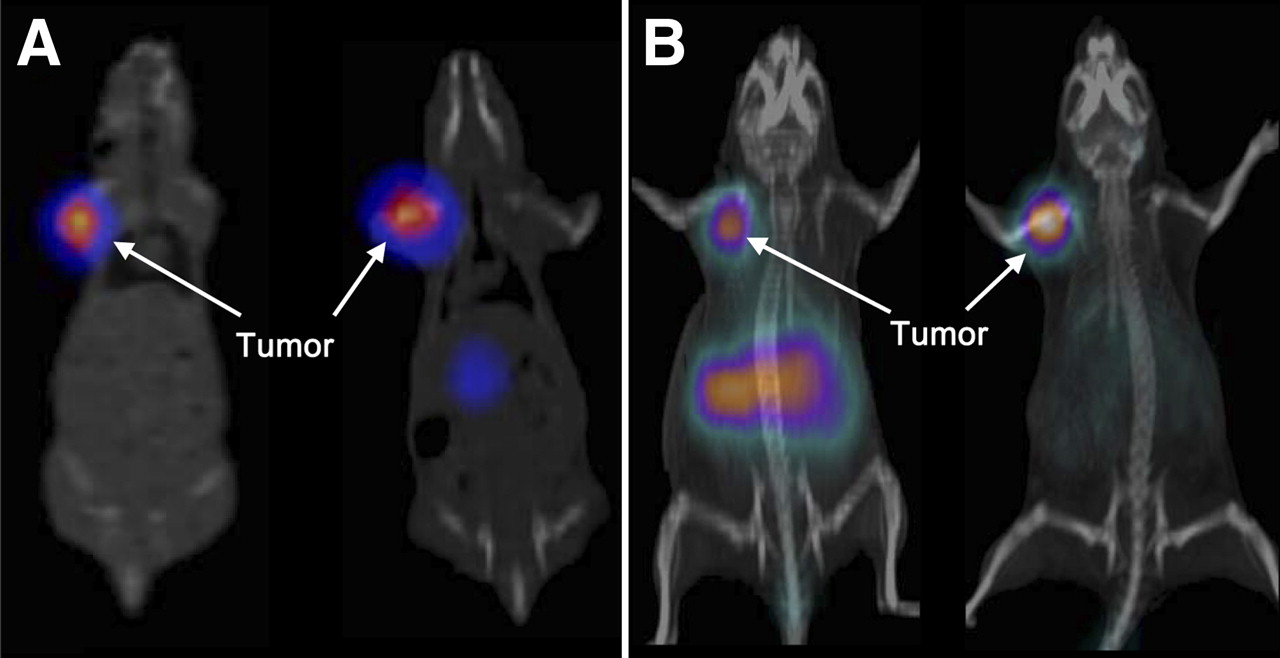
(A) SPECT/CT images of PC-3 tumor–bearing nude mice at 12 h after injection of 99mTc-N4-AR (left) and 111In-DOTA-AR (right). (B) PET/CT images of PC-3 tumor–bearing nude mice at 1 h after injection of 68Ga-NODAGA-AR (left) and 12 h after injection of 64Cu-CB-TE2A-AR (right).
DISCUSSION
Because of the overexpression of GRPr on a variety of major human tumors, development of radiolabeled bombesin peptides paves an excellent way for targeted imaging and radionuclide therapy of GRPr-positive tumors. Over the past 2 decades, several groups, including our laboratory, reported several radiolabeled agonist–based bombesin peptides (9,11,26,31,32). Although these agonists were believed to be ideal targeting ligands because of the internalization of the receptor–agonist complexes, their high and sustained uptake in the normal receptor–positive organs resulted in unfavorable pharmacokinetics. Recently, radiolabeled bombesin-based antagonists showed improved pharmacokinetics in animal models bearing PC-3 or LnCap tumors (17–20,33). In the bombesin family field, use of antagonists may have an additional benefit because agonists were shown to be mitogenic. The ideal targeting properties of bombesin antagonists such as high tumor uptake, fast washout from the normal organs, and improved stability have attracted our interest during the last few years and boosted our efforts toward clinical translation of these molecules for targeted imaging and radionuclide therapy. Among the several bombesin antagonists studied in our laboratory, the statine-based analogs showed good pharmacokinetics.
With this background, we designed the current study to develop clinically translatable bombesin antagonists by conjugating AR to different chelators. Conjugation of different chelators allows labeling of AR with a variety of radionuclides and subsequent application in different imaging modalities (SPECT/CT and PET/CT) but also in targeted radionuclide therapy after intelligent replacement of the imaging radionuclides with surrogate therapeutic radionuclides, arriving at theranostic pairs. We used different chelators and conjugation strategies for this purpose: DOTA, as a monoamide, coupled for 111In labeling for SPECT (111In3+ was shown to be a close surrogate of the β-emitters 177Lu3+ and 90Y3+ (11)); NODAGA for 67/68Ga labeling; CB-TE2A for 64Cu and its β-emitting surrogate 67Cu; and finally N4 for the labeling with 99mTc and possibly with its matched-pair 186/188Re β-emitters (17).
An important conclusion of our study is that even after N-terminal modifications (coupling of PEG4 as pharmacokinetic modulator plus chelator), all 4 peptide conjugates retained significant binding affinity to the GRPr. However, side-by-side comparison showed that conjugation of different chelators to the same bombesin antagonist peptide, AR, significantly influenced the receptor affinity. This may be due in part to the charge on the N terminus; positive charges appear to improve affinity if used along with agonists (34). At this time, there is no explanation for this improvement.
In addition, the 4 peptides were shown to hold the antagonistic potency of the parent peptide. Bombesin-triggered Ca2+ mobilization was successfully antagonized by all peptides; the half maximal effective antagonist concentration values correlated well with the IC50 values.
The in vitro assay showed that all 4 radioligands have a similar cellular uptake profile, for which a high amount of radioligand specifically binds to the receptors and induces internalization of receptor to a lesser extent (Fig. 2). The binding-dissociation experiments showed that on binding to the receptor (at 4°C for 2 h), increase in temperature (37°C) and exposure to fresh medium resulted in 2 such phenomena as dissociation and internalization of the radioligand (Fig. 2). The antagonist 99mTc-N4-AR binds strongly to the receptors, showing less dissociation than with the other radioligands, whereas, compared with other radioligands, the amount of 67Ga-NODAGA-AR dissociated is greater than 40%.
The pharmacokinetic data show similarities but also remarkable differences among the 4 radiopeptides. All show fast blood clearance and specific uptake in tumor and physiologically GRPr-expressing organs such as the pancreas, intestine, and adrenals. The uptake in the tumor and other GRPr-positive organs correlated not only with IC50 values but also with antagonist potency. The retention in the tumor seems to correlate with the rate of dissociation from the receptor in cultured cells.
The 4 radiopeptides may be grouped according to their tumor uptake; 64Cu-CB-TE2A-AR and 99mTc-N4-AR are outstanding in this regard. They show high tumor uptake at early time points, which stays or even increases over 4 h after injection. The tumor uptake of 99mTc-N4-AR is high even at 24 h. Compared with previously reported N4-conjugated bombesin antagonists, substitution of PEG4 for the gly-4-amino benzoyl spacer leads to significantly improved tumor–to–normal-tissue ratios (33). Also compared with the potent bombesin antagonist 99mTc-demobesin 1, 99mTc-N4-AR shows higher tumor–to–normal-organ ratios at all time points in the PC-3 tumor model (17). If the 186/188Re congener complex shows the same or similar pharmacokinetics, this therapeutic radiopeptide may be of relevance for targeted radionuclide therapy of GRPr-positive tumors.
64Cu-CB-TE2A-AR shows high uptake similar to that of 99mTc-N4-AR at 1 and 4 h, but faster washout was seen at 24 h. Among different chelating systems used for 64Cu labeling, the CB-TE2A chelator showed high in vivo stability of 64Cu2+ complexes, resulting in improved pharmacokinetics of the radioconjugates (35). Low liver uptake is considered an indication of high in vivo stability, which was also shown using the 64Cu-NOTA-8-Aoc-BBN(7-14) derivative (26). On the other hand, high uptake and retention in liver tissue is found when DOTA is chosen as chelator; Garrison et al. (35) showed that the bombesin agonist 64Cu-CB-TE2A-8-Aoc-BN(7-14) possesses high in vivo stability with significantly lower liver uptake than 64Cu-DOTA-8-Aoc-BN(7-14). The current study demonstrates that 64Cu-CB-TE2A-AR, compared with 64Cu-CB-TE2A-8-Aoc-BN(7-14), not only showed high in vivo stability but also high tumor uptake. Hence, the combination of bombesin antagonist with CB-TE2A ligand for the development of 64Cu-based PET probes combines advantages such as high tumor uptake, favorable pharmacokinetics, and improved in vivo stability of the radiometal complex. 64Cu-CB-TE2A-AR is the first 64Cu-labeled bombesin antagonist showing highly promising preclinical data. In addition, on the basis of these properties, one may conclude that 67Cu-CB-TE2A-AR is a promising therapeutic radiopharmaceutical.
Both 111In-DOTA-AR and 68Ga-NODAGA-AR showed lower uptake in the tumor and receptor-positive organs than did 64Cu-CB-TE2A-AR and 99mTc-N4-AR—a finding that is in accordance with the in vitro binding affinity results, in which CB-TE2A-AR and N4-AR, compared with DOTA-AR and NODAGA-AR, showed high affinity to GRPr. Although, showing relatively low tumor uptake, 68Ga-NODAGA-AR may still be interesting for clinical application as a PET agent because this conjugate is labeled with 68Ga at room temperature with high specific activity.
Conjugation of potent peptides to DOTA allows stable complexation with a variety of +3 metal ions, facilitating application of the resulting radioligand for both imaging and targeted radionuclide therapy. Compared with reported DOTA-conjugated bombesin antagonists, 111In-DOTA-AR shows improved tumor–to–normal-tissue ratios at all time points (18–20). Therefore, in addition to SPECT, DOTA-AR can be a useful targeted therapeutic agent, with immediate clinical translational properties because it can be easily labeled with 177Lu and 90Y.
The SPECT/CT and PET/CT images show a clear delineation of the PC-3 tumors. Image acquisition at 12 h after injection showed good clearance of the radioactivity from the abdominal organs and significant accumulation only in the tumor. The PET/CT image, acquired at 1 h after injection using 68Ga-NODAGA-AR, showed high abdominal activity, compared with the image acquired using 64Cu-CB-TE2A-AR at 12 h after injection. This finding shows in a remarkable way that the half-life of the radionuclide may be of high relevance in regard to a good image contrast.
Important parameters for good image contrast are the tumor–to–normal-tissue ratios, in particular the tumor-to-blood and tumor-to-muscle ratios. For all 4 radiopeptides, these ratios are already high at 1 h and increase over time. As the kidney uptake of radiopeptides is dose-limiting in radiotherapeutic applications, particular attention is given to the tumor-to-kidney ratio. The high values found here are outstanding and unchallenged by any other radiopeptide.
CONCLUSION
We have developed 4 different peptide-based radiovectors for the imaging of GRPr-positive tumors. All 4 radiovectors show promising in vitro and in vivo pharmacokinetic performances that may improve diagnostic imaging of cancers overexpressing these receptors. In particular, the encouraging performance of the 64Cu-CB-TE2A-AR motivated us to introduce this targeting vector into the clinic for use in patients with primary prostate cancer.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Novartis Pharma for analytic assistance and Sibylle Tschumi, Edith Rauber, and Valentina Rufener-Schirp for their expert technical help. This study was funded in part by Bayer Schering Pharma, the Swiss National Science Foundation, and the COST actions D38 and BM0607. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Nov. 11, 2011.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication June 10, 2011.
- Accepted for publication July 29, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A Vision for Gastrin-Releasing Peptide Receptor Targeting for Imaging and Therapy: Perspective from Academia and Industry
- Bombesin Antagonist-Based Radiotherapy of Prostate Cancer Combined with WST-11 Vascular Targeted Photodynamic Therapy
- Bombesin-Targeted PET of Prostate Cancer
- Approaches to Improve the Pharmacokinetics of Radiolabeled Glucagon-Like Peptide-1 Receptor Ligands Using Antagonistic Tracers
- Cerenkov Luminescence Imaging for Radiation Dose Calculation of a 90Y-Labeled Gastrin-Releasing Peptide Receptor Antagonist
- Preclinical Comparison of Al18F- and 68Ga-Labeled Gastrin-Releasing Peptide Receptor Antagonists for PET Imaging of Prostate Cancer
- N-Terminal Modifications Improve the Receptor Affinity and Pharmacokinetics of Radiolabeled Peptidic Gastrin-Releasing Peptide Receptor Antagonists: Examples of 68Ga- and 64Cu-Labeled Peptides for PET Imaging
- Interrogating Tumor Metabolism and Tumor Microenvironments Using Molecular Positron Emission Tomography Imaging. Theranostic Approaches to Improve Therapeutics
- Positron-emission Tomography (PET) Imaging Agents for Diagnosis of Human Prostate Cancer: Agonist vs. Antagonist Ligands