


Abstract
The αv-integrins, cell adhesion molecules that are highly expressed on activated endothelial cells and tumor cells but not on dormant endothelial cells or normal cells, present an attractive target for tumor imaging and therapy. We previously coupled a cyclic Arg-Gly-Asp (RGD) peptide, c(RGDyK), with 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA) and labeled the RGD-DOTA conjugate with 64Cu (half-life, 12.8 h; 19% β+) for solid tumor targeting, with high tumor-to-background contrast. The rapid tumor washout rate and persistent liver and kidney retention of this tracer prompted us to optimize the tracer for improved pharmacokinetic behavior. In this study, we introduced a polyethylene glycol (PEG; molecular weight, 3,400) moiety between DOTA and RGD and evaluated the 64Cu-DOTA-PEG-RGD tracer for microPET imaging in brain tumor models. Methods: DOTA was activated in situ and conjugated with RGD-PEG-NH2 under slightly basic conditions. αvβ3-Integrin-binding affinity was evaluated with a solid-phase receptor-binding assay in the presence of 125I-echistatin. Female nude mice bearing subcutaneous U87MG glioblastoma xenografts were administered 64Cu-DOTA-PEG-RGD, and the biodistributions of the radiotracer were evaluated from 30 min to 4 h after injection. microPET (20 min of static imaging at 1 h after injection) and then quantitative autoradiography were used for tumor visualization and quantification. The same tracer was also applied to an orthotopic U87MG model for tumor detection. Results: The radiotracer was synthesized with a high specific activity (14,800–29,600 GBq/mmol [400–800 Ci/mmol]). The c(RGDyK)-PEG-DOTA ligand showed intermediate binding affinity for αvβ3-integrin (50% inhibitory concentration, 67.5 ± 7.8 nmol/L [mean ± SD]). The pegylated RGD peptide demonstrated rapid blood clearance (0.57 ± 0.15 percentage injected dose [%ID]/g [mean ± SD] at 30 min after injection and 0.03 ± 0.02 %ID/g at 4 h after injection). Activity accumulation in the tumor was rapid and high at early time points (2.74 ± 0.45 %ID/g at 30 min after injection), and some activity washout was seen over time (1.62 ± 0.18 %ID/g at 4 h after injection). Compared with 64Cu-DOTA-RGD, this tracer showed improved in vivo kinetics, with significantly reduced liver uptake (0.99 ± 0.08 %ID/g vs. 1.73 ± 0.39 %ID/g at 30 min after injection and 0.58 ± 0.07 %ID/g vs. 2.57 ± 0.49 %ID/g at 4 h after injection). The pegylated RGD peptide showed higher renal accumulation at early time points (3.51 ± 0.24 %ID/g vs. 2.18 ± 0.23 %ID/g at 30 min after infection) but more rapid clearance (1.82 ± 0.29 %ID/g vs. 2.01 ± 0.25 %ID/g at 1 h after injection) than 64Cu-DOTA-RGD. The integrin receptor specificity of this radiotracer was demonstrated by blocking of tumor uptake by coinjection with nonradiolabeled c(RGDyK). The high tumor-to-organ ratios for the pegylated RGD peptide tracer (at 1 h after injection: tumor-to-blood ratio, 20; tumor-to-muscle ratio, 12; tumor-to-liver ratio, 2.7; and tumor-to-kidney ratio, 1.2) were confirmed by microPET and autoradiographic imaging in a subcutaneous U87MG tumor model. This tracer was also able to detect an orthotopic brain tumor in a model in which U87MG cells were implanted into the mouse forebrain. Although the magnitude of tumor uptake in the orthotopic xenograft was lower than that in the subcutaneous xenograft, the orthotopic tumor was still visualized with clear contrast from normal brain tissue. Conclusion: This study demonstrated the suitability of a PEG moiety for improving the in vivo kinetics of a 64Cu-RGD peptide tracer without compromising the tumor-targeting ability and specificity of the peptide. Systematic investigations of the effects of the size and geometry of PEG on tumor targeting and in vivo kinetics will lead to the development of radiotracers suitable for clinical applications such as visualizing and quantifying αv-integrin expression by PET. In addition, the same ligand labeled with therapeutic radionuclides may be applicable for integrin-targeted internal radiotherapy.
Angiogenesis, the formation and differentiation of blood vessels, plays a key role in tumor growth and metastasis spread and has become the new frontier for tumor control (1,2). Angiogenesis is a complex process involving extensive interplay among cells, soluble factors, and extracellular matrix components (3–11). Integrins are cell adhesion molecules known to be involved in multiple steps of angiogenesis and metastasis. The function of integrins during tumor angiogenesis has been studied most extensively for αvβ3-integrin and αvβ5-integrin, which are highly expressed on activated endothelial cells and tumor cells but not on quiescent vessels and normal cells (12). The expression of αvβ3-integrin is required for angiogenesis induced by fibroblast growth factor 2 and tumor necrosis factor α, and the expression of αvβ5-integrin is required for angiogenesis induced by vascular endothelial growth factor and transforming growth factor α (13). Monoclonal antibodies, peptides, and peptidomimetic antagonists of αv-integrins have been shown to block angiogenesis in both preclinical tumor models and phase I and II clinical trials by inhibiting endothelium-specific integrin survival signaling (14,15). The ability to noninvasively visualize and quantify the αv-integrin expression level during tumor growth and spread as well as during anti-integrin treatment will provide new opportunities to develop individualized therapeutic approaches, to select appropriate patients entering clinical trials for anti-integrin treatment, to establish optimized dosages and dose intervals for effective treatment on the basis of receptor occupancy studies, to detect early antiangiogenic responses and quantitatively evaluate treatment efficacy, and finally to cooptimize imaging and therapy via structure–activity studies.
Arg-Gly-Asp (RGD) peptide antagonists of αv-integrins have been labeled with different radionuclides for both imaging and therapeutic purposes. Notably, various investigators (16–20) labeled cyclic RGD peptides with 18F (half-life [t1/2], 119.7 min; 99% β+) for microPET imaging in various solid tumor models, and high tumor-to-background contrast was obtained. However, these radiotracers have some drawbacks: the labeling procedure is tedious and relatively inefficient, tumor uptake is modest, and tumor retention time is short. Although the tracers were able to localize αv-integrin-positive tumors, their utility in reflecting targeted radiotherapy is limited because no therapeutic fluorine isotope is available. Because of the existence of an imaging and therapy radionuclide pair (64Cu/67Cu) for diagnosing and treating cancer, 64Cu-labeled RGD peptides with successful tumor-targeting properties will eventually be able to determine dosimetry and tumor responses to the same ligand labeled with therapeutic amounts of 67Cu for αv-integrin-mediated internal radiotherapy.
Chen et al. (21) recently described the synthesis of a 64Cu-labeled RGD peptide and microPET imaging of orthotopic MDA-MB-435 breast cancer tumor xenografts, with high tumor-to-contralateral background contrast. However, this tracer also had a rapid tumor washout rate and showed persistent activity accumulation in the liver and kidneys. The unfavorable in vivo pharmacokinetics of this tracer limit the application of a 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA)-RGD conjugate for tumor imaging and radiotherapy. Haubner et al. (22) chose to modify the RGD peptide with a glycosylate moiety to increase hydrophilicity and thus reduce blood clearance and reduce hepatobiliary excretion. We have found that polyethylene glycol (PEG) is a suitable tool for modifying the RGD peptide, with improved in vivo kinetics.
Chen et al. (23) recently modified a cyclic RGD peptide, c(RGDyK), by introducing a monomethoxy PEG moiety (mPEG; molecular weight [MW], 2,000) into the lysine ε-amino group. The resulting RGD-mPEG conjugate showed drastically decreased renal uptake and slightly increased liver accumulation compared with the parental RGD peptide. In this study, we inserted a heterobifunctional PEG linker (MW, 3,400) between DOTA and c(RGDyK). The resulting DOTA-PEG-RGD conjugate then was radiolabeled with 64Cu (t1/2, 12.8 h; 19% β+) for microPET imaging studies with both subcutaneous and orthotopic U87MG glioblastoma models.
MATERIALS AND METHODS
Materials
c(RGDyK) was synthetically produced by solution cyclization of fully protected linear pentapeptide H-Gly-Asp(tert-butyloxy)-d-Tyr(tert-butyloxy)-Lys(butoxycarbonyl)-Arg(2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl)-OH, followed by trifluoroacetic acid (TFA) deprotection in the presence of the free radical scavenger triisopropylsilane (23). RGD-PEG conjugate was prepared by coupling of c(RGDyK) with t-butoxycarbonyl (t-Boc)-protected PEG-succinimidyl ester (MW, 3,400), followed by TFA cleavage (24). 64Cu was produced on a CS-15 biomedical cyclotron at the School of Medicine, Washington University, by the 64Ni(p,n)64Cu nuclear reaction (25). All reagents, unless otherwise specified, were of analytic grade and were commercially available. DOTA was purchased from Macrocyclics, Inc. N-Hydroxysulfosuccinimide (SNHS), 1-ethyl-3-[3-(dimethylamino)-propyl]carbodiimide (EDC), and Chelex 100 (50–100 mesh) were obtained from Aldrich.
Chromalux HB microplates were obtained from Dynex Technologies. 125I-Echistatin, labeled by the lactoperoxidase method to a specific activity of 74,000 GBq/mmol (2,000 Ci/mmol), was obtained from Amersham Biosciences. Echistatin was purchased from Sigma. Purified human αvβ3-integrin in a Triton X-100 formulation was obtained from Chemicon International.
Semipreparative reversed-phase high-pressure liquid chromatography (HPLC) was accomplished by use of a Waters 515 chromatography system with a 486 tunable absorbance detector. Purification was performed with a Vydac 218TP510 protein and peptide column (5 μm; 250 × 10 mm). The flow rate was 5 mL/min; the mobile phase was changed from 95% solvent A (0.1% TFA in water) and 5% solvent B (0.1% TFA in acetonitrile) (0–2 min) to 35% solvent A and 65% solvent B at 32 min. The analytic HPLC method was performed with the same gradient system but with a Vydac 218TP54 column (5 μm; 250 × 4.6 mm) and a flow rate of 1 mL/min. The absorbance was monitored at 218 nm. Radio–thin-layer chromatography (TLC) was performed by use of MKC18F plates (Whatman), Bioscan System 200, and Winscan software (version 2.2; BioScan Inc.). Reversed-phase extraction C18 SepPak cartridges were obtained from Waters.
Preparation of DOTA-PEG-RGD Conjugate
DOTA was activated in situ by EDC at a molar ratio for DOTA:EDC:SNHS of 10:5:4 (21). RGD-PEG conjugate (8 mg; 2 μmol) dissolved in 1,600 μL of water (5 mg/mL) was cooled to 4°C and added to a DOTA-sulfosuccinimydyl ester (OSSu) reaction mixture (10 μmol; calculated on the basis of SNHS), and the pH was adjusted to 8.5 with 1N NaOH. The reaction mixture was incubated overnight at 4°C. DOTA-PEG-RGD conjugate was purified by semipreparative HPLC. The sample containing the RGD conjugate was collected, lyophilized, and dissolved in H2O at a concentration of 1 mg/mL for use in radiolabeling reactions.
64Cu Radiolabeling
DOTA-PEG-RGD was labeled with 64Cu by the addition of 37–185 MBq (1–5 mCi) of 64Cu (5–25 μg of DOTA-PEG-RGD per 37 MBq of 64Cu) in 0.1N sodium acetate (pH 5.5) buffer and 45 min of incubation at 50°C. The reaction was terminated by the addition of 5 μL of ethylenediaminetetraacetic acid solution (10 mmol/L). The radiochemical yield was determined by radio-TLC with MKC18F TLC plates as the stationary phase and methanol:10% sodium acetate (70:30) as the eluent. 64Cu-DOTA-PEG-RGD was purified by use of a C18 SepPak cartridge with 85% ethanol as the elution solvent. Radiochemical purity was determined by radio-TLC or radio-HPLC. The ethanol was evaporated, the activity was reconstituted with phosphate-buffered saline, and the sample was passed through a 0.22-μm Millipore filter into a sterile multidose vial for use in animal experiments.
Octanol–Water Partition Coefficient
About 370 kBq of 64Cu-DOTA-c(RGDyK) or 64Cu-DOTA-PEG-c(RGDyK) in 500 μL of saline were added to 500 μL of octanol in an Eppendorf microcentrifuge tube (Brinkman). The 2 layers were mixed for 10 min at room temperature, the tube was centrifuged at 14,000 rpm for 5 min (model 5415C Eppendorf microcentrifuge; Brinkman), and the counts in 100-μL aliquots of both organic and inorganic layers were determined by use of a γ-counter (Packard). The experiment was repeated 3 times.
Solid-Phase Receptor-Binding Assay
The standard assay was performed as described previously with modifications (26). Microtiter-2 96-well plates (Dynatech Laboratories, Inc.) were coated with αvβ3-integrin (500 ng/mL; 100 μL per well) in coating buffer (Tris-HCl [pH 7.4] at 25 mmol/L, NaCl at 150 mmol/L, CaCl2 at 1 mmol/L, MgCl2 at 0.5 mmol/L, and MnCl2 at 1 mmol/L) for 16 h at 4°C. The wells then were blocked for 2 h with 200 μL of blocking buffer (coating buffer with 1% radioimmunoassay-grade bovine serum albumin). The plates then were washed twice with binding buffer (coating buffer with 0.1% bovine serum albumin) and incubated with 125I-echistatin (0.06 nmol/L) in the presence of various concentrations of RGD peptide (0.1 nmol/L–5 μmol/L) at room temperature for 3 h. After incubation, the plates were washed 3 times with binding buffer, and the radioactivity was solubilized with 2N boiling NaOH and subjected to γ-counting. Nonspecific binding of 125I-echistatin to αvβ3-integrin was determined in the presence of echistatin at 100 nmol/L. The 50% inhibitory concentrations were calculated by nonlinear regression analysis by use of the GraphPad Prism computer fitting program (GraphPad Software, Inc.). Each data point represents the average value for triplicate wells.
Biodistribution Studies
Animal experiments were conducted under a protocol approved by the University of Southern California Institutional Animal Care and Use Committee. Female athymic nude mice (nu/nu) obtained from Harlan at 4–6 wk of age were injected subcutaneously in the right front leg with 5 × 106 U87MG glioblastoma cells suspended in 150 μL of Eagle’s minimum essential medium. When the tumors reached 0.4–0.6 cm in diameter (14–18 d after implantation), the mice were used for biodistribution and microPET imaging experiments.
Details of the orthotopic (intracranial) xenotransplant model in nu/nu mice were described previously (27). In brief, 105 U87MG tumor cells in 1 μL of RPMI medium were injected through a 33-gauge injection needle over 10 min into the nude mouse (4–6 wk old) forebrain 1.5 mm lateral and 0.5 mm anterior to the bregma and at a depth of 2.5 mm by use of a continuous infusion pump. The needle was removed, the burr hole was closed with bone wax, and the skin incision was closed with dermal glue. Mice were kept under ketamine–xylazine anesthesia during the procedure. This tumor cell number resulted in the growth of tumors in all experimental animals and a highly reproducible growth rate. At 5 wk after injection, when the tumors reached 3 mm or more in diameter, the mice were used for microPET imaging studies.
Nude mice bearing subcutaneously xenografted human U87MG glioblastoma tumors were injected intravenously with approximately 370 kBq (10 μCi) of 64Cu-DOTA-PEG-RGD. Animals were euthanized at 30 min, 1 h, 2 h, and 4 h after injection. Blood, tumors, and major organs and tissues were collected and wet weighed, and counts were determined by use of a γ-counter. The percentage injected dose per gram (%ID/g) was determined for each sample. For each mouse, the radioactivity in the tissue samples was calibrated against a known quantity of the injected compound. Values are reported as mean ± SD for groups of 4 animals. The receptor-mediated localization of the radiotracer was investigated by injection of 64Cu-DOTA-PEG-RGD with c(RGDyK) at 10 mg/kg in a subcutaneous U87MG tumor model. Biodistributions were determined as described above at 1 h after injection for 4 mice per group.
microPET Imaging
PET was performed by use of a microPET R4 rodent model scanner (Concorde Microsystems Inc.). The scanner has a computer-controlled bed and 10.8-cm transaxial and 8-cm axial fields of view. It has no septa and operates exclusively in the 3-dimensional list mode. All raw data were first sorted into 3-dimensional sinograms; this step was followed by Fourier rebinning and 2-dimensional (2D) filtered backprojection image reconstruction by use of a ramp filter with the Nyquist limit (0.5 cycle per voxel) as the cutoff frequency. For the subcutaneous tumor model, each mouse was injected with 14.8 MBq (400 μCi) of 64Cu-DOTA-PEG-RGD via the tail vein, sacrificed at 1 h after injection by cervical dislocation under ketamine–xylazine anesthesia, and placed near the center of the field of view of the microPET scanner, where the highest image resolution and sensitivity are available. The mouse was scanned for 20 min and then subjected to whole-body autoradiography. The raw data were framed into one static frame without attenuation correction. For the orthotopic U87MG tumor model, a 20-min static image was obtained at 1 h after injection. The brain tumor was excised postmortem, fixed in buffered formalin, and embedded in paraffin for hexatoxylin–eosin staining.
Autoradiography
Autoradiography was performed by use of a Packard Cyclone Storage Phosphor Screen system and a Bright 5030/WD/MR cryomicrotome (Hacker Instruments). Immediately after microPET scanning, the sacrificed mice with subcutaneous U87MG tumors were frozen in a dry ice–isopropyl alcohol bath for 2 min. The bodies then were embedded in 4% carboxymethylcellulose (Aldrich) in water by use of a stainless steel mold. The mold was placed in the dry ice–isopropyl alcohol bath for 5 min and then into a −20°C freezer for 1 h. The walls of the mold were removed, and the frozen block was mounted in the cryomicrotome. The block was cut into sections of 50 μm, and the desired sections were digitally photographed and captured for autoradiography. The sections were transferred into a chilled film cassette containing a Packard Super Resolution Screen (spatial resolution, 0.1 mm) and kept there overnight at −20°C. Screens were laser scanned by use of the Packard Cyclone system.
Statistical Analysis
The data were expressed as mean ± SD. A 1-way ANOVA was used for statistical evaluation. Means were compared by use of the Student t test. A P value of <0.05 was considered significant.
RESULTS
DOTA Conjugation and 64Cu Labeling
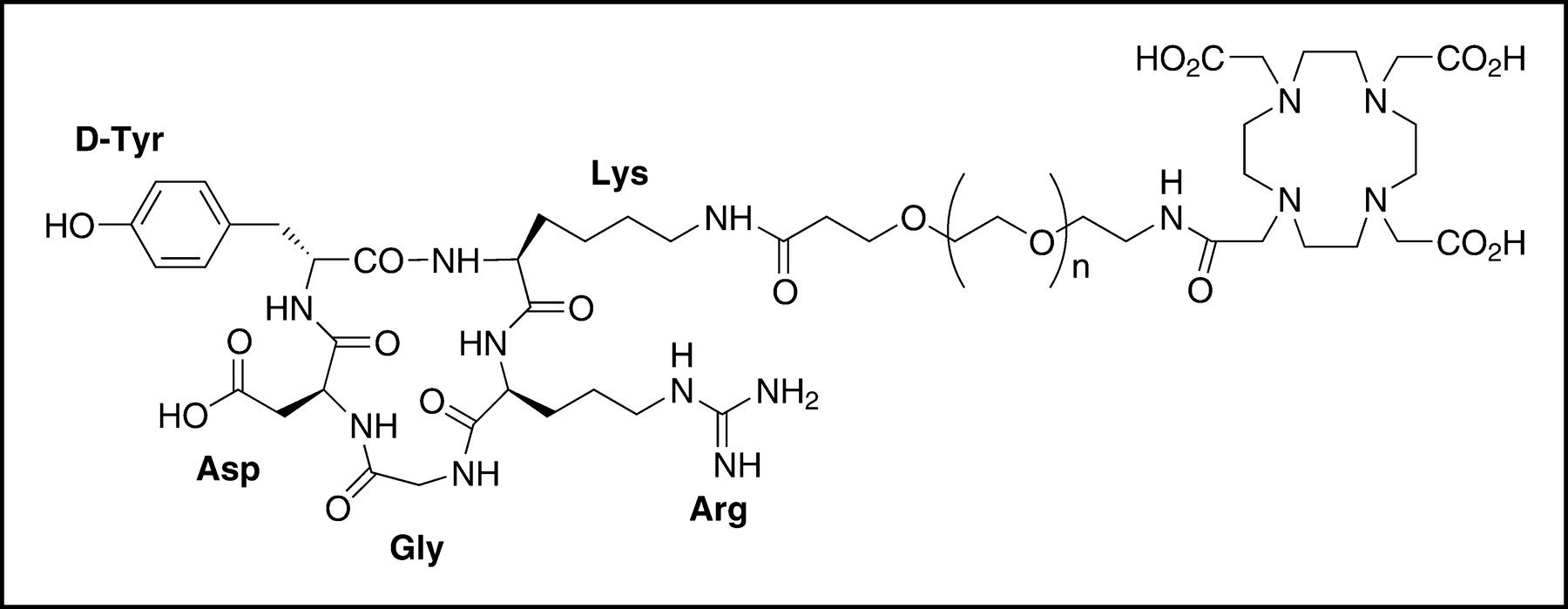
The synthesis of PEG-RGD may be performed by coupling of t-Boc-NH-PEG-CO2Su (MW, 3,400) with c(RGDyK), followed by TFA cleavage to unblock the t-Boc protecting group (24). A DOTA-PEG-RGD conjugate (Fig. 1) was produced in the presence of an excess amount of DOTA-OSSu activated in situ. The MW of the DOTA conjugate, evaluated by matrix-assisted laser desorption ionization—time-of-flight mass spectroscopy, increased from 4,000 to 4,400. The mass spectrometry peak was broad (spectrum not shown) because of the polydispersity of PEG (28). The increase in MW, of approximately 400, corresponds to the conjugation of the macrocylic chelator DOTA and is accompanied by the disappearance of amino group reactivity, demonstrated by the 6-trinitrobenzene sulfonic acid reaction (28). Contemporaneously, a slight change in the retention time (from 21.9 min for PEG-RGD to 21.5 min for DOTA-PEG-RGD) and a sharpening of the peak in reversed-phase HPLC elution were found. 64Cu-DOTA-PEG-RGD was prepared at radiochemical purities of about 50%, 80%, 95%, and ≥98% when 5, 10, 15, and 20 μg of peptide per 37 MBq of 64Cu were used. The specific activity of 64Cu-DOTA-PEG-RGD ranged from 14,800 to 29,600 GBq/mmol (400–800 Ci/mmol).
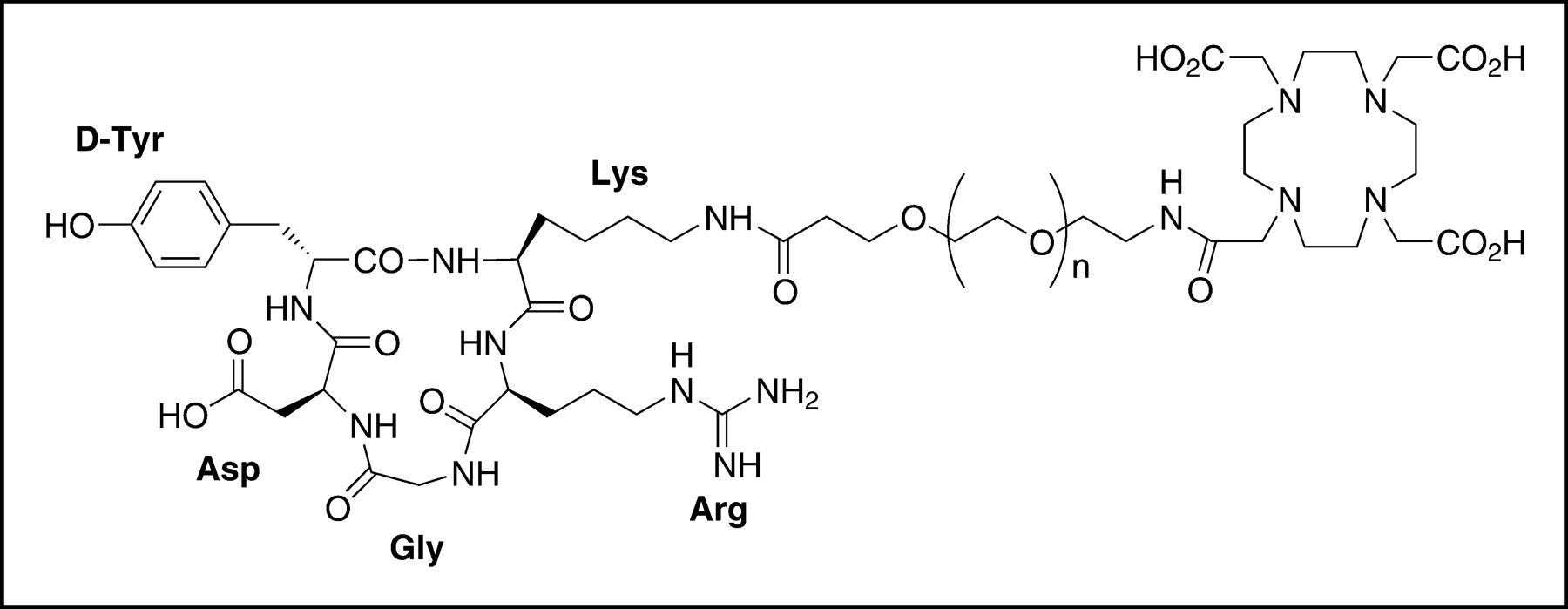
Schematic structure of DOTA-PEG-RGD. Heterofunctional PEG (MW, 3,400) is linked to the DOTA carboxylate group through the N terminus and to the lysine side chain ε-amino group of c(RGDyK) through the C terminus.
Competitive Displacement and Hydrophilicity Studies
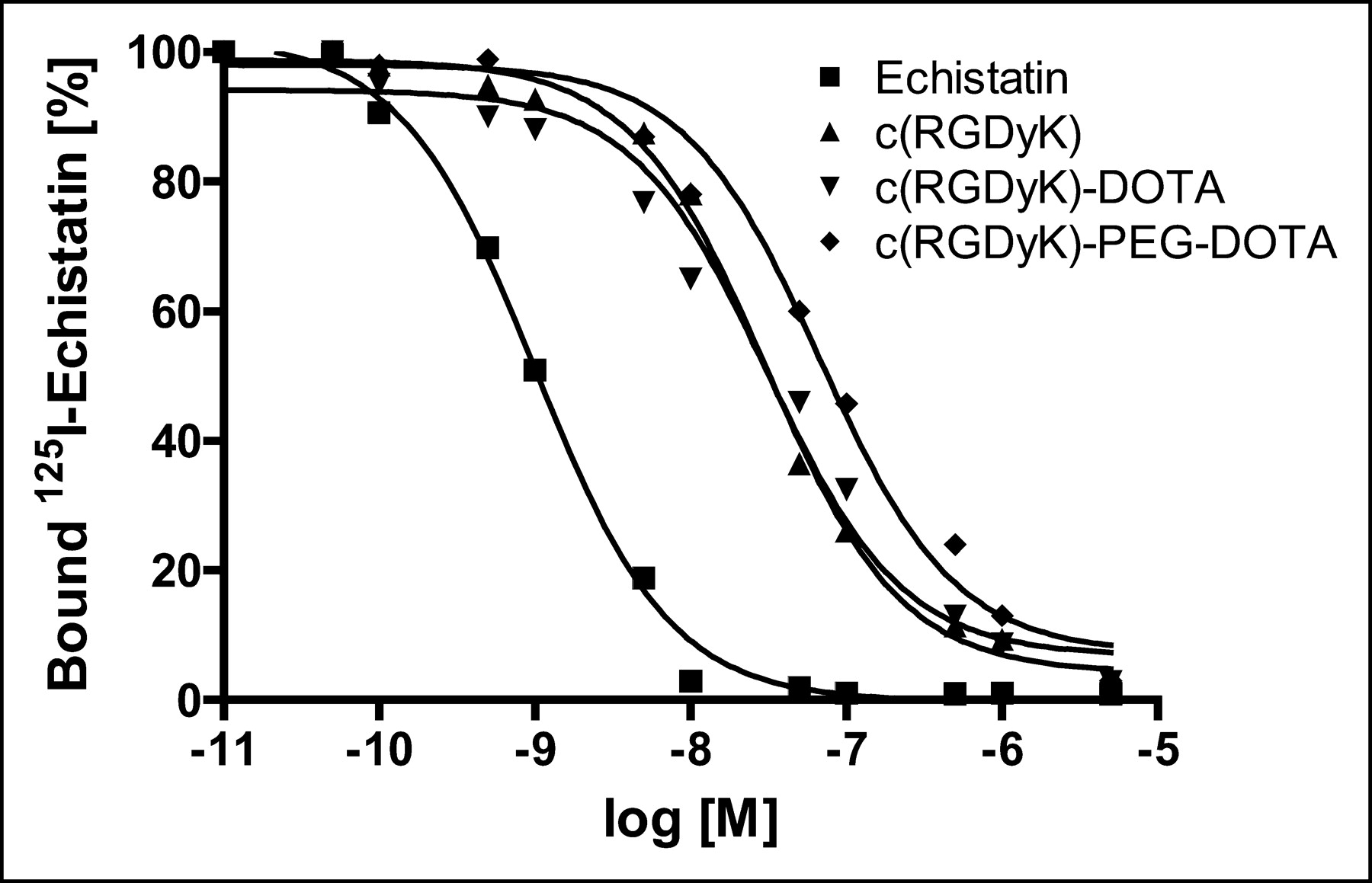
To compare the affinities of c(RGDyK)-DOTA and c(RGDyK)-PEG-DOTA for αvβ3-integrin, a competitive displacement study was performed (Fig. 2). The binding of 125I-echistatin to αvβ3-integrin was competed for by cold echistatin, c(RGDyK), c(RGDyK)-DOTA, and c(RGDyK)-PEG-DOTA in a concentration-dependent manner. All of the peptides were able to fully suppress the binding of the radioligand to purified human αvβ3-integrin. The concentrations required for half-maximal competition (50% inhibitory concentrations) were calculated to be 1.01 ± 0.23 nmol/L for echistatin, 30.3 ± 5.8 nmol/L for c(RGDyK), 31.2 ± 7.4 nmol/L for c(RGDyK)-DOTA, and 67.5 ± 10.5 nmol/L for c(RGDyK)-PEG-DOTA. Both the lead compound c(RGDyK)-DOTA and the pegylated analog c(RGDyK)-PEG-DOTA showed high hydrophilicity, as indicated from octanol–water partition coefficient measurements. LogP values for c(RGDyK)-DOTA and c(RGDyK)-PEG-DOTA were −2.8 ± 0.04 and −2.97 ± 0.30, respectively.

Competition between 125I-echistatin and unlabeled echistatin, c(RGDyK), c(RGDyK)-DOTA, and DOTA-PEG-c(RGDyK) for specific binding to purified αvβ3-integrin, as determined by a solid-phase receptor assay. All measurements were made in triplicate. M = molar concentration.
Biodistributions of 64Cu-DOTA-PEG-RGD
The distributions of radioactivity in selected mouse organs and U87MG glioblastoma xenografts at various times after the administration of 64Cu-DOTA-PEG-RGD are shown in Figure 3. The radiotracer exhibited a rapid decrease in radioactivity over time in blood and most organs. At early time points, the high kidney activity was evidently attributable to the elimination of radiolabeled peptide in the urine. Analogously, radioactivity accumulated in the liver could have been caused by the radioactivity remaining in the blood as well as by partial bile excretion of the agent. The highest tumor uptake of 64Cu-DOTA-PEG-RGD (2.74 ± 0.45 %ID/g) was found at 30 min after injection and decreased to 1.62 ± 0.18 %ID/g at 4 h after injection, suggesting that the 64Cu was present in the tumor up to at least 4 h after injection.
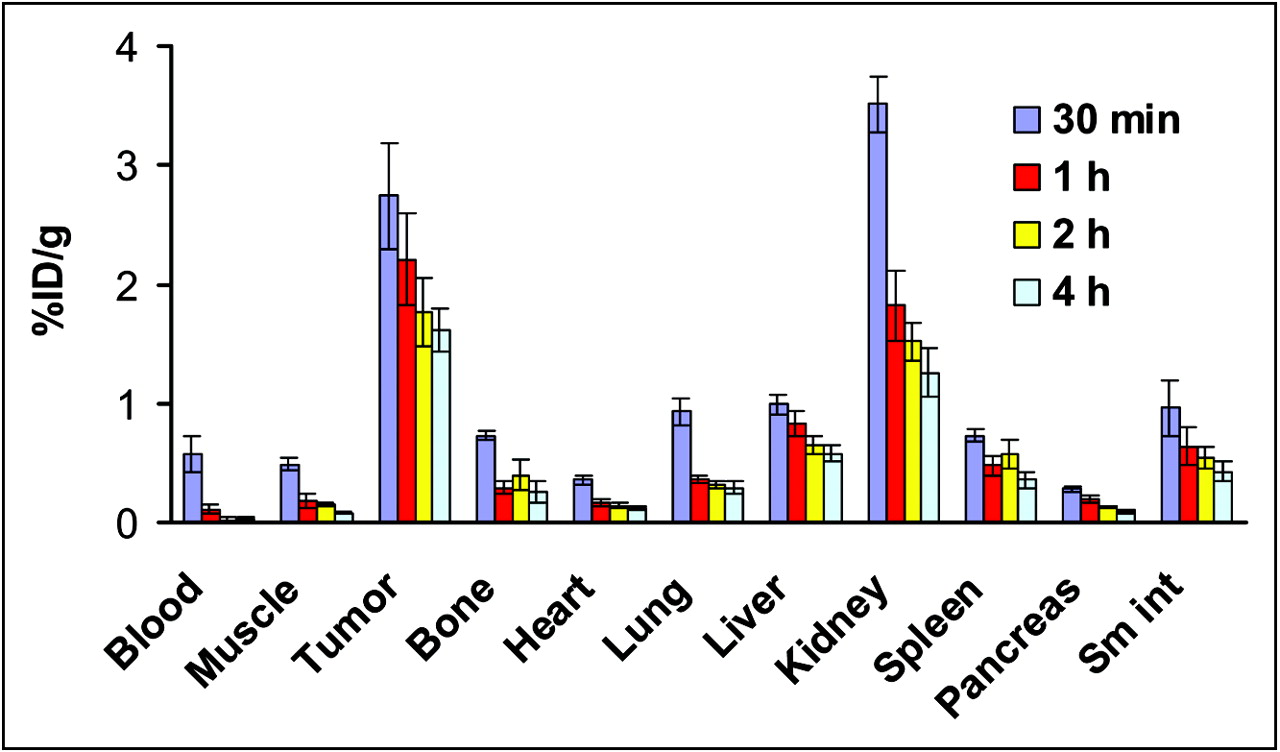
Biodistributions of 64Cu-DOTA-PEG-RGD in nude mice bearing subcutaneously xenotransplanted U87MG tumors. The data are reported as mean ± SD (n = 4). Sm int = small intestine.
A comparison of the kinetics of excretion of 64Cu-DOTA-PEG-RGD and 64Cu-DOTA-RGD in the blood, liver, kidneys, small intestine, and U87MG tumors is shown in Figure 4. The blood activity of 64Cu-DOTA-PEG-RGD (0.57 ± 0.15 %ID/g) was significantly higher than that of 64Cu-DOTA-RGD (0.35 ± 0.09 %ID/g) at 30 min after injection. The pegylated RGD peptide showed more rapid blood clearance than 64Cu-DOTA-RGD, as the blood activity was as low as 0.02 ± 0.02 %ID/g at 2 h after injection for 64Cu-DOTA-PEG-RGD; the blood activity for 64Cu-DOTA-RGD was 0.20 ± 0.03 %ID/g at 2 h after injection and remained unchanged at 4 h after injection. The kidney uptake of 64Cu-DOTA-PEG-RGD was higher than that of 64Cu-DOTA-RGD at early time points, although the pegylated RGD peptide showed more rapid renal clearance, resulting in significantly lower kidney activity accumulation at 4 h. The liver uptake of the pegylated RGD peptide was significantly lower than that of 64Cu-DOTA-RGD at all time points. Although the liver uptake of 64Cu-DOTA-RGD increased over time (2.57 ± 0.49 %ID/g at 4 h after injection), 64Cu-DOTA-PEG-RGD liver uptake decreased over time (0.58 ± 0.07 %ID/g at 4 h after injection). Intestinal activity accumulation of the pegylated RGD peptide was significantly higher than that of the lead compound at all time points (P < 0.001). Pegylation had a minimal effect on tumor uptake and clearance.

Comparison of biodistributions of 64Cu-DOTA-PEG-RGD (•) and 64Cu-DOTA-RGD (○) in nude mice. Error bars denote SDs (n = 4). Sm int = small intestine.
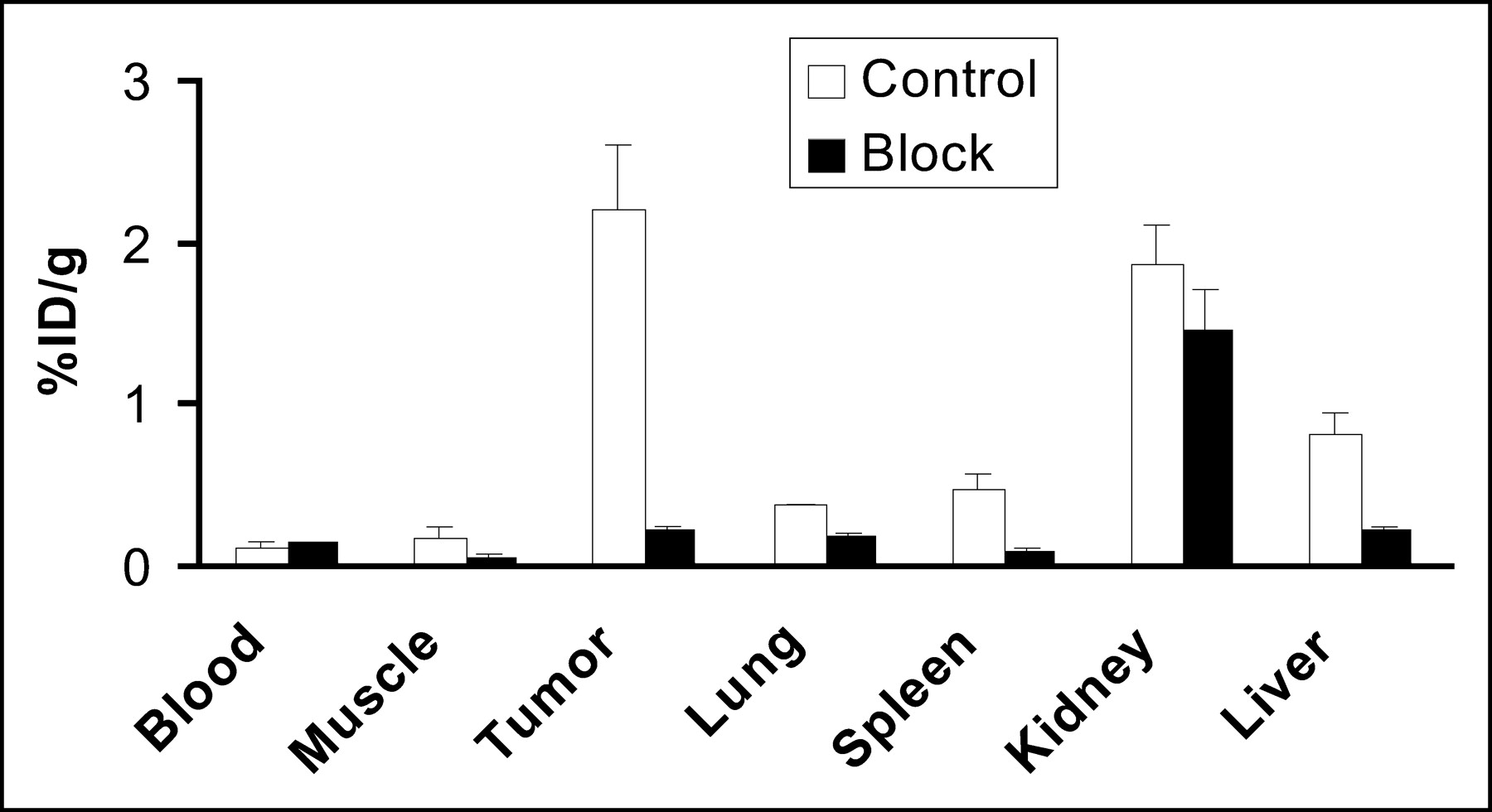
Coinjection of c(RGDyK) (10 mg/kg) with 64Cu-DOTA-PEG-RGD resulted in a significant decrease in radioactivity in all dissected tissues, except the kidneys (Fig. 5). Uptake in tumors was reduced most dramatically, from 2.21 ± 0.38 %ID/g to 0.22 ± 0.03 %ID/g. The blocking experiment showed that the uptake and retention of 64Cu-DOTA-PEG-RGD were receptor dependent in tumors as well as several normal organs, such as the liver, spleen, and lungs.
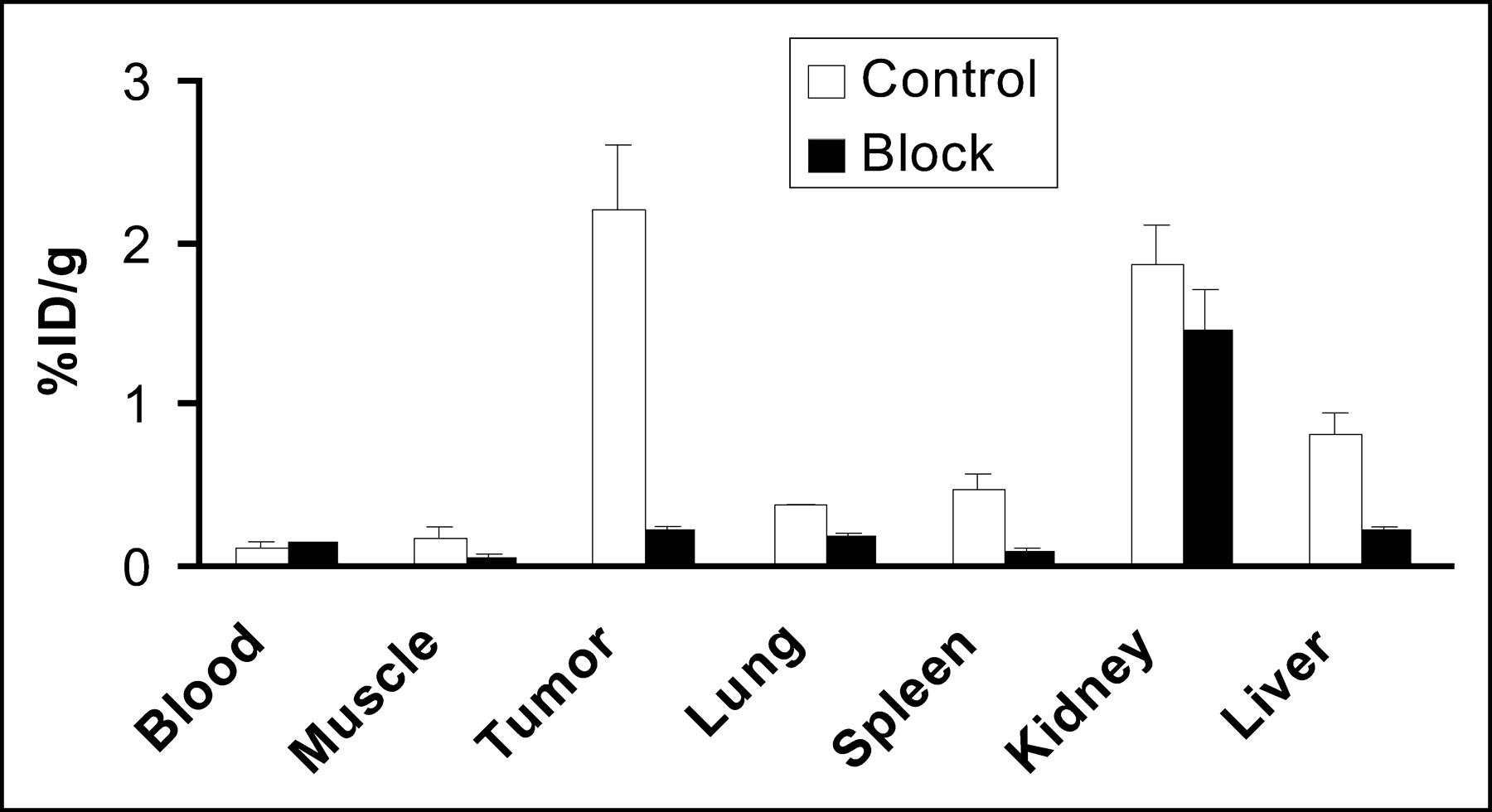
Biodistributions of 64Cu-DOTA-PEG-RGD in the absence and in the presence of c(RGDyK) at 10 mg/kg at 1 h after injection in athymic nude mice with subcutaneous U87MG glioblastoma tumors. Reduced uptake in tumor, liver, muscle, and spleen in the blocking experiment indicated αv-integrin-mediated localization of 64Cu-DOTA-PEG-RGD in these tissues. Error bars denote SDs (n = 4). Data are mean ± SD (n = 4).
microPET Imaging and Whole-Body Autoradiography
A 2D projection of the microPET image (20-min static single frame) of a female athymic nude mouse bearing an U87MG tumor on the right shoulder at 60 min after intravenous injection of 14.8 MBq (400 μCi) of 64Cu-DOTA-PEG-RGD is shown in Figure 6A. The tumor was clearly visible, with high contrast relative to the contralateral background. Prominent uptake was also observed in the kidneys, intestinal tract, and urinary bladder; these data agreed well with the data obtained from direct tissue sampling. No activity accumulation was observed in the normal brain, probably because of the presence of an intact blood–brain barrier and a low level of αv-integrin expression.
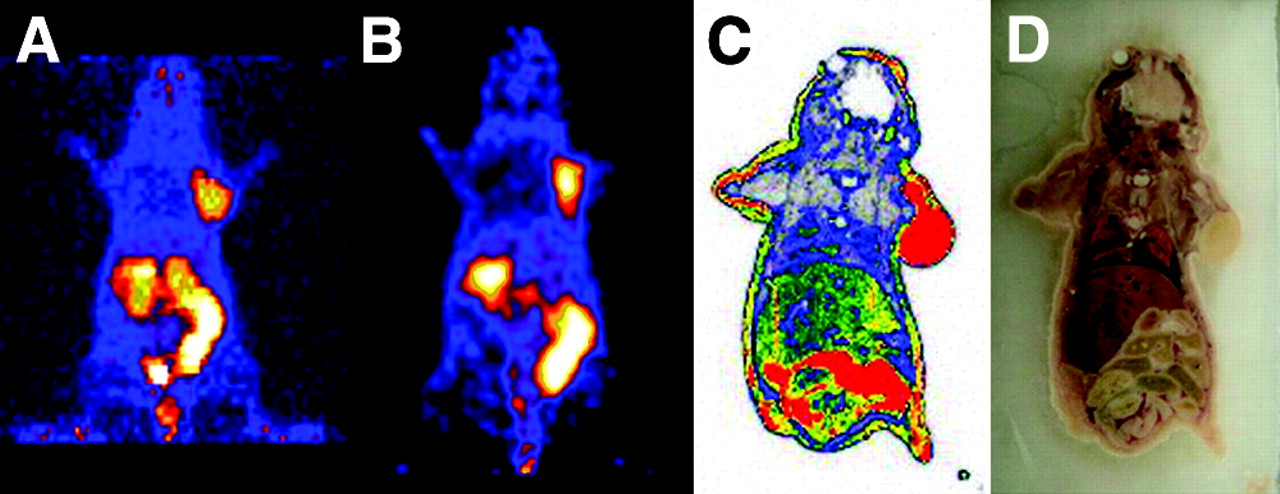
(A) 2D projection of U87MG tumor-bearing mouse at 60 min after injection of 14.8 MBq (400 μCi) of 64Cu-DOTA-PEG-RGD (20-min static image). (B) Coronal image of the same tumor-bearing mouse. (C) Digital autoradiograph of the section containing the tumor after microPET imaging. (D) Anatomic photograph of the section.
Quantitation of tumor and major organ activity accumulation in microPET scanning was realized by measuring regions of interest that encompassed the entire organ in the coronal orientation. Uptake in U87MG tumors, liver, and kidneys was calculated to be 2.6 ± 0.6, 0.8 ± 0.2, and 2.4 ± 0.5 %ID/g, respectively. These data were in good accordance with the biodistribution data obtained at 1 h after injection. To demonstrate that the uptake detected by microPET scanning of a living mouse corresponded to 64Cu-DOTA-PEG-RGD that had localized appropriately to an integrin-positive tumor after microPET scanning, the mouse was subjected to digital whole-body autoradiography. A comparison of the microPET image encompassing the U87MG tumor xenograft (Fig. 6B) and the corresponding autoradiographic section (Fig. 6C) clearly demonstrated the correlation between the regions of high uptake observed in the scan and autoradiographically detected tumor-associated radioactivity.
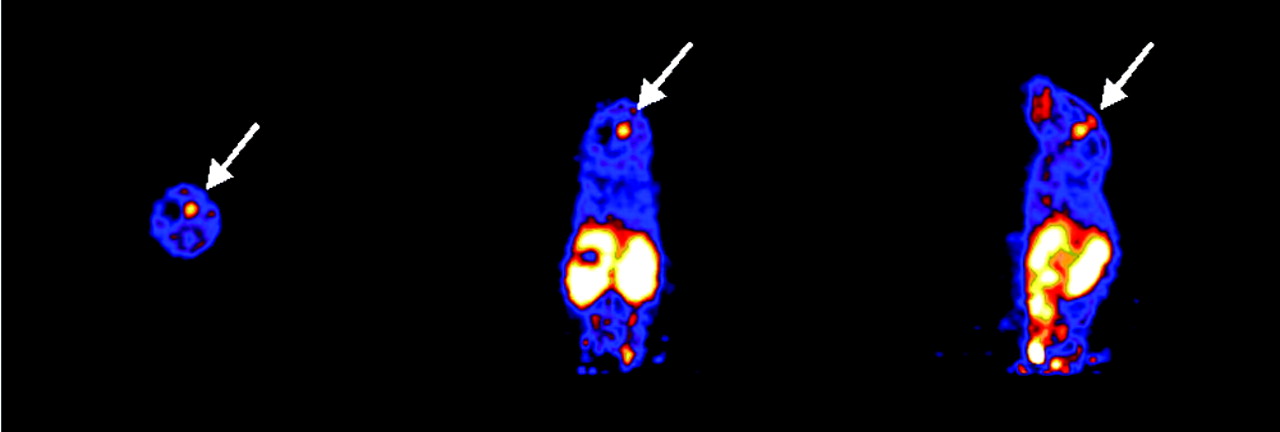
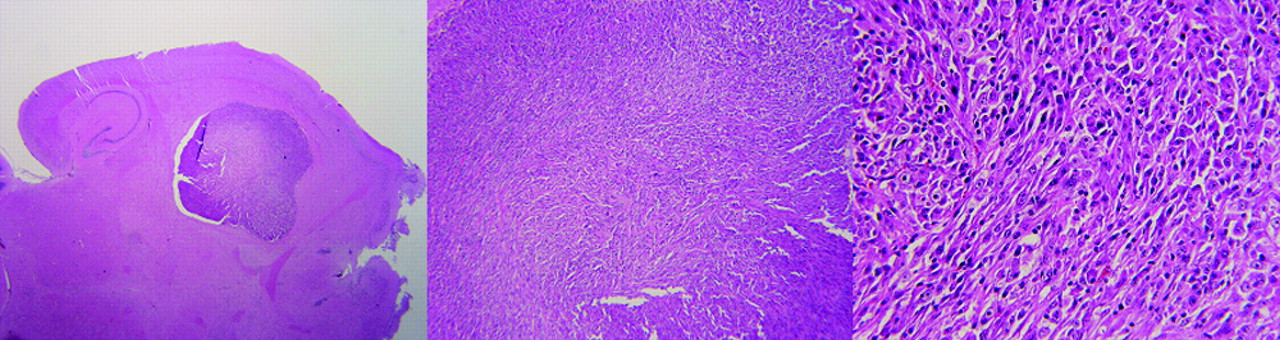
microPET imaging was also successful in visualizing an orthotopic U87MG glioblastoma implanted into the mouse forebrain (Fig. 7). Although the absolute tumor uptake (0.4 ± 0.1 %ID/g) was significantly lower than that of the subcutaneous tumor, the tumor-to-brain ratio still reached 4.0 ± 0.5 because of the extremely low background activity accumulation in the normal brain tissue. Histologic staining of the mouse brain (Fig. 8) removed at the end of the scan confirmed the presence and location of the U87MG tumor, which measured 2.9 × 2.8 mm.

Transaxial, coronal, and sagittal slices (from left to right) of a mouse containing an orthotopically implanted U87MG tumor (arrows). The tumor is about 3 mm in diameter; the tumor-to-brain ratio is 4.0 ± 0.5.
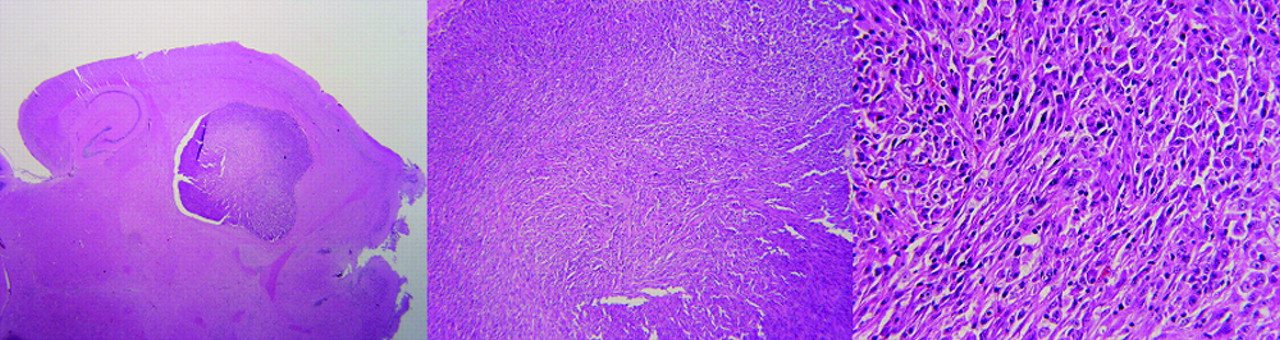
Histopathologic analysis of orthotopically injected U87MG brain tumor cells after microPET imaging. From left to right, panels show original, 4×, and 20× magnification. Tumor size and density of tumor cells were in accordance with volumetric and uptake information obtained from microPET scanning.
DISCUSSION
This study demonstrated that pegylation improves the in vivo kinetics of 64Cu-labeled RGD peptide and that 64Cu-labeled pegylated RGD peptide is able to image a subcutaneous U87MG tumor xenograft and to visualize an orthotopic brain tumor implanted into the mouse forebrain by microPET scanning.
The overexpression of αv-integrins (αvβ3 and αvβ5) on solid tumor cells and activated endothelial cells has aroused interest in the development of radiolabeled RGD peptides for imaging and therapy of cancer. Various investigators have shown that 18F-labeled RGD peptides can accumulate in different tumors, such as breast cancer (16,20), brain tumor (17,23), and melanoma and osteosarcoma (18,19). However, the introduction of a prosthetic 4-18F-fluorobenzoyl group to the lysine side chain ε-amino group resulted in unfavorable hepatobiliary excretion, as indicated by high uptake in the gallbladder and intestines; this situation may hamper the imaging of tumor lesions located in the lower abdomen and limit the activity dose that can be administered safely because of whole-body radiation exposure. Haubner et al. demonstrated that substituting a hydrophilic sugar moiety for the lysine resulted in predominant renal clearance for both 125I-labeled (22) and 18F-labeled (18,19) glycosylated RGD peptides. On the other hand, we found that the tumor-targeting ability and in vivo kinetics of an RGD peptide could be substantially improved by introducing a PEG moiety into the lysine side chain amino group. We coupled a c(RGDyK) peptide with mPEG (MW, 2,000) and labeled the c(RGDyK)-mPEG conjugate with 125I. The pegylated RGD peptide resulted in faster blood clearance and lower kidney uptake, without compromising the receptor-targeting ability, than c(RGDyK) (23). We also found that 18F-labeled pegylated RGD (PEG MW, 3,400) resulted in a longer tumor retention time and far less hepatobiliary excretion than the corresponding 18F-labeled RGD (24).
Because no therapeutic fluorine isotope is available, 18F-labeled RGD peptides will not be able to provide direct dosimetry information for integrin-targeted internal radiotherapy. 64Cu with 19% positron emission and 40% β-emission (maximum energy, 578 keV) efficiencies has the potential for both PET and therapy. Chen et al. previously conjugated c(RGDyK) with macrocyclic chelator DOTA and labeled the DOTA-RGD conjugate with 64Cu for PET of breast cancer tumors (21). This tracer showed significant liver uptake. Recent metabolic studies of 111In-diethylenetriaminepentaacetic acid (DTPA)-conjugated polypeptides indicated that when DTPA was attached to the lysine of the polypeptides, a 111In-DTPA adduct of lysine was generated as the final radiometabolite after lysosomal proteolysis of the parental peptides in the liver (29). We assume that the prominent liver retention after intravenous administration of 64Cu-DOTA-RGD seen here may be ascribed to the combined effects of the transchelation of 64Cu from DOTA to superoxide dismutase (30) in the liver and the persistent localization of the final radiometal metabolite 64Cu-DOTA-lysine within the tissue. Therefore, the practical use of 64Cu as an imaging and therapeutic isotope relies on the development of new chelating agents for Cu(II) that have greater kinetic stability as well as on the prevention of the delivery of negatively charged metabolites to liver nonparenchymal cells via scavenger receptor-mediated endocytosis (31).
Intrigued by the ability of pegylation to improve the tumor-targeting and in vivo kinetics of both 125I- and 18F-labeled RGD peptides, we extended our study to observe the impact of pegylation on 64Cu-DOTA-RGD. The results of an isolated, immobilized integrin receptor-binding assay indicated that introduction of the PEG moiety at the lysine side chain amino group had a modest influence on αvβ3-integrin affinity and selectivity. All of the RGD peptide ligands bound αvβ3-integrin with a significantly lower affinity than echistatin, which is folded in a series of irregular loops to form a rigid core stabilized by 4 disulfide bridges (32). It is also worth noting that the introduction of a PEG moiety with an MW of 3,400 significantly reduced the circulatory t1/2 of the pegylated radiotracer, in contrast to the results reported for many other pegylated proteins and polypeptides (33). Since the octanol–water partition coefficients of the cyclic RGD peptide and pegylated RGD peptide tracers were similar, the more rapid blood clearance of the pegylated RGD peptide could have been attributable to the fact that this compound showed reduced interactions with blood elements (plasma proteins or blood cells) rather than changes in hydrophilicity on modification.
Both 64Cu-DOTA-RGD and 64Cu-DOTA-PEG-RGD were cleared rapidly from the kidneys. In general, PEG shows little toxicity and is eliminated from the body intact either by the kidneys (for PEG of <30 kDa) or in the feces (for PEG of >20 kDa) (34–36); subsequent reabsorption occurs in the proximal tubular cells, where the pegylated peptide is internalized and degraded in the lysosomes. Following this pathway, the main radiolabeled catabolite of 64Cu-DOTA-PEG-RGD probably is 64Cu-DOTA-PEG-Lys. 64Cu-DOTA-Lys and 64Cu-DOTA-PEG-Lys have the same overall negative charge (−1), but 64Cu-DOTA-PEG-RGD had significantly lower liver uptake and more rapid liver clearance. We assume that the PEG polymer, along with the associated water molecules, acts like a shield to wrap the 64Cu-DOTA chelate and reduces the apparent negative charge, and the negative charge density thus reduces the apparent affinity of the radiometal metabolite for hepatic scavenger receptors. This notion agrees with the phenomenon in which an increase in the negative charge density of proteins by acylation efficiently increases liver uptake (37).
The features of rapid blood and renal clearance and limited hepatic and biliary excretion permit high signal-to-noise ratios for in vivo imaging soon after injection (1 h). The activities quantified in the mouse tumor, liver, and kidneys were comparable to those obtained by the direct tissue sampling technique. microPET imaging used with the orthotopic U87MG tumor model revealed a good tumor-to-brain ratio; the magnitude of uptake of the orthotopic tumor was about 5 or 6 times lower than that of the subcutaneous tumor. Whether the blood–brain barrier or different integrin expression characteristics of orthotopic brain tumors and subcutaneous tumors are responsible for this difference in uptake needs to be investigated further.
The receptor specificity of the RGD peptide was not significantly affected by the introduction of a PEG moiety and a 64Cu-DOTA radiolabel, because of the invariance of the “kinked” conformation of the pentapeptide (38). The unaltered tumor uptake and clearance and the favorable biokinetics make this radiopharmaceutical suitable for PET of solid tumors that overexpress αv-integrin and for integrin-targeted internal radiotherapy when therapeutic amounts of 64Cu- or 67Cu-labeled pegylated RGD peptide are applied. A radionuclide with PET and therapeutic capacities, such as 64Cu, could be very advantageous, because accurate individualized dosimetry could be obtained for each patient with PET before radiotherapy. The DOTA-PEG-RGD conjugate could also be used with 86Y (t1/2, 14.7 h; 33% β+) as a surrogate marker to detect the uptake and dosimetry of 90Y-labeled RGD.
We recognize that 2 important issues remain unresolved. The first issue is whether the magnitude of tumor uptake obtained from PET correlates well with the tumor integrin expression level. The ability to measure tumor integrin receptor density noninvasively could have great value in predicting the effectiveness of RGD peptide therapy in patients with brain tumors and other solid tumors that express αv-integrins. Our recent quantitative analysis of tracer kinetics with the reference tissue sampling technique, which assumes that the radiolabel is not irreversibly trapped within the reference region and that the rate of turnover of the exchanging spaces in both the tumor and the reference tissue is rapid compared with the duration of the time–activity curves (X. Chen, P.S. Conti, unpublished data, 2003), has suggested the feasibility of this reference method (39) for quantifying αv-integrin receptor density during tumor growth and in response to competition from a nonlabeled RGD peptide or anti-integrin drugs targeted to αv-integrins. Our future studies will focus on the validation of the results of such an analysis with traditional Western blots and in vitro receptor autoradiography.
The second issue is whether the tumor retention of a suitably labeled RGD peptide is attributable to interactions with αv-integrins expressed on the neovasculature, on the tumor cells, or on a combination of both cell types. The intrinsic resolution of PET will not allow this question to be answered directly. In this regard, it is possible that further advances in molecular biology may elucidate subclasses of αv-integrins present on endothelial cells versus tumor cells, thereby providing an approach for developing more specific diagnostic and therapeutic ligands in the future. In the interim, existing agents may be used to assess overall αv-integrin activity in tumors.
CONCLUSION
A 64Cu-labeled pegylated RGD peptide was developed for PET imaging of tumor αv-integrin expression. This new radiotracer was directly compared with 64Cu-DOTA-RGD in vivo, and the results suggested that DOTA-PEG-RGD is a suitable ligand for labeling radiometals that can form stable complexes with the DOTA moiety for both imaging and therapeutic applications. Optimization of this tracer by systematic investigation of the impact of PEG size and geometry and the application of different RGD peptide and peptidomimetic antagonists of αv-integrins and different chelating agents for Cu(II) may further improve the imaging and therapeutic potentials of the peptides.
Acknowledgments
This work was performed in part with contributions from National Institute of Biomedical Imaging and Bioengineering grant R21 EB001785, American Chemical Society grant ACS-IRG-580007-42, the Wright Foundation, Department of Defense (DOD) Breast Cancer Research Program (BCRP) Concept Award DAMD17-03-1-0752, DOD BCRP Idea Award BC030012, and National Cancer Institute (NCI) grant P20 CA86532. 64Cu was provided by Washington University Medical School with partial funding from NCI grant R24 CA86307.
Footnotes
Received Dec. 1, 2003; revision accepted Jun. 2, 2004.
For correspondence or reprints contact: Peter S. Conti, MD, PhD, Department of Radiology, University of Southern California, 1510 San Pablo St., Suite 350, Los Angeles, CA 90033.
E-mail: pconti{at}usc.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A Novel Engineered Small Protein for Positron Emission Tomography Imaging of Human Programmed Death Ligand-1: Validation in Mouse Models and Human Cancer Tissues
- A Novel Engineered Anti-CD20 Tracer Enables Early Time PET Imaging in a Humanized Transgenic Mouse Model of B-cell Non-Hodgkins Lymphoma
- Novel 64Cu- and 68Ga-Labeled RGD Conjugates Show Improved PET Imaging of {alpha}{nu}{beta}3 Integrin Expression and Facile Radiosynthesis
- Imaging of {alpha}v{beta}3 Expression by a Bifunctional Chimeric RGD Peptide not Cross-Reacting with {alpha}v{beta}5
- A Novel Neutrophil-Specific PET Imaging Agent: cFLFLFK-PEG-64Cu
- 111In-LLP2A-DOTA Polyethylene Glycol-Targeting {alpha}4{beta}1 Integrin: Comparative Pharmacokinetics for Imaging and Therapy of Lymphoid Malignancies
- Engineered Knottin Peptides: A New Class of Agents for Imaging Integrin Expression in Living Subjects
- Identification and Characterization of a Peptide with Affinity to Head and Neck Cancer
- Multimodality Probes: Amphibian Cars for Molecular Imaging
- Multimodality Molecular Imaging of Tumor Angiogenesis
- Dual-Function Probe for PET and Near-Infrared Fluorescence Imaging of Tumor Vasculature
- microPET of Tumor Integrin {alpha}v{beta}3 Expression Using 18F-Labeled PEGylated Tetrameric RGD Peptide (18F-FPRGD4)
- Small-Animal PET of Tumor Angiogenesis Using a 76Br-Labeled Human Recombinant Antibody Fragment to the ED-B Domain of Fibronectin
- Specific Targeting of Tumor Angiogenesis by RGD-Conjugated Ultrasmall Superparamagnetic Iron Oxide Particles Using a Clinical 1.5-T Magnetic Resonance Scanner
- PET Imaging of Colorectal Cancer in Xenograft-Bearing Mice by Use of an 18F-Labeled T84.66 Anti-Carcinoembryonic Antigen Diabody
- How molecular imaging is speeding up antiangiogenic drug development
- In vitro and In vivo Characterization of 64Cu-Labeled AbegrinTM, a Humanized Monoclonal Antibody against Integrin {alpha}v{beta}3
- Positron Emission Tomography As an Imaging Biomarker
- A Thiol-Reactive 18F-Labeling Agent, N-[2-(4-18F-Fluorobenzamido)Ethyl]Maleimide, and Synthesis of RGD Peptide-Based Tracer for PET Imaging of {alpha}v{beta}3 Integrin Expression
- Characterization and Development of a Peptide (p160) with Affinity for Neuroblastoma Cells
- Molecular Targeting with Peptides or Peptide-Polymer Conjugates: Just a Question of Size?
- microPET Imaging of Glioma Integrin {alpha}v{beta}3 Expression Using 64Cu-Labeled Tetrameric RGD Peptide
- Preclinical Evaluation of the Breast Cancer Cell-Binding Peptide, p160