


Abstract
N-[[4-[[[(2-ethylphenyl)amino]carbonyl]amino]phenyl]acetyl]-Nε-6-[(2E)-1-oxo-3-(3-pyridinyl-2-propenyl)]-l-lysyl-l-2-aminohexanedioyl-(1-amino-1-cyclohexane)carboxamide (LLP2A) is a high-affinity, high-specificity peptidomimetic ligand (inhibitory concentration of 50% = 2 pM) that binds the activated α4β1 integrin found on a variety of malignant lymphoid cell lines. To better determine whether this ligand holds promise for imaging and therapy in lymphoid malignancies, 6 LLP2A derivatives, as LLP2A-1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (LLP2A-DOTA) and LLP2A-DOTA-polyethylene glycol (LLP2A-DOTA-PEG), were designed, synthesized, and radiolabeled with 111In. Comparative pharmacokinetic studies in mice with Raji B-cell lymphoma xenografts were then complemented by small-animal PET of the lead molecular LLP2A format using 64Cu-LLP2A-11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (64Cu-LLP2A-CB-TE2A). Methods: LLP2A-DOTA and LLP2A-CB-TE2A were prepared using solid-phase synthesis; LLP2A-DOTA-PEG2,000, LLP2A-DOTA-PEG5,000, LLP2A-DOTA-PEG10,000, (LLP2A-DOTA)2PEG10,000, and (LLP2A-DOTA)4PEG10,000 were prepared by PEGylation. 111In radiolabeling of DOTA and 64Cu radiolabeling of CB-TE2A conjugates yielded 370–1,850 and 3,700–7,400 kBq/μg (10–50 and 100–200 μCi/μg), respectively. The pharmacokinetics of the six 111In radioconjugates were studied in vivo using biodistribution data (4 and 24 h) and whole-body autoradiography (24 h) in mice with Raji tumor xenografts. 64Cu-LLP2A-CB-TE2A was imaged (4 and 24 h) on a small-animal PET scanner in the same mouse model. Results: The highest tumor uptake in pharmacokinetic studies was obtained with LLP2A-DOTA and (LLP2A-DOTA)4-PEG10,000. For 111In-LLP2A-DOTA (1 nM) at 4 and 24 h after injection, ratios of tumor to blood and tumor to nontumor (normal) organ (T/NT) were 8 to 35:1 for all organs or tissue except the spleen, marrow, and kidney, which were between 2:1 and 1:1. Tetravalent (LLP2A-DOTA)4-PEG10,000 (1.1 nM) had tumor uptake similar to the univalent LLP2A-DOTA but higher liver, marrow, and kidney uptake. The excellent T/NT of LLP2A was also demonstrated by small-animal PET with 64Cu-LLP2A-CB-TE2A at both 4 and 24 h after injection; obvious spleen targeting was apparent, but little kidney or liver activity was observed. Conclusion: Of the conjugates investigated, the univalent, non-PEGylated ligand 111In-LLP2A-DOTA exhibited the best T/NT ratios and showed the greatest potential for imaging of α4β1 in human lymphoma. Furthermore, this univalent non-PEGylated LLP2A format, as 64Cu-LLP2A-CB-TE2A, demonstrated excellent tumor targeting by small-animal PET and warrants further investigation as an agent for the study of α4β1 expression in human lymphoid malignancies.
Radiolabeled peptides as radioligands not only are useful tools for cancer staging but also are being considered for prognostic and therapeutic applications (1–3). Imaging with SPECT or PET can provide exquisite localization and may evidence possible new therapies using radionuclides with appropriate particle ranges (on peptide or peptidomimetic compounds targeting cell surface receptors) (4,5). Somatostatin receptor–directed tumor imaging and therapy of the last decade has been recently followed by imaging with gastrin or cholecystokinin-2 (CCK-2) receptor as the target (4,6).
Various integrins have demonstrated unique features worthy of close scrutiny, and other interesting and provocative candidates for molecular targeted imaging have emerged from the studies. Integrins are a family of adhesion molecules consisting of non–covalent-bound transmembrane subunits; large extracellular segments pair to create heterodimers with distinct adhesion capabilities (7). In mammals, 24 different receptors are created by the subunit assembly of 18 α- and 8 β-subunits. Integrin signaling has now been shown to play key roles in cell–cell and cell–matrix interactions, tumor angiogenesis, and metastasis (8). Integrins expressed on endothelial cells modulate cell migration and survival during angiogenesis, whereas integrins expressed on carcinoma cells enhance metastasis by facilitating invasion and movement across blood vessels. The α4β1 integrin may provide a unique target for radioligand imaging, perhaps providing prognosis regarding therapy of neoplastic disease, although it has been recognized previously to have some role in inflammation. As a heterodimeric transmembrane receptor that recognizes 2 well-known ligands—vascular cell adhesion molecule-1 and fibronectin (9)—in adults, α4β1 is believed to be largely restricted to expression on hematopoietic cells regulating lymphocyte trafficking. Monoclonal antibodies to α4β1 integrin have been widely studied for their therapeutic effects in diseases of the immune system (10,11). However, there have been concerns using this target for monoclonal antibody–based therapy because of the complexity of inflammation factors that could be elicited by systemic administration of biologically active monoclonal antibodies and could result in relatively low efficacy and safety ratios. Recent reports demonstrating α4β1 expression in leukemia, lymphoma, melanoma, and sarcomas (12), however, support its involvement in growth and metastasis (12) and have prompted the evaluation of this integrin as a prognostic molecular imaging target using a new specific ligand.
Screening of one-bead-one-compound combinatorial libraries (13) composed largely of organic building blocks, d-, and unnatural amino acids identified several high-affinity ligands that bind to α4β1 integrin and are activated on malignant lymphoid cells. The peptidomimetic N-[[4-[[[(2-ethylphenyl)amino]carbonyl]amino]phenyl]acetyl]-Nε-6-[(2E)-1-oxo-3-(3-pyridinyl-2-propenyl)]-l-lysyl-l-2-aminohexanedioyl-(1-amino-1-cyclohexane)carboxamide (LLP2A) was demonstrated with high-stringency competitive screening and shown to have uniquely high affinity (14).
A competitive cell-adhesion assay to immobilize CS-1 fibronectin peptide determined that the inhibitory concentration of 50% of LLP2A to Jurkat cells was 2 pM (14). In addition, LLP2A was found to be protease-resistant including being highly stable in human plasma. Several human hematopoietic malignancies (e.g., malignant B- and T-cell lymphoma and leukemia) demonstrated preferential binding of LLP2A over normal lymphocytes, suggesting this ligand binds an activated state of this integrin (14). Preliminary fluorescent in vivo imaging demonstrated selective fluorescence uptake both in Molt-4 human T-cell leukemia implants and in K562 ovarian tumors when they were transfected with α4β1; no uptake was detected in negative K562 nontransfected tumors.
The quantitative determination of such integrin expression by radionuclide imaging may provide enhanced diagnostic and prognostic information for several lymphocyte malignancies, leading to individualized therapy choices and new algorithms for monitoring response. In this study, we sought the LLP2A molecular construct providing in vivo the highest ratios of tumor (T) to nontumor (NT; normal) organs.
Here we report studies that explore the characteristics of 7 forms of LLP2A. Six 111In-radioconjugates, with variations in molecular weight, PEGylation, and valency (parameters known to influence the pharmacokinetics of targeting radioligands), were studied (Table 1). LLP2A was synthesized with an 111In binding chelate: LLP2A-1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (LLP2A-DOTA) and LLP2A-DOTA conjugated to PEGs of various sizes and structures (LLP2A-DOTA-polyethylene glycol [LLP2A-DOTA-PEG]). The pharmacokinetics and biodistribution in nude mice bearing human lymphoma Raji xenografts were determined. Small-animal PET was then used to study the selected lead LLP2A construct, conjugated with 11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-TE2A), a 64Cu chelate, as 64Cu-LLP2A-CB-TE2A. Observations from the 111In-radioconjugate biodistribution studies were reinforced by whole-body autoradiography (WBAR) of the 111In-radioconjugates and the small-animal PET of the selected LLP2A construct radiolabeled with 64Cu.
Characteristics of LLP2A Derivatives in Mice
MATERIALS AND METHODS
Synthesis of LLP2A-DOTA, LLP2A-CB-TE2A, and PEGylated LLP2A-DOTA
Materials.
Rink amide 4-methylbenzhydrylamine resin (loading, 0.65 mmol/g) was purchased from GL Biochem. N-hydroxybenzotriazole and 1,3-diisopropylcarbodiimide (DIC) were purchased from Advanced ChemTech. Fmoc-Aad(OtBu)-OH was obtained from Chem-Impex International, Inc., Fmoc-Ach-OH was purchased from NeoMPS, Inc., and Fmoc-Lys(Dde)-OH [Dde, 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl] was purchased from EMD Biosciences. DOTA-mono-N-hydroxysuccinimide (NHS)-Tris (t-Bu) ester was purchased from Macrocyclics. The CB-TE2A chelate salt (CB-TE2A.2TFA) was synthesized as previously described (15). All PEG-NHS esters were purchased from Nektar Therapeutics AL Corp. N,N-dimethylformamide (DMF) was purchased from VWR. Dichloromethane (DCM), methanol (MeOH), diethyl ether, and acetonitrile (CH3CN) were purchased from Fisher. All other chemical reagents were purchased from Sigma-Aldrich. Analytic high-performance liquid chromatography (HPLC) analyses and semipreparative HPLC purification were performed on an HPLC system (System Gold; Beckman). Matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis was performed on a mass spectrometer (BIFLEX III; Bruker).
Synthesis of LLP2A-DOTA and LLP2A-CB-TE2A.
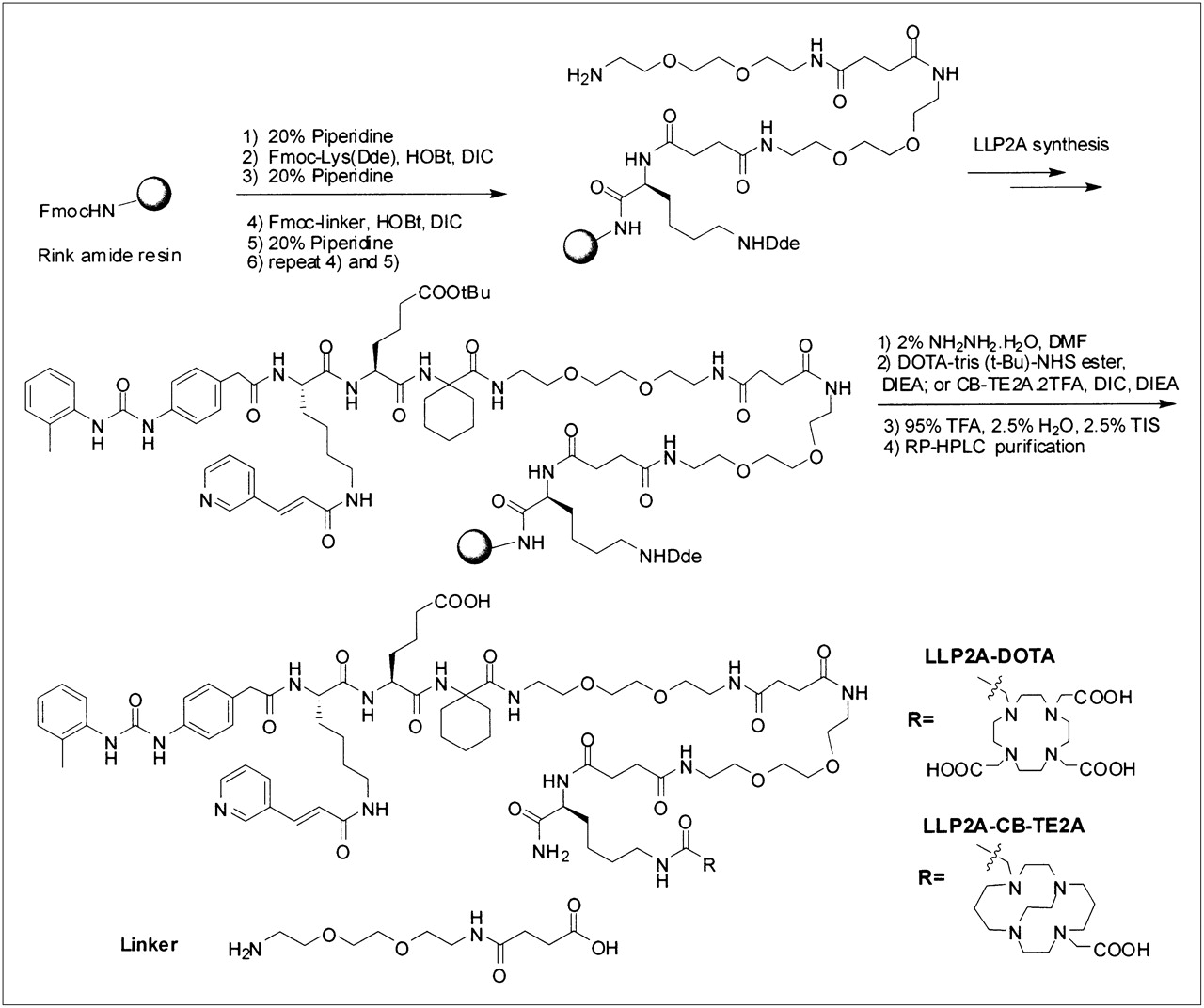
LLP2A-DOTA was designed to have DOTA attached to the side chain of Lys and 2 hydrophilic linkers between LLP2A and Lys(DOTA). The synthesis was performed on rink amide 4-methylbenzhydrylamine resin using a standard solid-phase peptide synthesis approach with Fmoc-tBu chemistry and N-hydroxybenzotriazole and DIC coupling. This synthesis is depicted in Figure 1. LLP2A is a peptidomimetic compound consisting of an organic moiety at the N terminus, a lysine derivative, and 2 amino acids (L-Aad and Ach): N-[[4-[[[(2-Methylphenyl)amino]carbonyl]amino]phenyl]acetyl]-Nε-6-[(2E)-1-oxo-3-(3-pyridinyl-2-propenyl)]-L-lysyl-L-Aad-Ach carboxamide (Aad: 2-aminohexanedioic acid; Ach: 1-amino-1-cyclohexane carboxylic acid).

Solid-phase synthesis of LLP2A-DOTA and LLP2A-CB-TE2A.
Fmoc-Lys(Dde)-OH was first coupled to the resin, then 2 linkers coupled to Lys (Dde) and LLP2A assembled on the N terminus of the linker as previously reported (14). The Dde protecting group was removed with 2% NH2NH2 in DMF twice (at 5 and 10 min) and the beads were washed with DMF, MeOH, and DMF again; this process was followed by the addition of DOTA-mono-NHS-Tris (t-Bu) ester (Macrocyclics) (3 Eq to resin) and N,N-diisopropylethylamine (DIEA) (6 Eq). The coupling reaction was conducted at room temperature overnight. The beads were thoroughly washed with DMF, MeOH, and DCM and then dried under vacuum for 1 h before a cleavage mixture of 95% trifluoroacetic acid (TFA):2.5% water:2.5% triisopropylsilane was added. Cleavage of compounds from the resin and removal of the protecting group were achieved simultaneously over 2 h at room temperature. The crude products were precipitated with cold ether and purified by semipreparative reversed-phase HPLC (Vydac column, 20 mm × 250 mm, 5 μm, 30 nm, C18), with a 45-min gradient from 100% aqueous medium (0.1% TFA) to 100% CH3CN (0.1% TFA). The flow rate was 5.0 mL/min. Ultraviolet detection was performed at 214 nM. The homogeneity of the compound was checked by analytic HPLC (Vydac column, 4.6 mm × 250 mm, 5 μm, 30 nm, C18), using a 25-min gradient from 100% aqueous medium (0.1% TFA) to 100% CH3CN (0.1% TFA), at a flow rate of 1 mL/min and ultraviolet detection at 214, 220, 254, and 280 nM. The purity was determined to be greater than 95%. The identity of the compounds was confirmed by MALDI-TOF MS. MS (MH+): 1,785.9.
The synthesis of LLP2A-CB-TE2A was similar to that for LLP2A-DOTA, except CB-TE2A was coupled to the side chain of lysine. The coupling of CB-TE2A was performed as previously reported using the CB-TE2A.2TFA salt with DIEA and DIC (15). In brief, CB-TE2A.2TFA salt (2.5 Eq to resin) was first dissolved in DMF. DIC (2.5 Eq to resin) and DIEA (5 Eq to resin) were added to this solution, and the solution was stirred for 30 min before adding it to the resin. The mixture was gently rotated overnight and filtered. The resin was washed with DMF, MeOH, and DCM and dried under vacuum. The compound was cleaved off the resin and purified by HPLC as described above. The purity was determined to be greater than 95%. The identity of the compounds was confirmed by MALDI-TOF MS. MS (MH+): 1,723.9.
Preparation of PEGylated of DOTA-LLP2A.
A d-Lys was added to the C terminus of DOTA-LLP2A, to facilitate the PEGylation of the ε-amino group. The synthesis of DOTA-LLP2A-d-Lys was the same as that of DOTA-LLP2A, except the first amino acid attached to the resin was Fmoc-d-Lys(Boc). PEGylation was achieved with PEG-NHS ester (Nektar Therapeutics AL Corp.) using the following protocol: 0.01 mmol of PEG-NHS ester was added to DOTA-LLP2A-d-Lys (2 Eq to each NHS) in 0.5 mL of dimethylsulfoxide, 0.1 mL of saturated NaHCO3, and 1 mL of water and rotated overnight at room temperature. The resulting solution was added to 50 mL of diethyl ether; the precipitate was collected, washed 3 times with diethyl ether, and dried. The white solid was dissolved in water and purified by gel filtration on a column (Superose 12; GE Healthcare). The sizing column was run in 30% acetonitrile in water with 0.1% TFA at a flow rate of 1 mL/min and detected at 254 nM. The product fraction was collected and lyophilized to give white powder. MALDI-TOF MS confirmed the structure. The 5 different forms of PEG-NHS—2,000-Da, 5,000-Da, and 10,000-Da mPEG-succinimidyl α-methylbutanoate (SMB); 10,000-Da SMB-PEG-SMB; and 10,000-Da 4-arm PEG-NHS—were used to generate LLP2A-DOTA-PEG2,000, LLP2A-DOTA-PEG5,000, LLP2A-DOTA-PEG10,000, (LLP2A-DOTA)2-PEG10,000, and (LLP2A-DOTA)4-PEG10,000, respectively (Table 1).
Radiolabeling and Quality Assurance
For 111In-LLP2A-DOTA, the 6 constructs shown in Table 1 (200–250 μg of each lot) were radiolabeled with carrier-free 111In (MDS Nordion) as 111InCl3 in 0.05 M hydrogen chloride (148–185 MBq [4–5 mCi], 15–20 μL). The final reaction mixture was adjusted to pH 5.5–6.5 using 0.1–0.25 M ammonium acetate, pH 5.3, and incubated for 30 min at 45°C−50°C. Ethylenediaminetetraacetic acid (0.1 M; 10–20 μL) was added to scavenge any unconjugated 111In.
The reaction mixtures of radiopeptidomimetic greater than 3.5 kDa were purified from the reaction mixture by molecular sieving chromatography or membrane dialysis (3.5-kDa cutoff) using 0.15 M phosphate-buffered saline (PBS) at pH 7 as dialyzer. Radiopeptidomimetics of less than 3.5 kDa were purified by HPLC (System Gold 128; Beckman Coulter) with a radioactive detector (Raytest USA), using C-18 or Superose 12 columns (GE Healthcare) eluted at 1 mL/min by 50% MeOH/H2O or PBS, respectively. The yield and quality assurance of the radiopeptidomimetics were evaluated by HPLC and thin-layer chromatography (TLC) using C-18 silica gel–precoated aluminum plates (EM Science) and cellulose acetate electrophoresis (CAE) (Gelman Sciences Inc.). TLC was eluted on 10% (w/v) ammonium acetate and methanol (1:1 v/v) solution, and CAE was performed on 0.05 M sodium barbital buffer at pH 8.6. The radiopeptidomimetic bands on CAE strips and TLC plates were exposed on phosphor imager plates and scanned using an imager (BAS 1800 II; Fuji Film). The data were converted as digital peaks by software (MultiGauge, version 2.1; Fuji Film). CAE, TLC, and HPLC analyses indicated that 111In-LLP2A-DOTA molecules, with or without PEG of various sizes and structures, were 85%−95% pure. Yields of radioactive ligand varied between 75% and 90%, and specific activities ranged between 666 and 1,850 kBq/μg (18 and 50 μCi/μg).
For 64Cu-LLP2A-CB-TE2A, 64Cu (Trace Life Sciences) as CuCl2 in 0.01 M hydrogen chloride and CB-TE2A-LLP2A were incubated in 0.14 M ammonium acetate, pH 7, at 95°C for 1 h as previously described (15). A small aliquot of the reaction solution was challenged in 10 mM ethylenediaminetetraacetic acid and evaluated by TLC, which demonstrated 100% radiochemical purity. The 64Cu-CBTE2A-LLP2A was formulated in PBS for injection.
Mouse Model
Raji cells, an American Type Culture Collection–certified cell line established from Burkitt lymphoma from a male human and obtained from that company, were grown in RPMI 1640 supplemented with 10% fetal calf serum. Female athymic BALB/c nu/nu mice (Harlan Sprague–Dawley) were maintained according to University of California animal care guidelines for mice on a normal diet ad libitum. At 6 wk of age, the mice received 4 Gy (400 rad) of external-beam irradiation to suppress immune responses to the Raji tumor xenografts; 3–5 d later, Raji cells (5 × 106) were injected subcutaneously on both dorsal flanks of the mice. Three weeks later, implants were measured in 3 orthogonal dimensions using a caliper; tumor volume was calculated using the formula for hemiellipsoids (16). At the time of LLP2A-conjugate injection, xenografts were well established with tumor volumes of 50–400 mm3. The pharmacokinetics were evaluated with 6–10 mice (10–18 tumors) in each group, and 3–5 mice (5–9 tumors) were studied at 4 or 24 h for each 111In radioconjugate (16).
Cell-Binding Studies
Direct live cell binding with 1 million Raji (α4β1 +) and HBT3477 (α4β1 −) cells was performed in parallel; these cells were incubated for 1 h with 1 ng of the 111In- or 64Cu-radiolabeled LLP2A construct, in a total volume of 150 μL of Tris-buffered saline with 1 mM MnCl3. Each study was performed in triplicate. The tubes were centrifuged and the cell pellet (bound) and supernatant (free) ligand counted for 1 min in the γ-well counter; the percentage bound to Raji and HBT cells was calculated. Because the percentage bound to HBT was relatively constant (3.5 ± 0.7), the percentage of the LLP2A constructs bound to Raji is shown without subtracting the percentage bound to HBT (Table 1). Binding of all the constructs was similar and reproducible, except for LLP2A-DOTA-PEG10,000, in which the binding was consistently less.
111In-labeled LLP2A-DOTA, LLP2A-DOTA-PEG10,000, (LLP2A-DOTA)2PEG10,000, and (LLP2A-DOTA)4PEG10,000 were also studied by competitive immunoassay (17). One million Raji cells per tube, in triplicate for each level, were resuspended in a solution of Tris-buffered saline, 1 mM MnCl3, and 5% fetal calf serum, with increasing amounts of radiolabeled peptidomimetic at 4 levels from 0.1 to 50 ng and in a total volume of 150 μL, and incubated for 1 h at room temperature. Cell pellets (bound counts) and supernatant (free counts) were collected and counted in a γ-well counter to determine percentage bound. The data were analyzed by Scatchard analysis to estimate the binding affinity and sites per cell (17). Negative cell binding performed in parallel with HBT cells was subtracted from that of the positive cells.
64Cu-LLP2A-CB-TE2A was evaluated by cell binding using the Lineweaver–Burke approach with increasing cells, as described by Lindmo (18).
Pharmacokinetics and Biodistribution
The pharmacokinetics of each of the 6 LLP2A-DOTA constructs were evaluated in mice (Table 1). The 111In-LLP2A-DOTA (740–1,850 kBq [20–50 μCi]), corresponding to a 1 nM (2 μg) injected dose, was compared with 10-nM doses of the same111In LLP2A-DOTA (20 μg); 20-μg doses for the remaining 5 LLP2A-DOTA constructs were studied in the same manner by being injected into the tail vein of mice bearing 1 or 2 Raji xenografts in the flanks. Blood and whole-body clearance and biodistribution data were obtained over the 24 h by previously described methods (Figs. 2–4⇓⇓) (16,19). The 111In dose was measured using a dose calibrator (CRC-12; Capintec, Inc.). To determine total-body clearances, the mouse was immediately counted after injection, and frequently over 24 h using 2 opposing isoresponsive sodium iodide detectors (Picker Nuclear). Blood clearances were determined by collecting blood samples from tail veins and quantitating the radioactivity in a γ-well counter (Packard). Decay-corrected radioactivity in the blood was expressed as percentage injected dose, using a weight-based theoretic blood volume. The data were best fit to a biexponential function. Tissue distribution data were obtained by sacrificing mice at 4 and 24 h after injection, weighing the tissues, and quantitating the radioactivity in the γ-well counter. The concentration in each sample was expressed as percentage injected dose per gram (%ID/g) (19).

Shown are blood (A) and whole-body (B) clearances of 111In-LLP2A-DOTA constructs in mice bearing Raji xenografts. Rapid blood clearance is demonstrated with only modestly increased blood area under curve for (LLP2A-DOTA)4-PEG10,000. Body-clearance rate correlated generally with retention in normal organs.

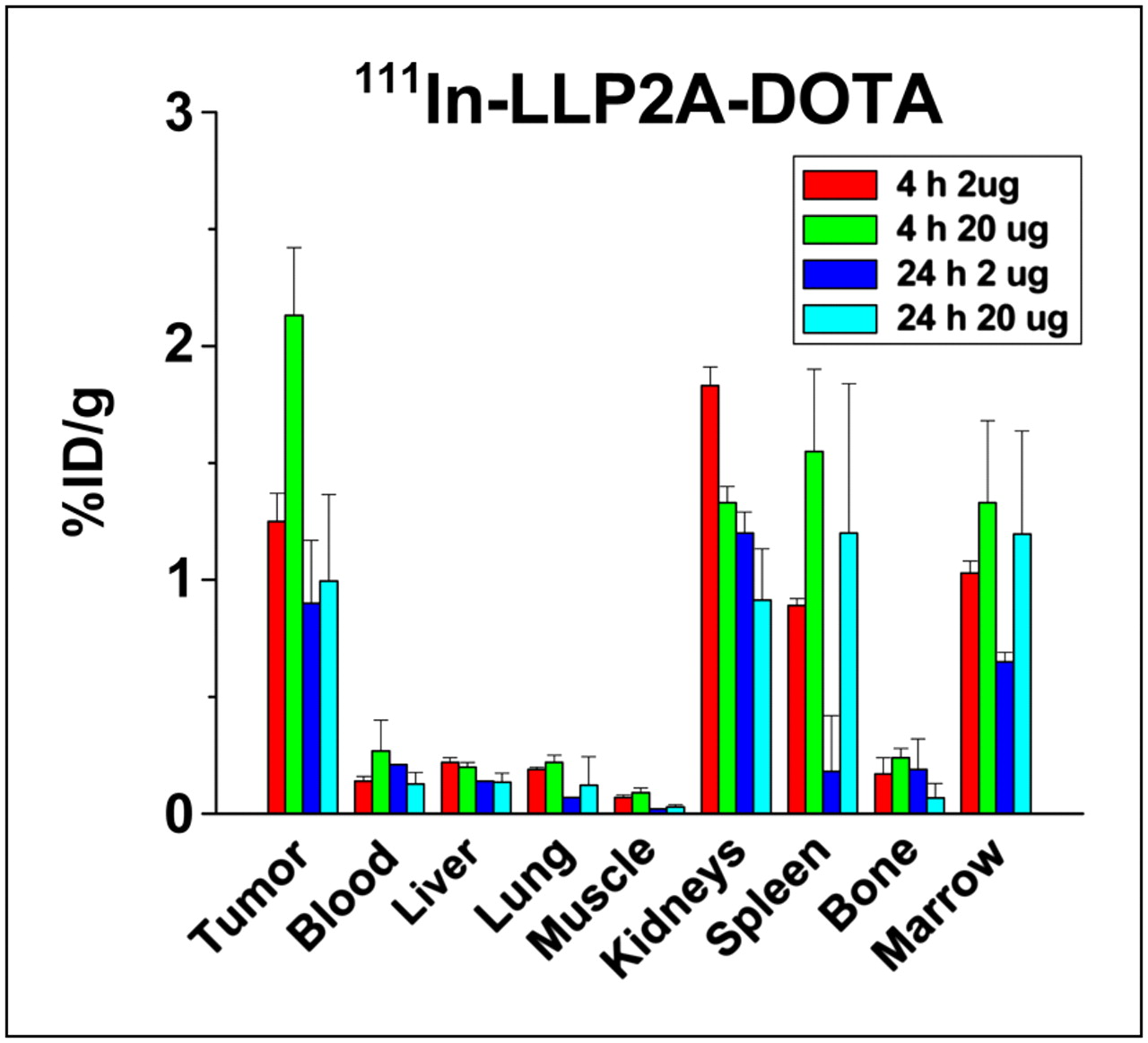
Biodistribution and tumor targeting of 111In-LLP2A-DOTA peptidomimetic injected at 2 μg (1 nM) and 20 μg (10 nM). Tumor uptake was generally decreased with higher dose, and spleen data suggest less uptake also. Liver, lung, and muscle are obviously unchanged. This comparison suggests that higher LLP2A microgram dose appears to preferentially cover tumor and lymphocyte α4β1 expression, decreasing binding of radiotracer 111In-LLP2A-DOTA but not affecting uptake in non–lymphoid tissues.

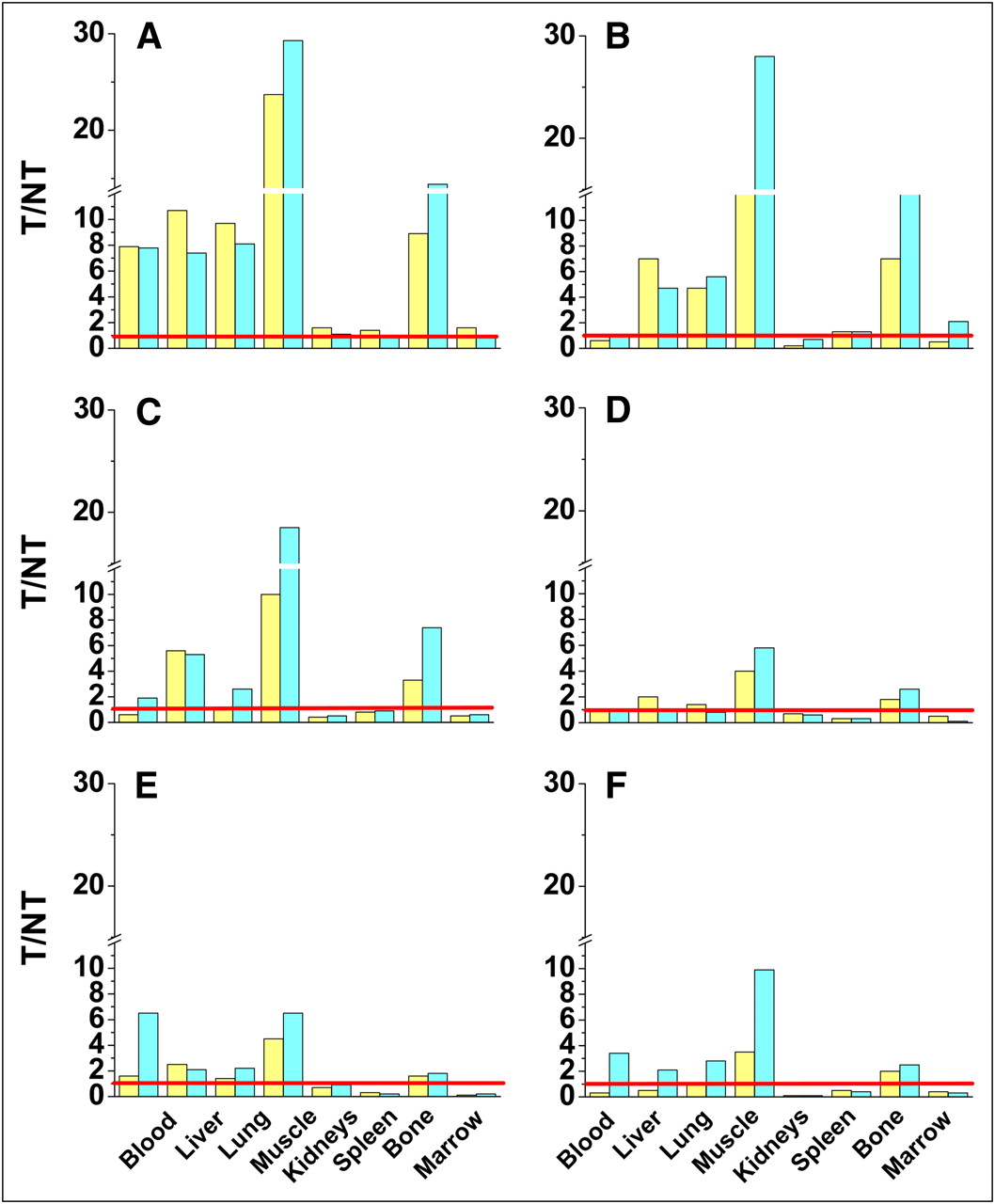
Biodistribution data is shown in T/NT ratios for all six 111In-LLP2A-DOTA constructs: LLP2A-DOTA (1 nM) (A), LLP2A-DOTA-PEG2,000 (B), LLP2A-DOTA-PEG5,000 (C), LLP2A-DOTA-PEG10,000 (D), (LLP2A-DOTA)2-PEG10,000 (E), and (LLP2A-DOTA)4-PEG10,000 (F). T/NT ratios for 111In-LLP2A-DOTA are notably superior.
WBAR
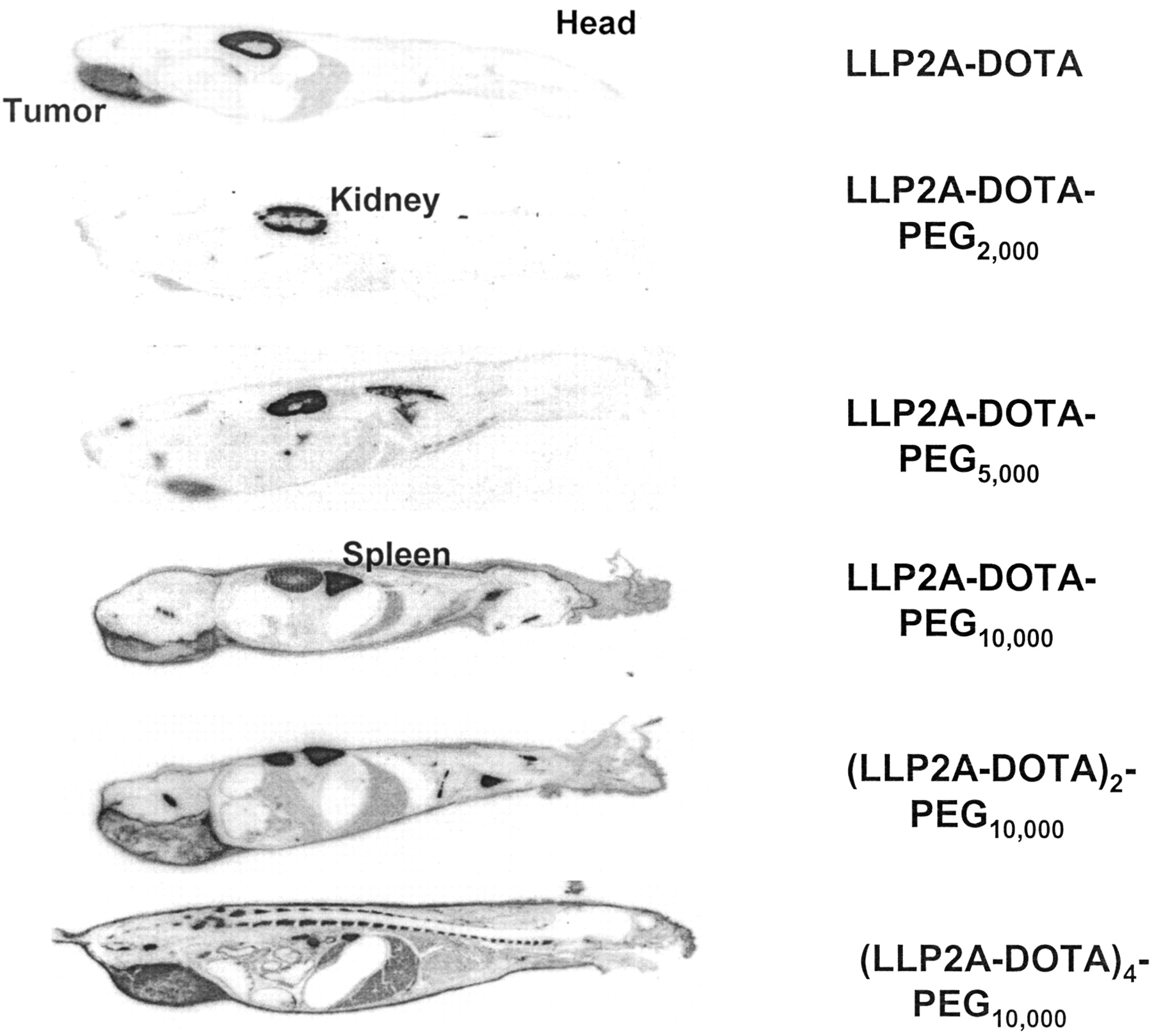
WBAR studies were conducted as previously described (19). At 24 h after injection of 3,700–7,400 kBq (100–200 μCi) (20 μg) of each 111In-LLP2A-DOTA construct, mice were anesthetized using an intravenous injection of aqueous solution (60 mg/100 μL) of sodium pentobarbital and fixed by flash-freezing in a hexane–dry ice bath and simultaneously embedded in 4% carboxymethylcellulose (Sigma). Fifty-micrometer-thick sagittal cryosections (−20°C) were cut with a Leica Cryopolycut (IMEB, Inc.). Sections showing the tumors, spleen, kidney, liver, and midline of the vertebral column were exposed to radiographic film sheets (AR; Kodak BioMax MS) (Fig. 5).

Whole-body autoradiographs of six 111In-LLP2A-DOTA constructs at 24 h after injection of 3,700–7,400 kBq (100–200 μCi) (20 μg). Tumor targeting is demonstrated to some degree with all 6 constructs, and wide variation in normal-organ uptake is apparent.
Small-Animal PET
Three mice were injected with 2 μg (3,700 kBq/μg [100 μCi/μg]) of 64Cu-LLP2A-CB-TE2A (intravenously) and imaged using a microPET Focus120 scanner (Siemens Medical Solutions). Data were processed with ASIPro software (Siemens Preclinical Solutions) (Fig. 6). Dynamic 15-min images were acquired at 4 and 24 h. Images were reconstructed using ASIPro and the maximum a posteriori iterative algorithm (18 iterations after 4 iterations 3-dimensional ordered-subset expectation maximization).
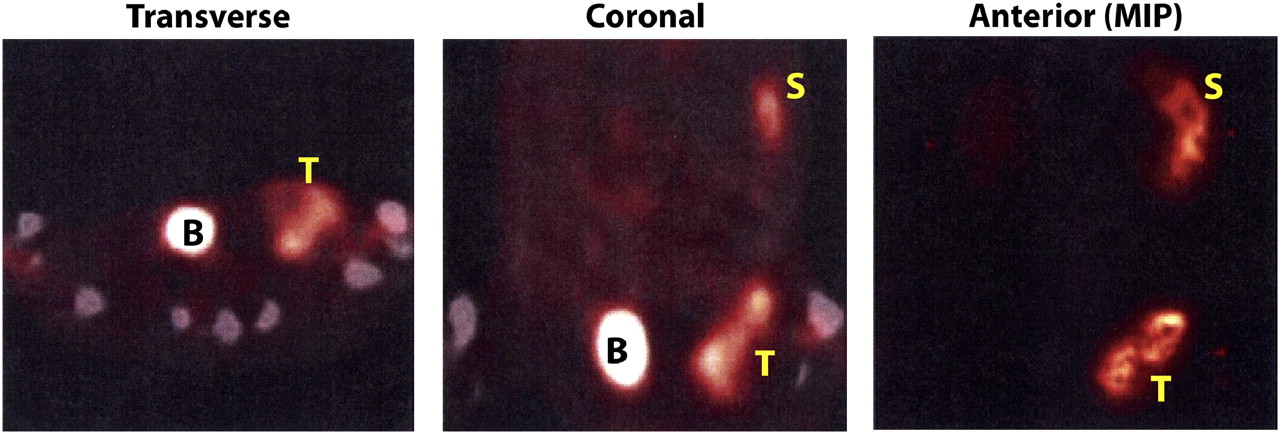
Small-animal PET and CT studies at 4 h after intravenous injection of 7,400 kBq (200 μCi) (2 μg) of 64Cu-LLP2A-CB-TE2A demonstrating excellent tumor targeting. Transverse section at mid pelvis and coronal section at mid abdomen and pelvis are both displayed fused with micro-CT images. Small-animal PET maximum-intensity-projection anterior image provides 24-h overview. Raji tumor (left flank) and spleen are easily seen; kidney and liver uptake appear minimal. Mouse was anesthetized with 3% isoflurane and maintained at 1.5%−2.0% isoflurane for duration of image acquisition. B = bladder; MIP = maximum-intensity projection; S = spleen; T = tumor.
RESULTS
Synthesis of LLP2A-DOTA, LLP2A-DOTA-PEG, and LLP2A-CB-TE2A
Milligram amounts of metal-free LLP2A-DOTA, LLP2A-DOTA-PEG, and LLP2A-CB-TE2A molecules were synthesized as described in “Materials and Methods.” Figure 1 depicts the approach used for the synthesis of the LLP2A-DOTA. MALDI-TOF MS analyses confirmed the structure of each of the 7 LLP2A constructs shown in Table 1.
Cell-Binding Studies
Cell-binding studies of Raji cells demonstrated 1.7 × 105 targets/cell for 111In-LLP2A-DOTA, an association constant of 2 × 108 M−1 by Scatchard analysis (17), and direct live cell binding of 25% of 1 ng/106 cells (Table 1). Similar live cell direct binding was demonstrated for all radiolabeled constructs, except for LLP2A-DOTA-PEG10,000, which had less. Scatchard analysis demonstrated that in competitive binding (LLP2A-DOTA)4PEG10,000 had an association constant of 2 × 109 M−1 (17), suggesting an increased effective affinity because of the 4 binding units per molecule. 64Cu-LLP2A-CB-TE2A Raji cell binding data by Lineweaver-Burke analysis indicated that the binding fraction approached 100% (18) and 34.7% binding of 1 ng/106 cells (Table 1).
Biodistribution Studies
Biodistribution and tumor targeting of the six 111In-LLP2A-DOTA constructs were evaluated in mice bearing Raji tumor xenografts. Blood and body clearances are given in Figure 2. Data analyzed from biodistributions at 4 and 24 h after injection are presented in Figures 2–4⇑⇑ as %ID/g for 111In-LLP2A-DOTA at 1 and 10 nM and T/NT comparisons for all 6 of the 111In-LLP2A-DOTA constructs.
All 111In-LLP2A-DOTA constructs target Raji tumor to some degree (Fig. 5; Supplemental Figs. 1 and 2; supplemental materials are available online only at http://jnm.snmjournals.org) but vary greatly in uptake by normal organs. The biodistribution of 111In-LLP2A-DOTA at 2 μg (1 nM) provided the highest tumor uptake and the best T/NT ratios. Kidney uptake, however, was at best only slightly less than tumor uptake, suggesting that renal tubular cells have moderate uptake and retention of at least this radiometal. The higher microgram dose of 111In-LLP2A-DOTA demonstrated that 10 nM of this peptidomimetic in the Raji mouse model could diminish the available tumor and spleen binding sites for the radioconjugate and thus decrease the %ID/g tumor uptake (Fig. 3).
111In-(LLP2A-DOTA)4-PEG10,000, the multivalent probe, had tumor uptake similar to 111In-LLP2A-DOTA, suggesting that 1.1 nM, with 4 times the relative binding ligands (4.4 nM binding units), did not interfere with the radioconjugate finding binding sites on tumor cells. 111In-(LLP2A-DOTA)4-PEG10,000 also had the best tumor retention at 24 h; however, the T/NT even at 24 h was still substantially less than that of 111In-LLP2A-DOTA because of the high normal-tissue uptake (Fig. 4; Supplemental Fig. 2). All 111In-LLP2A-DOTA constructs provide the T/NT ratios that were low for kidneys, spleen, and marrow, whereas T/NT for muscle is quite high. The highest accumulation in normal organs was observed with (LLP2A-DOTA)4-PEG10,000 in the kidneys (up to 45.25 ± 0.98 %ID/g at 24 h).
Imaging Studies
WBAR.
To provide visual support to the biodistribution data, radiolabeled LLP2A derivatives were visualized in mice by WBAR (20). Each of the six 111In-LLP2A-DOTA constructs was injected into 1 mouse at a dose of 3,700–7,400 kBq/20 μg (100–200 μCi/20 μg). Mice were sacrificed 24 h later and cryosectioned. Whole-body autoradiographic images for each of the 111In-LLP2A-DOTA constructs are shown in Figure 5. Autoradiographs of mice sections were selected to show a section through the tumor, liver, and kidney, except for (LLP2A-DOTA)4-PEG10,000, for which sections through the kidney were not shown because the high renal uptake hindered the visualization of any other organs. The uptake of radioactivity visualized for all 6 LLP2A-DOTA constructs, primarily in the kidneys, liver, and tumor, is consistent with the biodistribution data. High and homogeneous uptakes were visualized for the kidneys and liver (Fig. 5). The best tumor uptakes, visualized for LLP2A-DOTA and (LLP2A-DOTA)4-PEG10,000, support the conclusions obtained from the biodistribution studies.
Small-Animal PET.
The 64Cu-LLP2A-CB-TE2A images of 3 mice at 4 and 24 h after intravenous injection demonstrated that the tumor and spleen are the key targets imaged with this radioligand in this tumor mouse model. Figure 6 demonstrates the small-animal PET maximum-intensity-projection image and both a coronal body section and a transverse pelvic section at 4 h with fusion to the micro-CT images. Twenty-four-hour images were remarkably similar (data not shown). These imaging data corroborate the 111In-LLP2A-DOTA pharmacokinetic results, except the maximum-intensity projection demonstrated less observable kidney and bone marrow uptake than might have been expected.
DISCUSSION
Because radiolabeled peptide or peptidomimetic ligands that specifically target receptors on malignant tissue provide new options for cancer detection, prognosis, and therapy, substantial work has been done to evaluate the effects of PEGylation of such peptides to enhance T/NT (21,22). Some reports have shown that the length of the PEG polymer compared with its targeting ligand, however, may play a role inhibiting binding, and an increase in total molecular charge may result in enhanced renal tubular cell and liver (hepatocyte) uptake compared with non-PEGylated molecules (23). Other studies suggest PEGylation may decrease uptake in these organs (22,24). Even when T/NT ratios are increased with PEGylation (25), optimal formulations for peptide or peptidomimetic targeted radionuclide imaging or therapy have not frequently been developed.
Because integrins have recently become recognized for their possible wealth of prognostic information, the integrin αvβ3 has received particular notice. This integrin serves as a molecular marker of tumor angiogenesis imaging and has been extensively studied (7,8,22,26). Increase in signal to noise or T/NT information for αvβ3 has been demonstrated with multiligand constructs (26,27); other studies of tetravalent and dimeric arginine-glycine-aspartate peptides have demonstrated high liver and renal uptake and thus these performed less well than their monomeric radioconjugate constructs (28,29).
Information from these prior pharmacokinetic and imaging studies was given consideration in the design and study of the 6 distinct molecular constructs of LLP2A-DOTA presented in this article (Table 1). Although we hypothesized that α4β1 may provide a useful target for molecular imaging as a prognostic predictor of tumor biology and of response to therapy, particularly in B- and T-cell malignancies, the challenge for initiating the evaluation of that potential by clinical imaging trials is the development of a molecular-targeting format that maximizes α4β1 integrin binding while minimizing nontargeted uptake in other tissues.
PEGylation, as noted above, has become a recognized method for modifying the pharmacokinetics of small molecules (30) and generally results in an increase in circulating time and solubility and reduced antigenicity and susceptibility to protease (31). The LLP2A, a tripeptide peptidomimetic consisting of unnatural amino acids with an N terminus capped by an organic moiety, is naturally highly resistant to proteolytic degradation (14). Our goal in these investigations was to define the effect of various sizes of PEG and multivalency on pharmacokinetics, to select a format that could likely achieve good T/NT ratios in translational studies.
Conceptually, the LLP2A molecular variants were to fall into 1 of 2 categories: increase of molecular weight, obtained through PEGylation with linear univalent 2-, 5-, and 10-kDa PEGs, and increase of LLP2A valency achieved by creating a bifunctional LLP2A linked by a bifunctional linear PEG or a tetrafunctional LLP2A with a branched tetrafunctional (4-arm) PEG. Two modifications were made to LLP2A during its solid-phase synthesis. First, to facilitate radiolabeling, DOTA—a chelator that forms stable complexes with a variety of metals—was attached to LLP2A via a hydrophilic linker to form LLP2A-DOTA. Second, to facilitate PEGylation, a d lysine was added to the C-terminal end of LLP2A-DOTA (Fig. 1). The PEGs carried active hydroxysuccinimide esters (NHS) that formed stable amide linkages with the primary amines.
The pharmacokinetics and tumor-targeting efficacy of the 6 radiolabeled LLP2A-DOTA constructs were evaluated in mice bearing human lymphoma Raji B-lymphoma xenografts. The LLP2A peptidomimetic, selected from one-bead-one-compound libraries for its binding with high affinity to α4β1 integrins, had previously demonstrated promising tumor targeting as an LLP2A-streptavidin-dye conjugate in mice carrying human T-leukemia Molt-4 xenografts (14). Raji cells were used for these investigations in place of the less tumor-producing Molt-4, as Raji cells demonstrated equal or more α4β1 integrin targets in vitro (Table 1) and had previously shown more reproducible and consistent subcutaneous tumors in this well-studied nude mouse model (16). The studies were performed with radiolabeled products that all had high specific activity and were of good quality; all 6 constructs (2–17 kDa) were cleared rapidly from the blood with less than 10% remaining after 4 h (Fig. 2). Prolonged circulation time, a frequent result of PEGylation, was only modestly achieved until the PEGylated LLP2A molecular size was greater than 15 kDa, as can be seen as well by the range of the area under the curve (Table 1). The molecule with a 10-kDa PEG (LLP2A-DOTA-PEG10,000) demonstrated repeated decreased tumor cell binding in vitro (Table 1). This decrease is likely the result of the relatively long PEG physically interfering with binding as previously suggested by others (23).
Most of the radioactive univalent LLP2A-DOTA constructs cleared rapidly not only from the blood but also from the body, suggesting that these molecular constructs filtered freely through the kidney (Fig. 2). The LLP2A-DOTA at the 1 nM level provided the best tumor uptake and T/NT ratio (Figs. 3 and 4); tumor targeting and relative washout of the remaining nonlymphocytic tissues, except for the kidney, was also confirmed by the small-animal PET (Fig. 6).
When a 10-nM dose was injected, however, there appeared to be blocking of tumor (and arguably of spleen and marrow) radioconjugate uptake. This evidence of specific block or saturation effect of the tumor cells should not be surprising. Calculations of the 2 × 105 α4β1 sites defined in vitro by LLP2A on the Raji cells (Table 1) would suggest approximately 1013 sites on 100- to 300-mg mouse tumors; a 2% uptake of 10-nM LLP2A could mathematically result in the saturation of many of the most available targets. As demonstrated in other integrin studies, such a change in some organ uptake and not in others after target-blocking doses suggests specific targeting in the structures that could have uptake blocked (29). Lack of change in the liver and kidneys in this study supports the premise that the uptake in nonlymphoid organs is caused by a different mechanism; amounts in the 10-nM range likely had begun to specifically saturate the α4β1 targets on tumor cells and lymphocytes.
Rapid clearance and low tumor uptake of the 3 linear PEGylated constructs was observed (tumor range, 0.3–0.8 %ID/g at 4 h) (Supplemental Fig. 1). Because the injected nanomolar concentrations of 2 of these 3 constructs were similar and only 2 times those of the LLP2A, the low tumor uptake was considered likely to relate to the interference of the PEG. Because the circulation time had not been increased by these PEGylations, these constructs were not considered for further study.
The multivalent LLP2A variants were retained in the body longer than the other 4. In particular, the (LLP2A-DOTA)4-PEG10,000 increased renal and liver uptake may be due in part to the altered charge or lipophilicity from the tetrameric DOTA conjugate. Rapid phagocytosis of these preparations was not likely, as the quality assurance showed them to be monomeric. This 17-kDa branched tetravalent molecule also had somewhat prolonged blood clearance. The observed extended half-life in circulation of (LLP2A-DOTA)4-PEG10,000 suggests that the branched PEG may expand the Stoke radius of a PEG peptidomimetic conjugate. Because the nature of PEG already expanded this size beyond its actual mass (32), only the Stoke radius of the (LLP2A-DOTA)4-PEG10,000 molecule was likely to have been above the effective size-exclusion limit for first-pass glomerular filtration. Tumor uptake of the tetramer was similar to (and retention better than) that in the LLP2A 1-nM study, despite the calculations that the 4 units/molecule increased the ligand moles injected by a factor of 4, making it 4.4 nmol of LLP2A binding units, compared with the 1.1 molecular nanomoles. Although the tumor uptake and retention were relatively good by comparison, the enhanced tumor binding and effective avidity were not worth the trade-off with major increased renal and hepatic uptake and retention.
Thus, 111In-LLP2A-DOTA may provide the best planar or SPECT radiopharmaceutical to study the α4β1 target in patients. However, in the studies reported here, the promising 111In-LLP2A-DOTA data profile led to small-animal PET with 64Cu. 64Cu-LLP2A-CB-TE2A (33) demonstrated excellent imaging of the tumor targeting of the LLP2A by small-animal PET at both 4 and 24 h and warrants further investigation.
CONCLUSION
Although the tetrameric 111In-(LLP2A-DOTA)4-PEG10,000 appears to have the best retention in tumors, it would require successful modifications that decrease renal retention to be further considered for imaging or therapeutics. 111In-LLP2A-DOTA, at a dose not saturating the tumor α4β1 receptors, provides a good T/NT profile with most organs; the linear PEGylation with 2-, 5-, or 10-kDa PEG failed to extend its circulation time. The excellent targeting confirmed by small-animal PET using this lead LLP2A construct suggests that patient PET studies are likely to provide sensitive quantitative identification and follow-up of α4β1 expression in neoplastic tissues of patients with B- or T-cell lymphoid malignancies. Uptake detected in the spleen and marrow is likely related to α4β1 expression and therefore might also provide prognostic information for therapy in these diseases.
Acknowledgments
We thank the David Kukis Center for Molecular and Genome Imaging, University of California, Davis, for 64Cu radiolabeling and Gary Mirick and Aina Yuan, University of California, Davis Medical Center, Department of Internal Medicine, for performing cell-binding studies and pharmacokinetic studies, respectively. This work was supported by grant NCDDG U19 CA113298 from NCDDG and small-animal resource program grant U24 CA110804 from NCI.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication August 22, 2008.
- Accepted for publication January 6, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- First-in-Humans Evaluation of Safety and Dosimetry of 64Cu-LLP2A for PET Imaging
- Advances in Receptor-Targeted Radiolabeled Peptides for Melanoma Imaging and Therapy
- Chemically Programmed Bispecific Antibodies That Recruit and Activate T Cells
- Molecular Imaging of Very Late Antigen-4 ({alpha}4{beta}1 Integrin) in the Premetastatic Niche
- Halogenated Benzimidazole Carboxamides Target Integrin {alpha}4{beta}1 on T-Cell and B-Cell Lymphomas