


Abstract
18F-FDG PET and, more recently, PET/CT have been established as response biomarkers for monitoring cytotoxic or cytoreductive cancer therapies. With the advent of targeted cancer therapies, which are predominantly cytostatic, 18F-FDG PET is increasingly being used to monitor the therapeutic response to these agents as well. The impressive outcome of 18F-FDG PET studies in patients with gastrointestinal stromal tumors treated with imatinib mesylate brought to the forefront the use of this biomarker for assessing the response to targeted therapies. The use of 18F-FDG PET for this purpose has practical challenges, including quantitative analysis and timing of scans. This review provides a summary of clinical studies of targeted therapies done to date with 18F-FDG PET and provides guidance on practical issues to ensure the optimal interpretation of imaging data in drug development and for patient care.
During the last 2 decades, 18F-FDG PET has been extensively used as a biomarker to monitor responses to various chemotherapy agents. An early reduction in the PET signal, within days to weeks after the commencement of treatment, has been shown to correlate well with response and, in some cases, even survival (1). Although several small studies have demonstrated the utility of 18F-FDG PET in monitoring cytoreductive or cytotoxic treatment, there really have been no large trials to date. The use of 18F-FDG PET is not yet a standard approach for most tumor types, and more work clearly is needed to make it a standard of care.
The evolution of drugs directed at specific abnormalities that drive the malignant phenotype—the so-called targeted or mechanism-based drugs—has also taken place in the last 2 decades (Table 1) (2–6). These agents are predominantly cytostatic in nature; that is, they halt the growth of tumors rather causing significant tumor cell death. The promise of developing such targeted therapies is exemplified by the regulatory approval of the BCR-ABL and c-KIT inhibitor imatinib mesylate for chronic myeloid leukemia and gastrointestinal stromal tumors (GIST) (7–11), the vascular endothelial growth factor inhibitor bevacizumab in combination with chemotherapy for the treatment of colon cancer and non–small cell lung cancer (12), and the epidermal growth factor receptor (EGFR) inhibitor cetuximab in combination with chemotherapy for the treatment of metastatic colorectal cancer (13) or in combination with radiation therapy for the treatment of squamous cell carcinoma of the head and neck (14).
Cytostatic Agents Licensed or Currently Under Clinical Development
Because of the cytostatic or targeted properties of such agents, it is the contention of many oncologists and drug developers that the traditional endpoints used to evaluate cytotoxic therapies during early-phase clinical trials (phase I and II), that is, radiologic size changes and maximum tolerated dose, are insufficient and sometimes inappropriate for assessing the biologic activity of targeted therapies (15–18). In practice, however, these traditional methods are still used in current phase I and II trials despite existing knowledge. In a review of the literature on the subject of cytostatic agents in 2004, Parulekar and Eisenhauer (15) concluded that to enhance the use of nontraditional methods, more research would be needed to define suitable molecular measures of drug effects and the means to incorporate them into drug development. The use of PET to measure the therapeutic response has many advantages over biopsy- and surrogate tissue–based measurements, including the direct measurement of heterogeneous tumors and metastases repeatedly over time with reduced statistical bias (19).
It is against this background that we review the use of the only licensed and most widely available PET biomarker, 18F-FDG PET, for monitoring the treatment response in clinical trials of targeted therapies and for patient care. Compared with the number of citations regarding cytoreductive therapies, there are fewer citations on the use of 18F-FDG PET for the evaluation of targeted or cytostatic therapies. The impressive outcome of 18F-FDG PET studies in patients with gastrointestinal stromal tumors treated with imatinib mesylate brought to the forefront the use of this biomarker for assessing the response to targeted therapies. Therefore, we believe it is timely to review the literature on this subject and to provide a summary of the practical challenges and guidance to ensure the optimal interpretation of 18F-FDG PET data in the development of targeted therapies and for patient care. A review of applications follows an account of the biochemical mechanisms that regulate 18F-FDG uptake.
18F-FDG AS PET TRACER
18F-FDG is a glucose analog that is taken up into tumor cells by glucose transporters. Within cells, it is phosphorylated by hexokinase to 18F-FDG phosphate which, because of the charge on the molecule, is trapped within cells; unlike glucose 6-phosphate, 18F-FDG phosphate is not a substrate for further glycolytic metabolism, and its level of dephosphorylation to 18F-FDG is low (1,20). Most tumors express high levels of glucose transporters together with high activities of hexokinase and therefore show high levels of 18F-FDG uptake (21,22). In breast cancer, for instance, 18F-FDG uptake has been found to correlate with the microvasculature for delivering nutrients, GLUT-1 for the transport of 18F-FDG into cells, hexokinase for the entrance of 18F-FDG into glycolysis, the number of tumor cells per volume, the proliferation rate (also reflected in necrosis), the number of lymphocytes (not macrophages), and hypoxia-inducible factor 1α for the upregulation of GLUT-1 (22).
High levels of glycolysis and low levels of gluconeogenesis are hallmarks of tumor cells (23). The generally accepted hypothesis is that most anticancer drugs decrease 18F-FDG uptake because of a reduction in cell viability through increased cell killing or cell cycle blockade (24). An alternative mode of action involves the direct inhibition of glucose transport or phosphorylation. Prenen et al. (25) demonstrated that imatinib mesylate acts, at least in part, by downregulating glucose transporter recruitment to the plasma membrane; similar mechanisms have also been postulated for phosphatidylinositol 3-kinase inhibitors (26). In general, drugs that directly target the glucose uptake mechanism are expected to cause a rapid reduction in tumor 18F-FDG uptake within hours to days; this effect is related to pharmacodynamics rather than cell viability changes per se.
Preclinical imaging studies and ex vivo tissue analysis have provided confidence that 18F-FDG PET may be a useful pharmacodynamic or response biomarker for many targeted therapies. For example, a decrease in 18F-FDG uptake (55% after 48 h) was reported for lung cancer xenografts treated with gefitinib (EGFR blocker) (27). Furthermore, rat glioma xenografts treated with hypoxia-inducible factor 1α inhibitor YC-1 showed a significant decrease in 18F-FDG uptake after 3 d of treatment (28). In addition, an early reduction in 18F-FDG uptake (24 h) was reported for a GIST xenograft model after treatment with imatinib mesylate (29). These studies suggested that many cytostatic drugs may act earlier than or at least have the same timing window for response assessment as cytoreductive therapies (weeks to months) (1,24).
We do not know whether 18F-FDG will be useful for all cytostatic agents. Recent preclinical studies with some targeted therapies, including mitogenic extracellular kinase (30) and heat shock protein 90 inhibitors (31) in xenograft models, have demonstrated that 18F-FDG PET may be less sensitive as an early marker of the response to therapy; other radiotracers, including 3′-deoxy-3′-18F-fluorothymidine (30) and 68Ga-labeled anti–human epidermal growth factor receptor 2 (HER2) (31), respectively, may be more sensitive in these settings. These findings support the need (for cytostatic agents) to design an imaging paradigm involving preclinical testing before clinical imaging (32).
Next, we provide an overview of studies that have explored the use of 18F-FDG PET as a response biomarker in clinical trials or for patient care.
18F-FDG PET IN CLINICAL STUDIES OF TARGETED ANTICANCER AGENTS
A PubMed search was performed with search terms such as “cytostatic,” “targeted therapy,” and “FDG PET” as well as names of individual agents. This search retrieved cytostatic agents in various phases of development (Table 1). A few of them have been licensed for clinical use. Notable among these are imatinib mesylate, trastuzumab, and bevacizumab.
c-KIT Inhibitors
No other targeted agent has generated as much interest in response monitoring with 18F-FDG PET as imatinib mesylate (Table 2). Imatinib mesylate is now licensed for the treatment of GIST as well as for the first-line treatment of chronic myeloid leukemia. A PubMed search performed with the terms “imatinib,” “FDG,” and “GIST” retrieved 31 studies in which 18F-FDG PET was used to assess the response to therapy. The majority of GIST have activating mutations in the genes for either KIT (75%−80%) or PDGFR (5%−10%), 2 closely related receptor tyrosine kinases (52).
Summary of 18F-FDG PET Studies Conducted for Monitoring Responses to Cytostatic Therapies
Early clinical studies with imatinib mesylate for GIST revealed remarkable responses on 18F-FDG PET (Fig. 1). 18F-FDG PET responses occurred as early as 24 h after treatment and certainly within 1–2 wk (7,41,53–56). A variety of imaging methods were used in the assessment of 18F-FDG responses in these studies; these included visual changes, maximum standardized uptake values (SUVmax), and European Organization for Research and Treatment of Cancer (EORTC) recommendations. It is possible that consistency in the reporting of responses in these studies was achieved because of the large changes in 18F-FDG uptake characteristic of the tumor type and drug. The prevailing hypothesis is that early changes in 18F-FDG uptake are attributable to the effect of the drug on the glucose uptake mechanism: GLUT transporter expression and hexokinase activity (25,26). In some of these studies, tumors that showed a rapid resolution of positive 18F-FDG PET results subsequently showed a decrease in size on follow-up CT at 8 wk (7); therefore, 18F-FDG PET predicted the response on CT (7,34). An early decrease in the PET signal SUVmax (EORTC guidelines) after the commencement of imatinib mesylate treatment was also associated with longer progression-free survival (92% vs. 12%) (55). Furthermore, international (EORTC) PET response criteria (24) were compared with CT Hounsfield units in this setting (34). A lack of change in 18F-FDG uptake or an increase in 18F-FDG uptake was found to correlate with progression and poor survival (34).
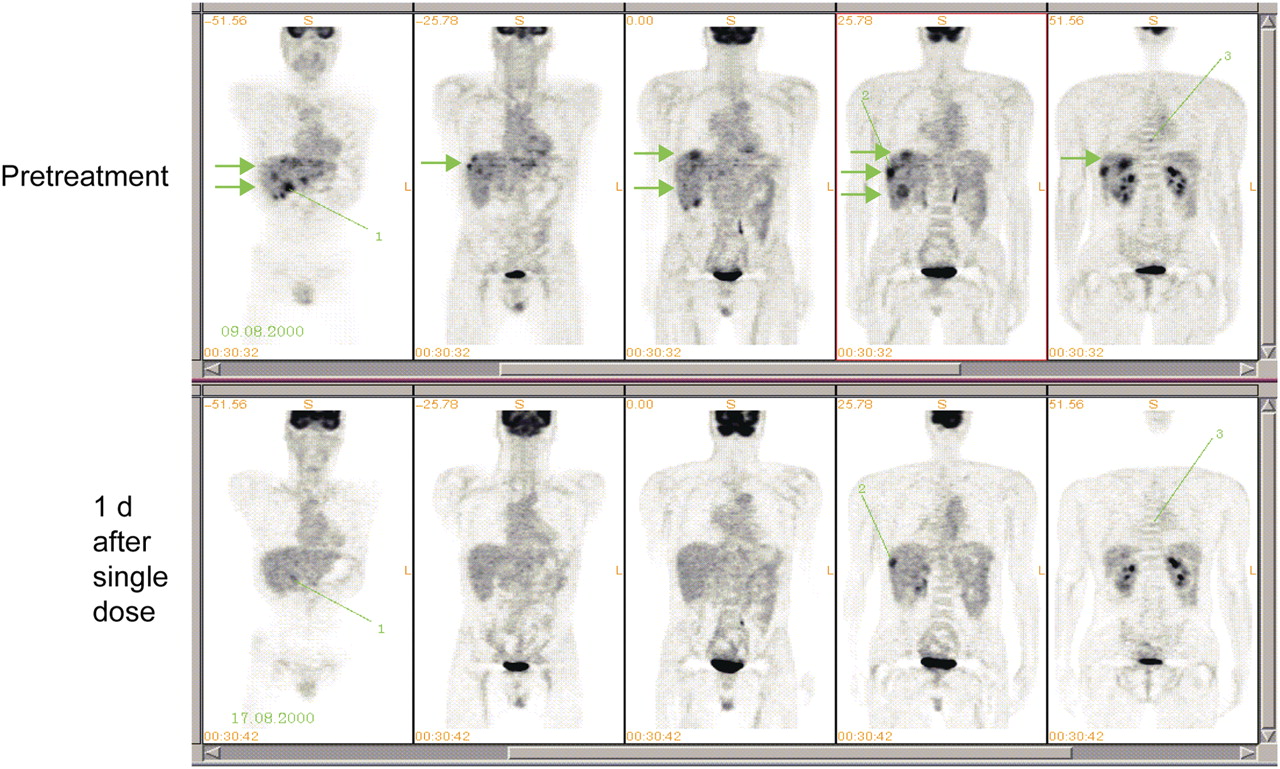
Multiple coronal 18F-FDG PET images of patient with GIST before (top) and 1 d after (bottom) treatment with imatinib mesylate. Arrows show liver metastases that rapidly changed on imaging; lines show lesions that did not. (Courtesy of Heikki Joensuu, Turku PET Center, Department of Oncology, Helsinki University Central Hospital, Helsinki, Finland.)
The successes of 18F-FDG PET in the early development of imatinib mesylate had an impact on guidelines for the management of GIST. The European Society of Medical Oncology Guidance Working Group recommended that both tumor size changes and tumor density changes on CT or consistent changes on MRI should be considered in response evaluations for GIST (57). 18F-FDG PET was recommended for equivocal cases or when the early prediction of a response is highly desirable, such as with preoperative cytoreductive therapies (57). The use of combined PET/CT allows the aforementioned CT criteria and PET metabolic activity to be measured in a single setting (58). Resistance to imatinib is a growing problem, with the most common mechanism of resistance involving specific mutations in the genes for the kinase domains of KIT or PDGFR. In this situation, other targeted agents, such as sunitinib, are available (52,59); serial 18F-FDG PET may be useful for monitoring the reversal of drug resistance in this setting.
EGFR Inhibitors
The EGFR kinase inhibitors gefitinib, elotinib, and cetuximab and, more recently, the EGFR/HER2 dual kinase inhibitor lapatinib have evoked interest because these agents block membrane-bound receptor tyrosine kinases that have important roles in tumor growth, resistance to apoptosis, and metastatic potential (60–62). A response to gefitinib was demonstrated for breast cancer in the neoadjuvant setting, with rapid decreases in the Ki67 index (63). High-profile failures were also reported; for instance, the drug used in combination with chemotherapy for lung cancer failed to improve overall survival in phase II and III trials (64,65). Experimental studies predicted the utility of 18F-FDG PET in monitoring the biologic activity of EFGR antagonists. For instance, rapid reductions in 18F-FDG uptake—as early as 2 h in cultured gefitinib-sensitive cells and within 48 h (by 55%) in gefitinib-sensitive xenografts—were reported by Su et al. (27); this effect was not seen in gefitinib-resistant cells (27). The decreases in 18F-FDG uptake were attributed largely to reduced translocation of GLUT-3 to the membrane (27). Despite this encouraging preclinical report, we found only one small clinical study in which 18F-FDG PET was used to monitor the response to gefitinib. In 5 patients, early changes in the 18F-FDG SUVmax at 2 d were associated with a progression-free interval of 12 mo (46). However, no conclusions could be drawn because of the small sample size.
No 18F-FDG studies with erlotinib have been reported.
Because the antitumor effect of lapatinib is due in part by its anti-EGFR effect (66), we performed a search of the use of 18F-FDG in this setting. A single study of 29 patients revealed the utility of 18F-FDG PET (47). In that study, a partial response to lapatinib in a patient with trastuzumab-resistant (HER2 and HER3 positive; estrogen receptor and progesterone receptor negative) breast cancer was associated with a 60% decrease in the 18F-FDG SUVmax, stable disease was associated with small to moderate (6%−42%) decreases in the 18F-FDG SUVmax, and 2 of 3 patients with progressive disease showed increases in the 18F-FDG SUVmax. In the patient who showed a partial response, the emergence of resistance was detected with 18F-FDG PET 2 mo before changes were seen on CT. In the patient whose SUVmax decreased despite disease progression on CT, the selected targeted lesions were assessed as stable disease by CT, but a new lesion appeared 2 mo after the start of treatment (47). These studies highlighted the need for consensus guidelines, such as those described by the EORTC (24), for reporting 18F-FDG responses in patients receiving targeted therapies so that small studies from different institutions can be compared.
Finally, a phase II trial of cetuximab in combination with leucovorin–5-fluorouracil–irinotecan for advanced gastric or gastroesophageal junction adenocarcinoma was performed with 18F-FDG PET and CT as endpoints for assessing efficacy (48). 18F-FDG PET and CT scans were obtained at baseline and after 6 wk of therapy; some patients had 6 more PET scans at 6-wk intervals. 18F-FDG PET was used to classify patients as metabolic responders and nonresponders. The results showed that 18F-FDG PET could correctly differentiate responders (who had a median time to progression of 16 mo) from nonresponders (who had a median time to progression of 11 mo) (48).
Phosphatidylinositol 3-Kinase–Mammalian Target of Rapamycin (mTOR) Axis Inhibitors
The phosphatidylinositol 3-kinase–mTOR axis is known to regulate glucose homeostasis in mammalian cells (67–69). Therefore, it has been postulated that 18F-FDG PET will be useful in monitoring the response to pathway inhibitors in this setting. Preclinical studies demonstrated rapid decreases in hexokinase activity after treatment with the mTOR inhibitor rapamycin; this finding could explain the reduced 18F-FDG uptake in mouse tumors treated with this drug (70). Despite this promise, the published literature lacks examples of clinical 18F-FDG PET studies of mTOR inhibition. This observation may reflect the small numbers of drug candidates in this class undergoing clinical evaluation with 18F-FDG PET as an endpoint. However, preliminary reports on small cohorts of patients have been presented at international meetings. For instance, Nogova et al. (71) demonstrated the utility of 18F-FDG PET as a pharmacodynamic biomarker of mTOR inhibition by everolimus (RAD001; Novartis Pharmaceuticals). In that study, a 1.4%−89.1% change in the SUVmax was reported for 8 patients at day 8, with partial recovery in 4 patients at day 28. Some of these changes could be classified as partial metabolic responses according to the EORTC guidelines. Furthermore, serial 18F-FDG PET responses in 19 patients with gastrointestinal, uterine, and neuroendocrine carcinomas and sarcomas treated with rapamycin were classified as partial metabolic responses (53%) and stable metabolic responses (47%) (72). In that study, changes in the 18F-FDG SUVmax were correlated with AKT activity but not with tumor proliferation or clinical outcome. Therefore, more research on the application of 18F-FDG PET in this setting is needed. In particular, it will be useful to differentiate effects on drug targets (AKT and translocation of glucose transporters to the cell membrane) from effects on cell viability and to determine how these affect clinical outcome.
Angiogenesis Inhibitors
These agents, which cause disruption of the abnormal vasculature formed by tumors, have generated immense interest during the last 2 decades. Unlike the situation for other cytostatic agents, it is difficult to find surrogate normal tissue biomarkers for assessment of the responses to antiangiogenic agents, although levels of circulating vascular endothelial growth factor have been used (73). At present, dynamic contrast-enhanced MRI and radiolabeled cyclic arginine-glycine-aspartic acid (RGD) peptide ligands are being evaluated as biomarkers for angiogenesis inhibition in tumors (74–76).
The antiangiogenic agent bevacizumab (Avastin; Roche) has generated remarkable responses when used in combination with chemotherapy for the treatment of colorectal liver metastases (12). In a study in which colorectal liver metastasis patients were treated with neoadjuvant bevacizumab and irinotecan and underwent PET/CT, complete PET responses were observed after 4 cycles of treatment (44). In addition, 18F-FDG PET/CT correctly predicted necrosis at pathology for 70% of patients, whereas CT alone did so for 35% of patients (44). In an early clinical trial of recombinant human endostatin, tumor blood flow and 18F-FDG uptake were determined by PET for 25 patients on days 28 and 56; both parameters generally decreased with increasing drug doses, but the effects were complex and, in some analyses, nonlinear (45). Blood flow increased at lower doses (30–60 mg/m2/d) but fell below the baseline by approximately 20% at doses of 120 mg/m2/d or more, with no further reduction at higher drug doses (45). Interestingly, the 18F-FDG SUV continued to increase through a dose of 180 mg/m2/d, before decreasing at doses of 300 mg/m2/d or more (45).
Because 18F-FDG shows high levels of extraction in tissues, changes in perfusion (decreases attributable to vascular pruning or increases attributable to reduced interstitial pressure and vascular normalization (77)) are likely to occur with antiangiogenic drug therapy and affect any static imaging protocol. The use of dynamic imaging may overcome this limitation and allow better interpretation of 18F-FDG data, such as to what extent changes in the 18F-FDG PET signal are attributable to effects on transport or phosphorylation.
These studies demonstrated that antiangiogenic drug therapies may have a complex, possibly multiphasic effect on 18F-FDG uptake and that dynamic analytic methods may be required for assessing responses. It may also be prudent to use multiple imaging approaches, such as blood flow measurements, as described by Herbst et al. (45), or hypoxia measurements, to fully understand the effects of drug therapy on tumor biology. Furthermore, measurement of the pharmacodynamic effects of antiangiogenic drug therapies with 18F-FDG at an earlier time point (within days) may be more appropriate than measurement at the end of cycle 1 or 2 of therapy, which may be more appropriate for monitoring changes in cell viability.
Endocrine Therapies
Although we have focused mainly on the utility of 18F-FDG PET for more recently discovered cytostatic agents, it is worth considering the literature on the oldest cytostatic agents—endocrine therapies—and their use in breast cancer. Breast cancer is associated with increased glucose metabolism because of the overexpression of GLUT-1 and hexokinase activity (78). A multivariate analysis (79) showed that a high SUV in primary breast cancer (SUV of >4), together with axillary node involvement on PET, was a highly significant independent prognostic factor for disease-free survival. Baseline 18F-FDG uptake was also correlated with prognostic markers in breast cancer, albeit with variable results (80).
Therapy of breast cancer is dominated by the use of estrogen receptor (ER) antagonists, such as tamoxifen and fulvestrant, or by the depletion of estrogens with aromatase inhibitors (63,81,82). ER expression determines sensitivity to endocrine therapy (82). For example, in patients receiving extensive pretreatment for metastatic breast cancer, ER levels determined by 16α-18F-fluoro-17β-estradiol PET predicted reductions in 18F-FDG uptake (after 1–3 cycles of treatment) and objective responses (83). 18F-FDG PET has been used in other studies to predict endocrine responses in breast cancer.
Studies with the single agent tamoxifen demonstrated an increase in tumor 18F-FDG uptake at approximately 1 wk after treatment in some patients (49,50,84). This effect also occurred with aromatase inhibitors, such as letrozole, at early time points (85). The increase in 18F-FDG uptake after therapy—the so-called “metabolic flare” reflecting hormone-induced changes in tumor metabolism—has been used as a pharmacodynamic endpoint for an early response (49,50). For instance, a metabolic flare after an estradiol challenge and then PET with 18F-FDG showed that a flare of more than 12% from the baseline was correlated with a response (percentage change of more than 20.9%) to any hormonal treatment in breast cancer and was also associated with better overall survival (P = 0.0062) (49). Another study showed that 18F-FDG uptake increased by 28.4% at about 1 wk after the commencement of tamoxifen therapy in patients who were responders; the value was only 10% in nonresponders (50). Because of the existence of 2 different types of 18F-FDG modulation, trials involving endocrine therapy should be appropriately designed to determine the biologic effects of drug therapies. For instance, to avoid the flare response, that is, to detect effects on cell viability, it is imperative to monitor responses after one or more cycles of therapy.
Androgen Receptor Blockade
Although a reduction in the serum prostate-specific antigen (PSA) level has been used in many studies as an indicator of a response to antiandrogen blockade in prostate cancer, it has been shown that a decrease in glucose uptake by prostate cancer cells precedes the decrease in the PSA level (86). The reason is that the PSA level in the circulation decreases only after prostate cancer cells have undergone apoptosis, a late event. A single clinical study reported the utility of 18F-FDG PET done at baseline and 1–5 mo after antiandrogen therapy with goserelin (51). 18F-FDG uptake decreased to 66.4% of that at baseline in all 10 patients studied, concomitantly with a reduction in the PSA level (51).
DISCUSSION
18F-FDG has been used extensively for the evaluation of cytoreductive therapies. Several guidelines have been developed to permit quantitative or at least semiquantitative assessments of changes, notably, the EORTC guidelines (24) and the National Cancer Institute guidelines (87). It is expected that guidelines for patient preparation and acquisition, reconstruction, and image analysis protocols will be broadly similar for cytoreductive therapies and cytostatic agents. For instance, it is just as essential to perform a baseline scan for cytostatic therapies. These guidelines need to be revised, however, to take into account the unique mechanisms of action of targeted therapies. Key among these is the issue of the optimal timing of PET scans after treatment. The time courses of changes in 18F-FDG uptake differ among therapeutic classes. Some of the effects are related to pharmacodynamics, whereas others are associated with reduced tumor cell viability (e.g., the assessment of responses to cytoreductive therapies). For example, imatinib mesylate decreases tumor 18F-FDG uptake within hours to days of the commencement of treatment, whereas endocrine therapies, such as tamoxifen, increases 18F-FDG uptake within the same time frame. In general, effects occurring from hours to days after the initiation of treatment reflect pharmacodynamics (e.g., a direct effect on glucose transporter expression or hexokinase activity). Effects occurring after approximately 2–3 wk or after 1–3 cycles of treatment are more characteristic of reduced cell viability.
Because the effects of targeted therapies on 18F-FDG kinetics are not always known a priori, we hypothesize that longitudinal preclinical studies with appropriate disease models and in which changes in 18F-FDG uptake are compared with molecular biochemical changes ex vivo could provide insights into mechanisms of action and expected clinical 18F-FDG profiles of novel agents. For most targeted therapies, the information from the literature review presented here (Table 2) will support a baseline scan followed by an early posttreatment scan, within 1 wk (pharmacodynamic effects), and a scan after 1 or 2 cycles of therapy (cell viability effects). For instance, with antiangiogenic therapies, it may be useful to evaluate drug effects at multiple time points to allow the assessment of drug effects on vascular pruning, normalization, and resultant cell viability changes (77,88,89). More research is required to support this suggestion. The optimal timing for posttreatment scanning for cytostatic agents therefore will be somewhat different from that proposed for cytoreductive therapies (2 wk) by the EORTC (24).
One issue that is difficult to resolve at present is the magnitude of change that can be considered significant. The EORTC guidelines suggested a threshold of 25% for a partial response. This proposed limit was met in a study of patients receiving imatinib mesylate for GIST (7); changes on PET were correlated with clinical outcomes. We do not expect correlations between early changes on 18F-FDG PET and clinical outcomes for all targeted therapies. With cytostatic agents, it is important to understand the cause of the change in 18F-FDG uptake rather than purely basing the interpretation of change on test–retest reproducibility (90). Several new therapeutic agents may affect glucose transporter expression or hexokinase activity directly; in contrast, with cytoreductive therapies, the change is largely attributable to a reduction in cell viability (24). The different mechanisms of action may lead to differences in the correlation of changes in 18F-FDG uptake with clinical outcomes. Therefore, it is not known whether the same EORTC response criteria will be appropriate for all classes of molecularly targeted therapeutic agents, particularly in the early assessment of pharmacodynamics, as these changes may not predict clinical outcomes. This topic should be reviewed further as more data on cytostatic agents become available.
It is expected that most of the biologic effects of targeted therapies will be predictable from preclinical studies, such that a clinical trial is an extension of the preclinical proof of concept. Despite their expected (theoretic) effects on glucose metabolism, some drug classes may not affect 18F-FDG uptake significantly. An example is the inability of 18F-FDG PET to predict responses in tumors with BRAF mutations treated with the mitogenic extracellular kinase inhibitor PD0325901 (30); preclinical studies demonstrated that 3′-deoxy-3′-18F-fluorothymidine PET was a better marker of therapeutic responses than 18F-FDG PET. In addition to assessing metabolism, it is probably prudent to examine the impact of therapy on perfusion, at least in a subset of patients. Given the available data, we suggest that all studies with antiangiogenic or antivascular therapies or studies involving drugs with potential antiangiogenic effects should be undertaken initially with a dynamic imaging protocol to enable the dissection of perfusion effects from true metabolism effects (91).
CONCLUSION
18F-FDG PET and PET/CT are useful endpoints for assessing responses to targeted therapies. The biologic basis of changes in 18F-FDG uptake may be more complex than those for traditional cytoreductive therapies. This factor may affect the timing of posttreatment scans and the clinical significance of the magnitude of changes. Preclinical studies with appropriate disease models may help to determine the optimal timing for imaging and the biologic relevance of the changes seen.
Acknowledgments
Eric O. Aboagye's laboratory is funded by Cancer Research U.K. (grant C2536/A5708) and the U.K. Medical Research Council (U1200.02.005.00001.01). Kaiyumars B. Contractor is supported by Cancer Research U.K. (grant C37/A5610).
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- 1.↵
- 2.↵
- 3.
- 4.
- 5.
- 6.↵
- 7.↵
- 8.
- 9.
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- Received for publication November 12, 2008.
- Accepted for publication January 28, 2009.





{kind=link}
Jump to section
Related Articles
Cited By...
- Phase I Study of Rapid Alternation of Sunitinib and Regorafenib for the Treatment of Tyrosine Kinase Inhibitor Refractory Gastrointestinal Stromal Tumors
- Variability of Proliferation and Diffusion in Different Lung Cancer Models as Measured by 3'-Deoxy-3'-18F-Fluorothymidine PET and Diffusion-Weighted MR Imaging
- Safety, Pharmacokinetics, Pharmacodynamics, and Antitumor Activity of Dalantercept, an Activin Receptor-like Kinase-1 Ligand Trap, in Patients with Advanced Cancer
- Resveratrol Suppresses Cancer Cell Glucose Uptake by Targeting Reactive Oxygen Species-Mediated Hypoxia-Inducible Factor-1{alpha} Activation
- Comparison of EORTC Criteria and PERCIST for PET/CT Response Evaluation of Patients with Metastatic Colorectal Cancer Treated with Irinotecan and Cetuximab
- Acute Cytotoxic Effects of Photoimmunotherapy Assessed by 18F-FDG PET
- Measuring Oncogenic Signaling Pathways in Cancer with PET: An Emerging Paradigm from Studies in Castration-Resistant Prostate Cancer
- 18F-FDG PET as a Surrogate Biomarker in Non-Small Cell Lung Cancer Treated with Erlotinib: Newly Identified Lesions Are More Informative Than Standardized Uptake Value
- Multimodal Imaging with 18F-FDG PET and Cerenkov Luminescence Imaging After MLN4924 Treatment in a Human Lymphoma Xenograft Model
- Small-Animal PET of Tumor Damage Induced by Photothermal Ablation with 64Cu-Bis-DOTA-Hypericin
- Low-Dose 18F-FDG PET/CT Enterography: Improving on CT Enterography Assessment of Patients with Crohn Disease
- The future of imaging: developing the tools for monitoring response to therapy in oncology: the 2009 Sir James MacKenzie Davidson Memorial lecture
- Assessing Tumor Response to Therapy