


Visual Abstract

Abstract
This phase I trial aimed to assess the feasibility and toxicity of combining the poly(adenosine diphosphate–ribose) polymerase inhibitor olaparib with 177Lu-DOTATATE in patients with somatostatin receptor–positive tumors, with the goal of enhancing treatment efficacy through the inhibition of tumor cell DNA repair mechanisms. Methods: Eighteen patients were enrolled, mostly with pancreatic or small intestinal neuroendocrine tumors or atypical lung carcinoids. Patients received a standard dose of 177Lu-DOTATATE (7,400 MBq) for up to 4 cycles, combined with escalating doses of olaparib (50–300 mg twice a day [BID]). The primary objective was to evaluate toxicity using National Cancer Institute Common Toxicity Criteria version 5.0. Secondary objectives included time to progression, overall survival, response rate, and dosimetry variables. Results: The combination of olaparib and 177Lu-DOTATATE was generally well tolerated. Five patients did not complete the 4 cycles because of progression, noncompliance, and carcinoid crisis after the first 177Lu-DOTATATE infusion. Among the remaining patients, thrombocytopenia was the primary dose-limiting toxicity, observed in 3 patients at the 300-mg dose level. Other toxicities were mild, predominantly low-grade bone marrow suppression, nausea, and fatigue. Conclusion: This study demonstrates that combining olaparib with 177Lu-DOTATATE is feasible, with toxicity primarily related to thrombocytopenia. On the basis of the findings, we recommend a starting dose of 200 mg BID for future studies, with the potential to escalate to 300 mg BID depending on patient tolerance. Further investigation in larger, randomized trials is warranted to assess the clinical efficacy of this combination and optimize dosing strategies.
- peptide receptor radionuclide therapy
- PRRT
- 177Lu-DOTATATE
- somatostatin receptors
- PARP inhibitor
- neuroendocrine tumors
Peptide receptor radionuclide therapy (PRRT) with 177Lu linked to a somatostatin analog is a mainstay strategy for patients with low- to intermediate-grade metastatic tumors expressing somatostatin receptors. 177Lu-DOTATATE was formally approved for gastroenteropancreatic neuroendocrine tumors (GEPNETs) after the NETTER-1 trial, which demonstrated increased progression-free survival compared with somatostatin analogs (1) but no survival advantage with longer follow-up (2). This targeted radiation therapy may provide a prolonged treatment effect, sometimes lasting for several years, but some patients experience suboptimal responses or early relapse, and ultimately all patients will progress, highlighting the need for improved treatment strategies.
Strategies to optimize treatment efficacy include modifying the number of treatment cycles or adjusting the administered activity based on the absorbed doses to critical organs, such as the kidneys (3–5). In addition, because 177Lu-DOTATATE is generally well tolerated, there is potential to explore combination therapies. Feasibility studies have been performed with 177Lu-DOTATATE combined with chemotherapy agents such as capecitabine and temozolomide (6–8) but indicated increased toxicity with the combination (9), as well as with the mTOR inhibitor everolimus (10).
One limitation of 177Lu-DOTATATE is the tumor cells’ ability to repair DNA damage. 177Lu decays primarily by emitting β-particles, which penetrate up to 1.5 mm in tissue, predominantly causing single-strand breaks in the DNA. However, the primary cell-killing effect of ionizing radiation is due to double-strand breaks, because these are more challenging to repair than single-strand breaks. Poly(adenosine diphosphate–ribose) polymerase 1 (PARP-1) inhibitors are pharmaceuticals that inhibit the repair of DNA damage (11). Combining a PARP inhibitor with 177Lu-DOTATATE could sensitize tumor cells to β-irradiation and increase the likelihood of tumor cell death by preventing effective repair of radiation-induced damage (12,13).
This combination strategy is theoretically appealing and has been preclinically investigated by Nonnekens et al. (13), who demonstrated that cells expressing somatostatin receptors could be synergistically sensitized to PRRT when combined with the PARP inhibitor olaparib. This combination resulted in increased cell death and reduced cellular proliferation compared with PRRT alone, primarily because of a greater number of double-strand breaks, leading to genomic instability (13).
To optimize the sequence of this combination, biokinetic modeling was conducted (14). The published results suggest that to maximize the tumor–to–normal tissue absorbed dose ratio, olaparib should be administered approximately 24 h after the start of the 177Lu-DOTATATE infusion, thereby minimizing the risk of bone marrow toxicity. Olaparib administration should continue for up to 4 wk, after which the irradiation dose to the tumor becomes minimal and no additional effect from PARP inhibition is expected. This approach also allows normal tissue time to recover from nonspecific systemic side effects associated with olaparib.
These findings informed the design of a phase I clinical trial aimed at assessing the toxicity and feasibility of combining 177Lu-DOTATATE with olaparib (177Lu-DOTATATE and olaparib in somatostatin receptor positive tumors, NCT04375267). Here, we report the primary objective of the study, which focuses on the assessment of toxicity.
MATERIALS AND METHODS
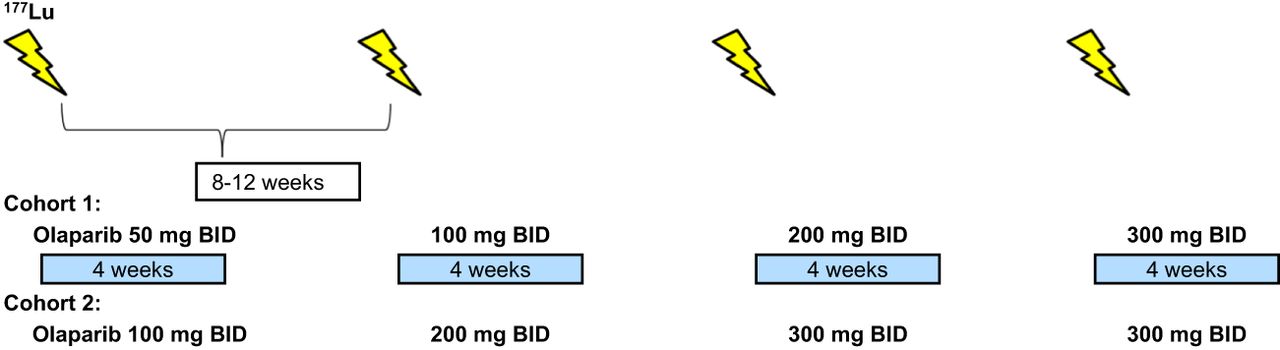
This prospective phase I trial evaluated the combination of a standard dose of 177Lu-DOTATATE (7,400 MBq for up to 4 infusions) with individual dose escalation of olaparib (Fig. 1). The study was approved by the Swedish Ethical Review Authority, and all patients provided informed consent before study-specific procedures. The primary objective was to assess the toxicity and feasibility of the combination, measured by Common Terminology Criteria for Adverse Events version 5.0 (National Cancer Institute). Secondary objectives included time to progression, overall survival, time to death from any cause, response rate, and dosimetry variables.

Schematic overview of study design.
Eligible patients had NETs or other tumors expressing somatostatin receptors, as confirmed by 68Ga-DOTATATE PET, including grade 2–3 GEPNETs, other NETs after standard therapy, or inoperable meningiomas not suitable for external radiotherapy. Patients were required to have documented disease progression within the past 14 mo and to have regional or distant metastases, or unresectable localized disease. Tumors had to be measurable according to RECIST 1.1, and patients needed a performance status of 0–1 with a life expectancy greater than 6 mo. Patients were required to be more than 18 y old (no upper age limit) and have adequate organ function, including a glomerular filtration rate greater than 50 mL/min. Key exclusion criteria included grade 1 GEPNETs, previous treatment with 177Lu-DOTATATE, antitumoral treatment (chemotherapy, tyrosine kinase inhibitors, and interferon) within 4 wk, or persisting toxicity from prior treatment. Patients with significant heart disease (New York Heart Association class III–IV) or extensive liver metastases with impaired liver function (>grade 1 of the Common Terminology Criteria for Adverse Events) were also excluded.
Pretreatment work-up included a CT scan of the thorax and abdomen, 18F-FDG PET/CT, 68Ga-DOTATATE or 68Ga-DOTATOC PET within 6 wk, iohexol clearance testing, and comprehensive blood sampling. The 177Lu-DOTATATE infusion was administered according to clinical practice with amino acid support (Vamin; Fresenius Kabi), with 1,500 mL given intravenously over 6–8 h starting 30 min before the radiopharmaceutical. Antiemetics, such as corticosteroids and 5-hydroxytryptamine type 3 inhibitors, were given, and 7,400 MBq of 177Lu-DOTATATE was infused intravenously over 30 min. Starting on day 2 (24 h after infusion), patients received 4 wk of oral olaparib twice a day (BID).
The study planned to include 18 patients. Cohort 1 started with olaparib at 50 mg BID for the first cycle, with dose escalation in subsequent cycles to 100, 200, and 300 mg BID if no dose-limiting toxicities (DLTs) occurred (Fig. 1). 177Lu-DOTATATE infusions were repeated every 8–12 wk for 4 cycles. A DLT was defined as any treatment-related reaction of grade 3 or higher of the Common Terminology Criteria for Adverse Events deemed attributable to olaparib and affecting treatment continuation (excluding short-term grade 3 nausea because of the 177Lu-DOTATATE infusion). If a DLT occurred at the 50-mg dose level, olaparib was discontinued, and the patient continued with 177Lu-DOTATATE alone. If a DLT occurred at higher doses (100, 200, or 300 mg), the subsequent cycle was deescalated by 1 dose level. Depending on cohort 1 outcomes, the starting dose in subsequent cohorts could be increased to 100 mg BID.
SPECT imaging for dosimetry was performed after each cycle. Patients were monitored with weekly blood tests and follow-up visits at 2 and 4 wk after infusion. CT scans of the thorax and abdomen was performed after 2 and 4 cycles and every 3 mo thereafter. 68Ga-DOTATATE or 68Ga-DOTATOC PET was performed at 3 and 12 mo after the last 177Lu-DOTATATE infusion and then annually. If the 18F-FDG PET/CT scan was positive at baseline, it was repeated at the same intervals.
RESULTS
In total, 18 patients were included in the study. Most patients had pancreatic NETs (n = 7), small intestinal NETs (n = 4), or atypical lung carcinoids (n = 4). All patients had a performance status of 0–1, with a median age of 65 y (range, 42–84 y) and a Ki-67 index between 7% and 50%. Detailed patient characteristics are presented in Table 1.
Patient Characteristics
All patients in the first cohort were escalated to 200 mg or more of olaparib, and the starting dose was increased to 100 mg BID for the second cohort. Subsequent cycles were dosed at 200 and 300 mg BID. Three patients had progressive disease after the first 2 cycles and were removed from the study. Three patients did not escalate to the maximum dose despite no DLT: 1 because of grade 1 abdominal pain, 1 because of prolonged grade 2 thrombocytopenia, and 1 because of grade 2 nausea and fatigue. One patient was noncompliant, and another experienced a carcinoid crisis after the first 177Lu-DOTATATE infusion, leading to study discontinuation. Consequently, 10 patients completed escalation of olaparib: all reached 200 mg BID without a DLT, and 7 reached 300 mg BID. Three patients developed grade 3 thrombocytopenia at the 300-mg dose level (Table 2), which resolved to grade 0–1 within 6 mo. Two of these patients had bone metastases.
Cycles, Escalated Dose of Olaparib, and DLT
Reported adverse events occurring in at least 3 patients are listed in Table 3, and additional rare events in fewer than 3 patients are listed in Supplemental Table 1 (supplemental materials are available at http://jnm.snmjournals.org). The most common adverse events, aside from bone marrow toxicity, were low-grade nausea and fatigue. Lymphocytopenia rated at least grade 3 was common but not classified as a DLT, because it is typically associated with PRRT. Four severe adverse events were reported, all requiring hospitalization or extended hospital stay, but nonattributable were to olaparib; these adverse events were infection, traumatic fall, congestive heart failure, and carcinoid crisis. The carcinoid crisis occurred after the first infusion of 177Lu-DOTATATE; this patient did not receive olaparib, and no further study-related treatment was administered.
Adverse Events in at Least 3 Patients
Longer follow-up is needed to determine efficacy and other secondary endpoints, but RECIST assessment at 6 mo follow-up demonstrated a disease control rate of 69% (11/16, excluding the 2 patients who were removed from study because of noncompliance and carcinoid reaction). Three patients with pancreatic NETs (2 patients with grade 3 and 1 patient with a high grade 2) had progressive disease after just 2 cycles, 1 patient had progressive disease after 4 cycles, and 1 patient had progressive disease at 6 mo follow-up. Twelve patients were still participating in the study at 6 mo follow-up and had partial remission (3 patients), stable disease (8 patients), and progressive disease (1 patient).
DISCUSSION
This phase I trial demonstrates the feasibility of combining the PARP inhibitor olaparib with 177Lu-DOTATATE. On the basis of our findings, we recommend that future studies begin with a dose of olaparib at 200 mg BID, escalating to 300 mg in subsequent cycles if bone marrow function permits.
PARP inhibition is a promising strategy for enhancing PRRT efficacy. As monotherapy, PARP inhibitors generally do not harm normal cells but are effective against DNA repair–deficient cancer cells, such as BRCA-mutated breast cancer cells (15). When combined with β-emitters, the ability of PARP inhibitors to impair DNA repair may increase the frequency of tumoricidal double-strand breaks. Preclinical data suggest a class effect across different PARP inhibitors, with several studies reporting synergistic effects. In addition to the synergy observed with olaparib plus 177Lu-DOTATATE mentioned earlier (13), similar effects have been observed with olaparib and 131I-meta-iodobenzylguanidine (16), 177Lu-DOTATATE and talazoparib (17), and other PARP inhibitors (18).
To our knowledge, no clinical data have been published on PARP inhibitors in combination with PRRT, but several studies are ongoing (Table 4). A preliminary report has been presented as a conference abstract from a phase I–II dose-escalation trial of olaparib (50, 100, 200, and 300 mg). This trial uses a classic 3 + 3 phase I design, rather than individual dose escalation, and olaparib was administrated for approximately 4 wk, starting 2 d before 177Lu-DOTATATE infusion (19). In addition, 4 other trials are recruiting for a Dutch phase I trial (NCT05870423) in locally advanced or metastatic NETs, using a 3 + 3 design with olaparib escalation from 100 to 300 mg that starts 3 d before 177Lu-DOTATATE and continues for 2 wk after infusion, and an Australian phase I trial (NCT05053854) in grade 2 GEPNETs, evaluating escalated doses of talazoparib administrated on days 2–6 after 177Lu-DOTATATE infusion. In addition, an ongoing phase I–II trial with an initial escalation phase of olaparib for 4 wk per cycle (NCT04086485) is in progress, and a recently launched phase II trial of 177Lu-DOTATATE and olaparib in children and adolescents (NCT06607692) is also under way (Table 4).
Ongoing Studies with 177Lu-PRRT and PARP Inhibitors
In some of these trials with olaparib, the PARP inhibitor is administrated before 177Lu-DOTATATE infusion. In contrast, we delayed olaparib administration (similar to the Australian trial with talazoparib) based on biokinetic modeling, which suggested that early administration might increase bone marrow toxicity. Thrombocytopenia, a known side effect of olaparib, was observed in 3 of 10 fully evaluable patients at the 300-mg dose level, although it was transient. Two of these patients had bone metastases. Other toxicities were generally mild, predominantly grade 1–2 bone marrow suppression, with some more severe lymphocytopenia as expected with PRRT. Because thrombocytopenia was the only DLT observed, further research on bone marrow dosimetry is warranted.
PARP inhibition is just one of many possibilities to intensify treatment with 177Lu-DOTATATE or 177Lu-DOTATOC. Given the excellent tolerability of 177Lu-DOTATATE as monotherapy, there are numerous avenues for combination therapy or treatment customization. Individualized approaches might adjust the number of cycles or the activity per cycle based on the radiation dose to the kidneys, a strategy supported by promising prospective data, although no randomized trials have been conducted yet (3–5). Another option is linking 177Lu to a somatostatin antagonist (20,21), but there is some concern with regard to bone marrow toxicity. Alternatively, replacing 177Lu with α-emitters such as 231Bi, 225Ac, or 212Pb offers a potential advantage; a phase I trial showed 212Pb-DOTAMTATE to be well tolerated and effective (22). β-emitters such as 161Tb, which also emit Auger electrons, represent another promising option (23–26).
Combination trials integrating chemotherapy (e.g., temozolomide and capecitabine) and everolimus with PRRT have been performed (6–8,10), although no published studies have shown superiority over PRRT alone. The START-NET trial (NCT05387603) is a 3-armed randomized trial that compares standard PRRT with 4 cycles of 177Lu-DOTATOC and an individualized approach based on kidney dosimetry; the third arm adds capecitabine for 18F-FDG–positive tumors. In addition, at least 4 other trials are under way for PRRT with 177Lu and a comparator in a randomized setting. The COMPETE trial (NCT03049189) compares 177Lu-edotreotide with standard-dose everolimus in GEPNETs, and a phase II trial compares 177Lu-DOTATOC with everolimus in bronchial NETs (NCT04665739). Another phase II trial compares 177Lu-DOTATOC with capecitabine plus temozolomide in pancreatic NETs (NCT05247905), whereas the COMPOSE trial (NCT04919226) compares 177Lu-edotreotide with the standard of care (capecitabine plus temozolomide; folinic acid, fluorouracil, and oxaliplatin; or everolimus) in grade 2–3 GEPNETs. Efficacy in this latter group has been demonstrated with 177Lu-DOTATATE in the NETTER-2 trial (27), although the comparator was a somatostatin analog, a treatment rarely used alone in highly proliferative grade 2–3 tumors.
Although this phase I trial establishes the feasibility of combining the PARP inhibitor olaparib with 177Lu-DOTATATE, several limitations must be acknowledged. There are so far inadequate efficacy data, and the small sample size, inherent to early-phase trials, limits the ability to detect rare adverse events. In addition, the trial’s design, with individual dose escalation, might not fully represent the tolerability and safety profile across broader patient populations. The short follow-up period restricts the assessment of long-term efficacy and toxicity, especially bone marrow function and potential delayed adverse effects. In particular, myelodysplastic syndrome and acute myeloid leukemia, which have been associated with both PARP inhibitors and PRRT, need to be monitored closely with long-term follow-up.
CONCLUSION
This phase I trial demonstrates the feasibility and tolerability of combining the PARP inhibitor olaparib with 177Lu-DOTATATE in patients with somatostatin receptor–positive tumors. The findings suggest that starting olaparib at 200 mg BID, with potential escalation to 300 mg BID, is a viable strategy that warrants further exploration. The observed bone marrow toxicity, particularly thrombocytopenia, underscores the need for careful patient selection and monitoring, as well as further research into optimizing bone marrow dosimetry. Although this combination approach holds promise for enhancing the efficacy of PRRT, larger randomized trials are necessary to determine its clinical benefit over monotherapy and to refine the optimal dosing schedule.
DISCLOSURE
This work was supported by research grants from the Swedish Cancer Society, the King Gustav V Jubilee Clinic Cancer Research Foundation, the Swedish Federal Government under an ALF agreement, and a grant for the radiopharmaceutical from Advanced Accelerator Applications/Novartis. Peter Bernhardt serves or has served as a consultant for ITM, Affibody AB, and Akiram Therapeutics and owns shares in Theravison AB. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can the combination of olaparib and 177Lu-DOTATATE enhance treatment efficacy in patients with somatostatin receptor–positive tumors while maintaining manageable toxicity?
PERTINENT FINDINGS: The combination was generally well tolerated, with thrombocytopenia being the primary DLT at a dose level of 300 mg BID of olaparib. The study recommends starting at 200 mg BID, with potential escalation depending on patient tolerance.
IMPLICATIONS FOR PATIENT CARE: This combination therapy offers a promising approach for enhancing treatment in patients with somatostatin receptor–positive tumors. However, careful monitoring of bone marrow function is essential, and further research is needed to optimize dosing and confirm clinical efficacy in larger trials.
ACKNOWLEDGMENT
We thank research nurse Jenny Tiberg at the Clinical Trial Unit, Department of Oncology, Sahlgrenska University Hospital in Gothenburg.
Footnotes
Published online Feb. 27, 2025.
- © 2025 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication October 2, 2024.
- Accepted for publication January 6, 2025.
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.