


Abstract
Straightforward radiosynthesis protocols for 18F-labeled radiopharmaceuticals are an indispensable but often overlooked prerequisite to successfully perform molecular imaging studies in vivo by PET. In recent years, thanks to the expansion of the 18F chemical toolbox, structurally diverse and novel clinically relevant radiopharmaceuticals have been synthesized with both high efficiency and ready implementation. This article provides an overview of recent 18F-labeling methodologies, specifically for B-18F, Si-18F, Al-18F, and iodine (III)-mediated radiofluorination via the spirocyclic iodonium ylide technology.
For nearly 60 y, radiochemists have devised numerous methodologies to introduce 18F into organic compounds to support the development and clinical translation of PET radiopharmaceuticals (1). Radiochemists have long sought simplified radiochemical methods to preferably introduce the 18F radionuclide in a kit-like manner, similar to the production of 99mTc-based imaging agents for SPECT. Here, we review 4 promising new radiochemistries based on B-18F, Si-18F, Al-18F formation and spirocyclic iodonium ylide (SCIDY) use coming close to that premise.
BORON–18F RADIOCHEMISTRY
In seeking new methods, several teams have eschewed C–F bond formation in favor of other heteroatom–fluorine bond-forming strategies. Ting et al. hypothesized that an organotrifluoroborate, readily prepared in water under mildly acidic conditions (pH 2–3), would afford late-stage, one-step labeling in a manner similar to radiometal chelation (2). Advantages of RBF3s include hydrophilicity (as anionic salts, they would enhance in vivo clearance for increased-contrast images compared with more lipophilic organofluorides that retard clearance), bioorthogonality (the strong B–F bond, being bioorthogonal, would be metabolically stable), nontoxicity (little in the vast literature would suggest that these are acutely toxic (3)), and the potential for tripling the molar activity (Am) of the fluoride used (because the pool of fluoride ions would condense on one third as many molecules to give the trifluoroborate) (Fig. 1A) (4). In addition, since peptides are deprotected in acid (often, concentrated hydrofluoric acid) and are high-performance liquid chromatography–purified in acidic medium, organotrifluoroborate synthesis would pose few problems to the structural integrity of most peptides being labeled.

(A) RBF3 radiofluorination. (B) Structure of 18F-AMBF3-rhodamine-bisRGD. DMF = dimethylformamide.
A concerted effort to apply both synthetic methods and physical organic chemistry led Perrin’s group to address the molecular basis for ensuring the kinetic stability of RBF3 in water, ultimately leading to several synthons that could be fluorinated relatively rapidly and in reasonable radiochemical yields (RCYs) while providing sufficient stability in vitro and ultimately in vivo (5,6). Early radiofluorination methods toward this goal provided preliminary signs of success with the 18F labeling and PET imaging of biotin, marimastat (7), arginylglycylaspartic acid (RGD) (8), bombesin (9), and bisRGD (10). Although going beyond simple proof of concept to show in vivo stability and target specificity with blocking controls, in certain cases bone uptake was seen, and in others, tumor uptake values were low. Furthermore, synthesis was still not readily user-friendly, often requiring the addition of carrier 19F-fluoride, and tumor uptake values were too low (<2% of the injected dose per gram). These observations were attributed to the extremely polar nature of the organotrifluoroborate and to the metabolic instability of the linker arm in the case of a bombesin analog.
With promising work from the Schirrmacher lab on isotope exchange for 18F labeling of silylfluorides—work that was recapitulated on boron first by Gabbai (11)—Perrin et al. applied this technique to existing organotrifluoroborates while also establishing a range of other useful organotrifluoroborate prosthetic compositions. Although there is no thermodynamic driving force for isotope exchange, this technique provides exceptional simplicity, because the precursor is identical to the product. This advantage has been particularly useful in the context of organotrifluoroborates in terms of precursor production, isolation, shelf stability, and characterization because organotrifluoroborates are stable, easily isolated, and characterized by 19F NMR spectroscopy. Use of isotope exchange on several new prosthetics, which were grafted onto new ligands, enabled successful labeling of several molecularly complex peptides, including Tyr3-octreotate (TATE), bradykinin, bombesin, and a fluorescent dimeric RGD (12). In the case of TATE, high tumor uptake was observed (≤16% of injected dose per gram), with very high contrast ratios (13). Excitingly, this method also enabled the radiolabeling of a fluorescent-dimeric-RGD (Fig. 1B) (14). Tumor uptake increased 10-fold over that found in a previous study with a dimeric-RGD-organotrifluoroborate construct.
SILICON–18F RADIOCHEMISTRY
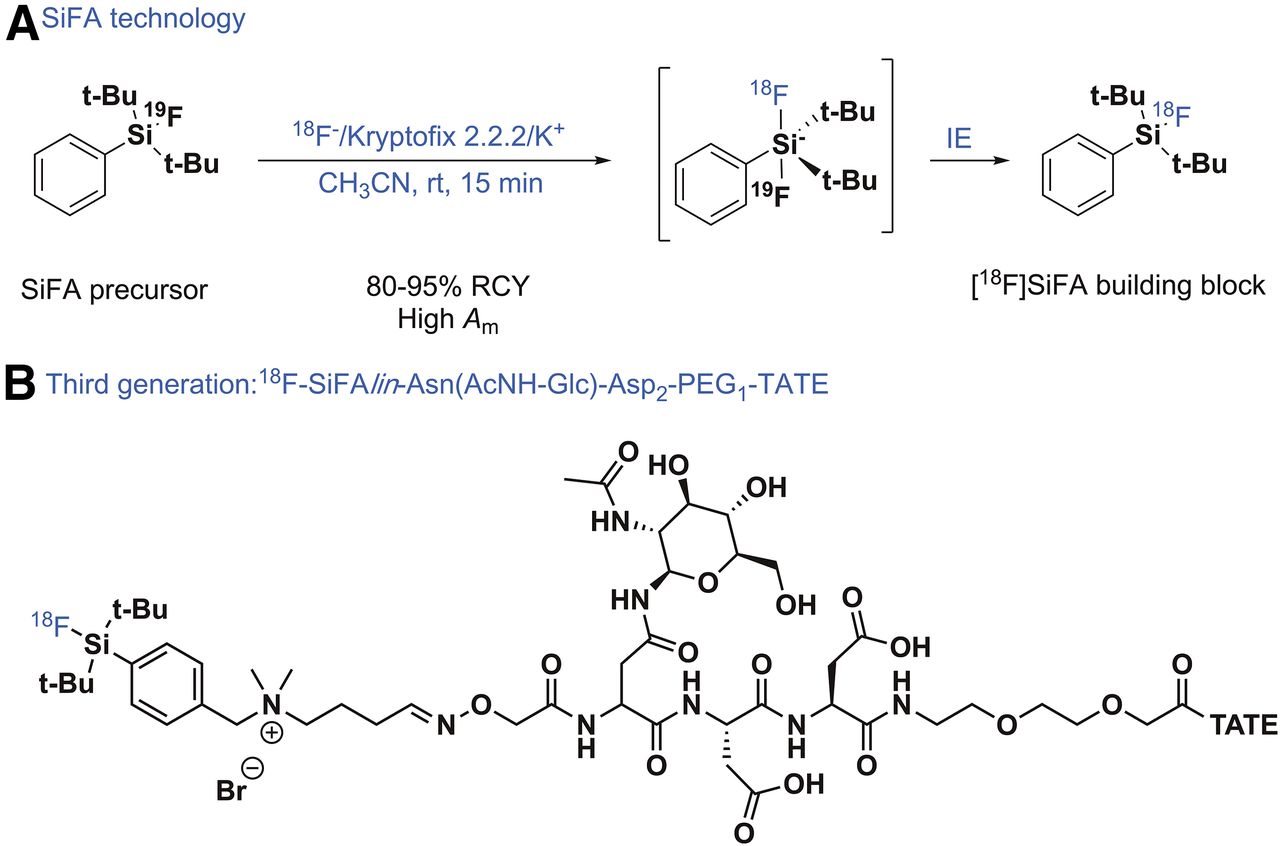
In the early days of silicon-fluoride-acceptor (SiFA) radiochemistry, the 18F labeling of the SiFA building block (Fig. 2A) worked surprisingly well and proved that revisiting isotopic exchange as a strategy to obtain radiotracers of high Am was a good choice despite the historical prevalent bias against isotopic exchange (isotopic exchange usually does not yield 18F-radiotracers of high Am) (15). Isotopic exchange of the nonradioactive 19F with anionic 18F− in various solvents ranging from acetonitrile to water gave not only access to simple labeled SiFA model compounds but also to a SiFA-modified TATE, a peptide well known to diagnose somatostatin receptor–positive tumors. This first SiFA-Tyr3-TATE was 18F-labeled in high RCYs and with surprisingly high Am for an isotopic exchange reaction (16). However, when injected into an AR42J tumor–bearing rat, a major drawback of the SiFA building block became apparent, namely its inherently high lipophilicity originating from its unique chemical structure, leading to an almost exclusive accumulation in the liver. The 2 tertiary butyl-groups, shielding the 18F–silicon bond from being exposed to water and becoming hydrolyzed in vivo, unfortunately are a prerequisite sine qua non to impart chemical stability to the 18F-SiFA structure. The high lipophilicity has serious implications for in vivo imaging simply because lipophilic compounds are siphoned out efficiently by the liver and accumulate there, extracting the radiotracer from the blood and making it unavailable for target engagement. At this point, the SiFA methodology was just an example of a new niche radiochemistry unambiguously failing in vivo as a result of SiFA’s chemical nature adversely influencing its biodistribution. An obvious solution to this problem was either changing the SiFA structure toward lower lipophilicity while still ensuring hydrolytic stability or adding hydrophilic modules counteracting SiFA’s lipophilicity. Wängler et al. chose an acetylated amino-sugar (AcNH-β-Glc) attached to an asparagine (Asn) side chain to add a hydrophilic moiety to the SiFA-TATE (17). The 18F-SiFA-Asn(AcNH-β-Glc)-PEG-Tyr3-TATE accumulated far more favorably in AR42J tumors in a mouse xenograft model. Eight percent of the injected dose per gram was taken up by the tumor, albeit there was a still-high liver uptake. Apparently, the reduction of lipophilicity was a step in the right direction although not yet drastic enough. This desired step was achieved by introducing SiFAlin, a positively charged SiFA building block, 2 highly hydrophilic Asn side chains, and the aforementioned AcNH-β-Glc (18). The LogD of this conjugate dropped to −1.21, being in the same range as 68Ga-DOTATATE. In a comparative small-animal PET/CT study of 68Ga-DOTATATE and 18F-SiFAlin-Asn(AcNH-Glc)-Asp2-PEG1-TATE (Fig. 2B) in an AR42J tumor xenograft model, the SUV of both compounds was almost identical, at 5.50 versus 7.80, respectively (19). This conjugate is currently being evaluated in a toxicity assessment, and pending positive results, a first-in-human PET study is planned in a healthy control group and in patients bearing a somatostatin receptor–positive tumor. A SiFA compound for prostate-specific membrane antigen imaging in LNCaP tumor–bearing mice has recently been reported in a symposium abstract (20). Besides the SiFA approach, Honer et al. have introduced an impressive preclinical line-up of 18F-silicon compounds (21).
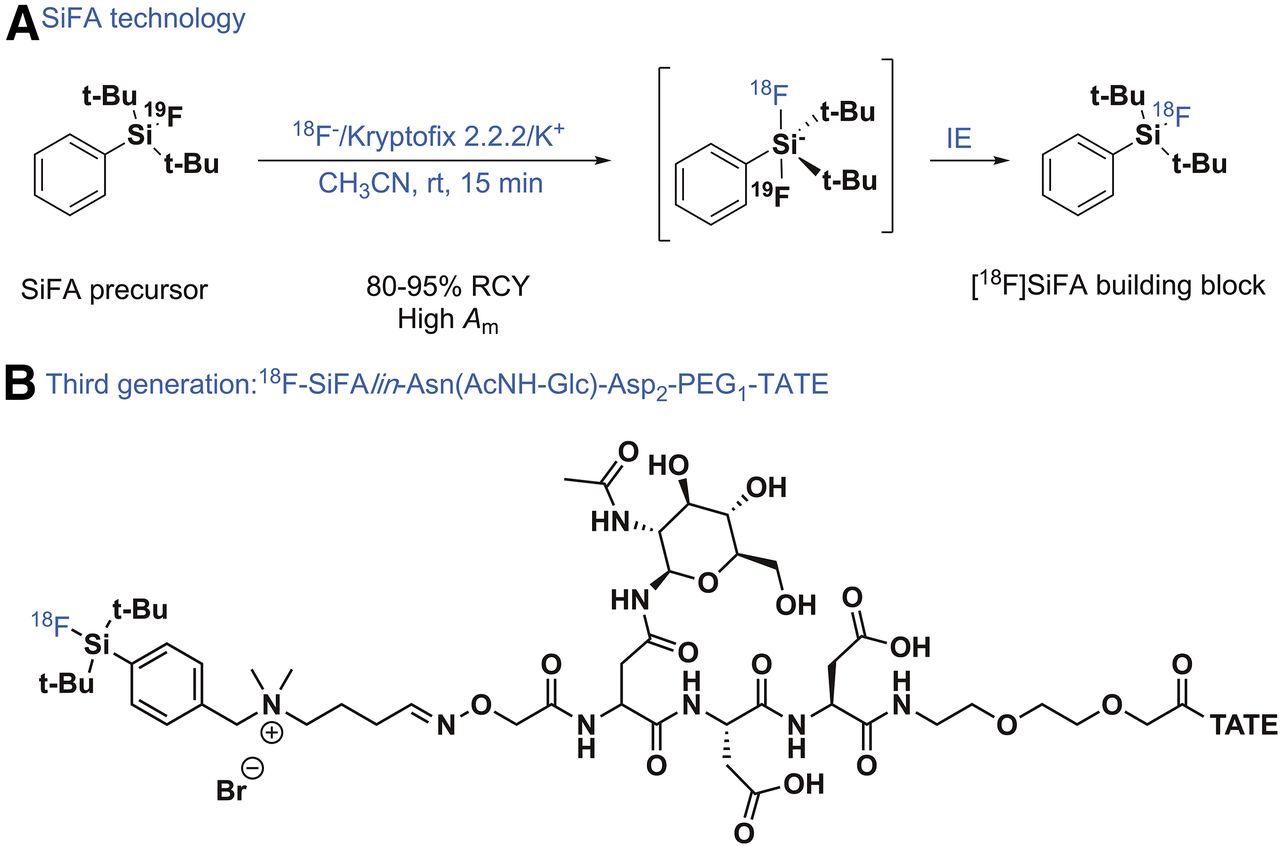
(A) SiFA labeling concept based on isotopic exchange. (B) Third generation of SiFA-derivatized TATE for diagnosis of somatostatin-expressing tumors. rt = room temperature.
ALUMINUM–18F RADIOCHEMISTRY
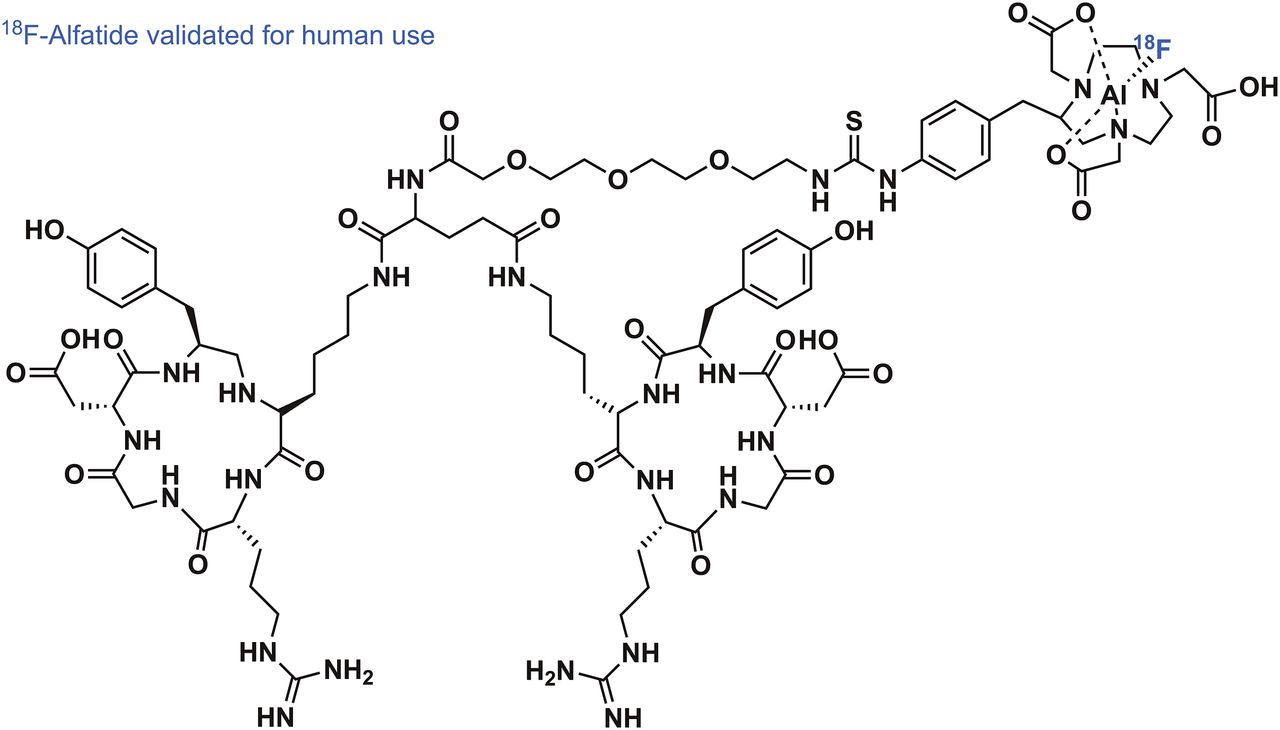
The Al–18F radiochemistry, first introduced by McBride et al. in 2009 (22), admittedly came a long way in a short time—from the disclosure of the basic radiochemical concept to the first reported applications in human PET imaging (23). The labeling mechanic of the Al–18F radiochemistry is rooted in the preformation of aluminum mono-18F-fluoride cation (18F-AlF2+) under aqueous conditions. The subsequent chelation of 18F-AlF2+ by well-known chelators such as pentetic acid, 1,4,7-triazacyclononane-N,N′,N″-triacetic acid (NOTA), and 1,4,7-triazacyclononane-N,N′-diacetic acid (NODA), all familiar from radiometal complexation chemistry, has invigorated strong clinical translational efforts to simply mimic 68Ga or 64Cu labeling of established radiometal-based PET tracers with 18F. The driving force for the efficient formation of 18F-AlF2+ is the exceptionally high binding energy between aluminum and fluorine of more than 670 kJ/mol. This radiochemistry works fully under aqueous conditions and has been thoroughly reviewed alongside its preclinical applications (24). Pentadentate chelators, which donate 3 nitrogen and 2 oxygen atoms to the binding of 18F-AlF2+, have been used first for PET tracer development of peptide and protein nature. These first-generation chelators for 18F− complexation, however, required high reaction temperatures of more than 100°C, potentially detrimental to the integrity of some compounds of higher molecular weight. Although high temperatures can be a general drawback in radiolabeling, impressive first-in-human PET imaging of integrin ανβ3–expressing tumors with RGD peptides derivatized with NOTA validated the use of 18F-AlF2+ in clinical PET imaging. The RGD derivative 18F-AlF-NOTA-PRGD2 (18F-alfatide) could be labeled in high RCYs of more than 40% (decay-corrected) and with a radiochemical purity of more than 95% within 20 min (Fig. 3) (23). Integrin-expressing lung tumors in several patients could be clearly delineated using 18F-alfatide, with a tumor-to-muscle and tumor-to-blood ratio of 2.9 and 5.87, respectively. After these encouraging results, a pilot study by Gao et al., aimed at imaging integrin ανβ3 in suspected lung cancer patients, further used 18F-alfatide successfully in a clinical setting (25). Seventeen patients with lung cancer were accurately identified among true-negative (hamartoma) as well as false-positive (chronic inflammation and inflammatory pseudotumor) cases. The question of whether 18F-alfatide PET/CT imaging can predict the short-term outcome of concurrent chemoradiotherapy in patients with advanced non–small cell lung cancer was investigated by Luan et al. (26). Responders and nonresponders to chemotherapy could be distinguished with high sensitivity, specificity, and accuracy, confirming that 18F-alfatide could be a useful tool in therapy response prediction. The electronically uncharged nature of 18F-AlF-NOTA complexes prompted the investigation of 18F-alfatide II, a derivative of 18F-alfatide, extended by one more polyethylene glycol unit, for imaging brain metastasis (27). In 9 patients with a total of 20 metastatic brain tumors, it was demonstrated that 18F-alfatide II exhibited a tumor-to-background ratio superior to that of 18F-FDG. Even though there is no direct clinical advantage to the use of 18F-alfatide derivatives in comparison to an already-established RGD tracer such as 18F-galacto-RGD, the ease of synthesis and reliable labeling methodology make 18F-alfatide derivatives a serious competitor. This view was further substantiated recently by Zhang et al., who evaluated the use of 18F-alfatide PET/CT to predict sensitivity to chemotherapy in patients with recently diagnosed glioblastoma (28). Twenty-five patients diagnosed with glioblastoma multiform underwent surgical resection. 18F-alfatide PET/CT successfully delineated the residual lesions of the glioblastoma. Baseline scans, as well as follow-up scans after 3 wk, were available and confirmed the predictable power of 18F-alfatide PET/CT for response evaluation after chemotherapy. Among others (29), recent advancements in chelator development resulted in the introduction of racemic H3RESCA, a new chelator complexing 18F-AlF2+ at room temperature. Besides extensive preclinical evaluation, a first-in-human study using a HER2-targeting H3RESCA-derivatized Affibody (PEP04314; Affibody AB) has been reported (30).
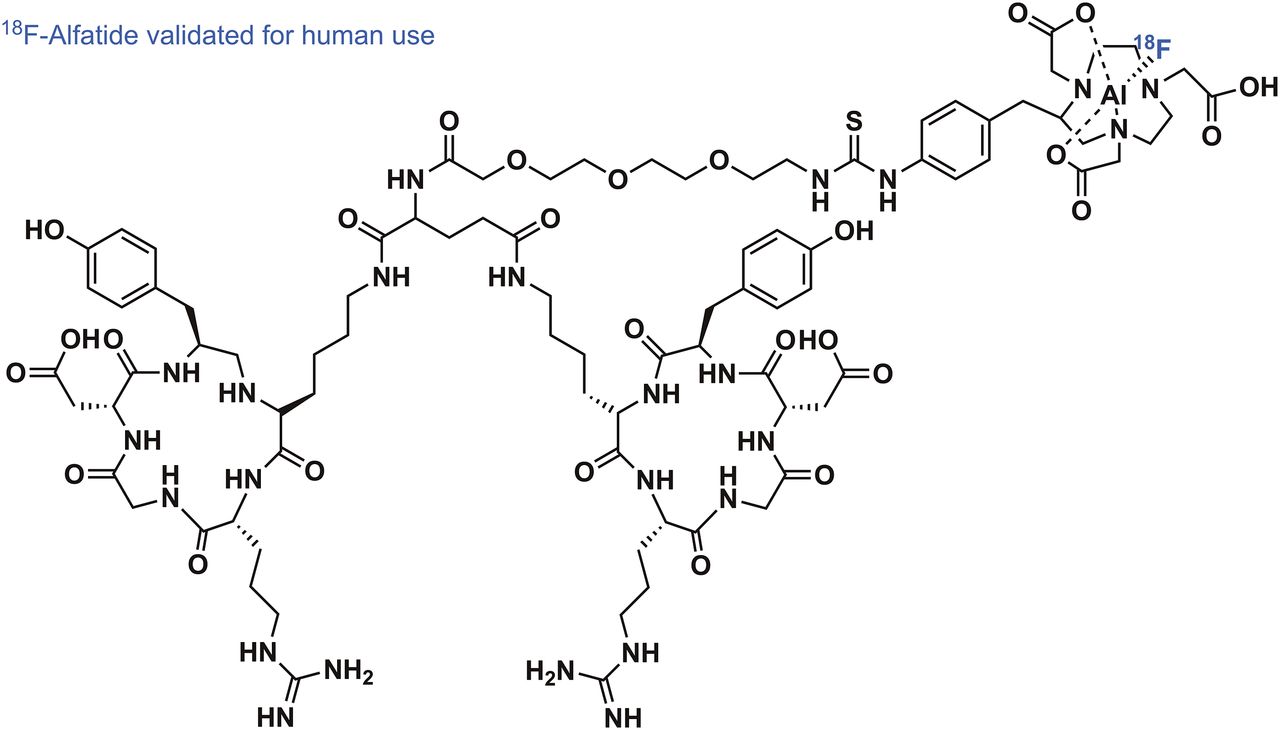
Structure of 18F-alfatide, clinically used radiotracer for imaging of integrin-expressing tumors.
IODINE(III)-MEDIATED RADIOFLUORINATION: SCIDY TECHNOLOGY
Since 2014, there were several reports of a 1-step, regioselective method for 18F incorporation via hypervalent iodine(III)-mediated precursors to afford 18F-arenes from 18F-fluoride in high RCYs and high Am (31–33). The novel technology relies on SCIDY technology to produce labeling precursors that regioselectively introduce 18F-fluoride ion into nonactivated aromatic rings with wide substrate scope under metal-free conditions (Fig. 4A). The technique involves stable, easily purified SCIDY precursors, is readily implemented with standard workup procedures, is easily adopted in any laboratory, and has since been applied for producing 18F-radiopharmaceuticals for PET imaging studies in humans. Notably, 18F-FPEB (3-fluoro-5-[(pyridin-3-yl)ethynyl]benzonitrile), an important radiopharmaceutical for imaging metabotropic glutamate receptor 5 in human that is challenged by low RCYs, was validated using the SCIDY-radiofluorination approach. A dramatically increased RCY for 18F-FPEB—from 1%–2% to 20% (a 10-fold increase)—was realized, and the product, which had an Am of 666 GBq/μmol, represents the first radiopharmaceutical prepared by such technologies (Fig. 4B) (34).

SCIDY-based radiofluorination and its translation into clinically relevant PET tracers. (A) Concept of 18F-radiofluorination. (B) 18F-FPEB for clinical PET imaging. (C) 18F-lorlatinib synthesized using SCIDY.
Two promising spirocyclic auxiliaries, namely, cyclopentyl (33) and adamantyl (35), demonstrated excellent incorporation yields on challenging electron-neutral or -donating arenes. SCIDY methodology proved to be efficient for radiolabeling a diverse range of nonactivated functionalized arenes and heteroarenes, with remarkably broad functional group compatibility. Several 18F-PET tracers and building blocks were successfully synthesized from 18F-fluoride for the first time, or the corresponding radiosyntheses were significantly simplified and improved via SCIDY methods. For example, the first synthesis of 5-18F-fluorouracil from 18F-fluoride was achieved in 11% RCY (33), and there are new 18F building blocks such as fluoroaromatic azides (36,37) that have wide applications in click-radiosynthesis. The conceptual advantages of excellent regioselectivity, and the viability of incorporating 18F into a wide array of nonactivated (hetero)arenes, make this methodology suitable for routine radiopharmaceutical production. Furthermore, a number of PET radiopharmaceuticals for routine clinical research, including 18F-lorlatinib (38), 18F-FDPA (39), 18F-6-fluoro-l-m-tyrosine, and 18F-mFBG (35), which are challenging to prepare using conventional labeling methods, have been isolated in high RCYs by this methodology (Fig. 4C). The promise of these newly developed reactants for radiofluorination is demonstrated for the wide range of nitrogen- and oxygen-containing heterocycles and drug fragments that have been labeled with 18F-fluoride, including basic amine functional groups, which have been a longstanding challenge in 18F-radiochemistry.
CONCLUSION
Amid the inherent technical and chemical challenges associated with efficiently delivering innovative 18F-radiopharmaceuticals, the methodologies described here represent major advances that will likely have an impact on translational imaging research as further steps toward the clinic are taken in coming years.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Dec. 28, 2017.
- © 2018 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication October 11, 2017.
- Accepted for publication December 24, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.