


Visual Abstract

Abstract
Efflux transporters of the adenosine triphosphate–binding cassette (ABC) superfamily, such as P-glycoprotein (P-gp/ABCB1) and breast cancer resistance protein (BCRP/ABCG2), are highly expressed at the blood–brain barrier (BBB), where they contribute to maintaining brain homeostasis. P-gp may serve as an imaging biomarker to assess the contribution of BBB functionality rather than integrity to the onset or progression of various neurologic diseases. Considerable efforts have been made to develop radiolabeled P-gp substrates to assess cerebral P-gp activity with PET. However, initially developed radiotracers have limited clinical utility as they lack sensitivity to detect moderate, physiologically relevant changes in cerebral P-gp activity. Learning from this molecular imaging area has called for specific criteria, different from those classically used for other central nervous system targets, for developing and selecting suitable PET tracers to study ABC transporter activity at the BBB in different neurologic diseases.
The blood–brain barrier (BBB) is the primary interface between the bloodstream and the central nervous system (CNS) (1,2). Over the past decade, the concept of the BBB has evolved from a structural barrier defined by tight junctions to a dynamic functional interface regulating blood–brain exchanges through specialized transporters (3). In endothelial cells forming the BBB, efflux transporters of the adenosine triphosphate–binding cassette (ABC) superfamily, such as P-glycoprotein (P-gp/ABCB1) and breast cancer resistance protein (BCRP/ABCG2), constitute energy-dependent systems to maintain brain homeostasis (4,5). P-gp function is now considered an essential component of the brain’s detoxification system, managing endogenous neurotoxic substrates such as amyloid-β peptides, steroids, bile acids, and metabolites, in addition to its well-established role in restricting the brain’s exposure to xenobiotics (4). Evidence from preclinical models of neurologic diseases and patients’ postmortem brain tissue samples suggests that the expression levels or activity of P-gp are altered (increased or decreased) in neurologic disorders, such as Alzheimer disease, amyotrophic lateral sclerosis, Creutzfeldt–Jakob disease, epilepsy, ischemic stroke/hypoxia, multiple sclerosis, or Parkinson disease (1,4). From a molecular perspective, the regulation of P-gp function in endothelial cells is often associated with the neuroinflammatory, oxidative, or glutamatergic pathways that may be altered in neurologic diseases (4,6). Consequently, P-gp function may serve as a biomarker to assess the contribution of BBB functionality (rather than physical integrity constituted by tight junctions) to the onset or the progression of neurologic diseases. Compared with P-gp, the importance of BCRP-mediated efflux at the BBB has received less attention, although both transporters display similar expression levels in humans, overlapping substrate specificities, and functional redundancy (6).
RADIOTRACERS TO ASSESS P-GP ACTIVITY AT THE BBB
Among available translational neuroimaging methods, PET bears great potential to image ABC transporters at the BBB. Most work has so far focused on P-gp. The development of radiolabeled P-gp substrates whose brain entry is restricted by P-gp–mediated efflux has afforded several radiotracers to assess P-gp activity in vivo (Fig. 1). This led to a large body of preclinical and clinical research, which considerably improved our knowledge on the role of P-gp at the BBB in health and disease (7). In contrast, the development of radiotracers based on small-molecule P-gp inhibitors (e.g., [11C]tariquidar) for mapping the density of P-gp at the BBB has been without success so far (8), and alternative approaches may therefore be required.

Chemical structures of radiolabeled P-gp substrates.
Initial Assumptions and Selection Criteria for Effective Radiotracers
In 2009, Kannan et al. formulated 3 criteria that need to be fulfilled by a radiolabeled P-gp substrate to effectively image cerebral P-gp activity (9). These criteria comprise a high signal magnitude, that is, a great imaging contrast between conditions of full and absent transporter activity; radiochemical purity of the brain PET signal, that is, absence of brain-penetrant radiolabeled metabolites; and selectivity for P-gp over BCRP (9).
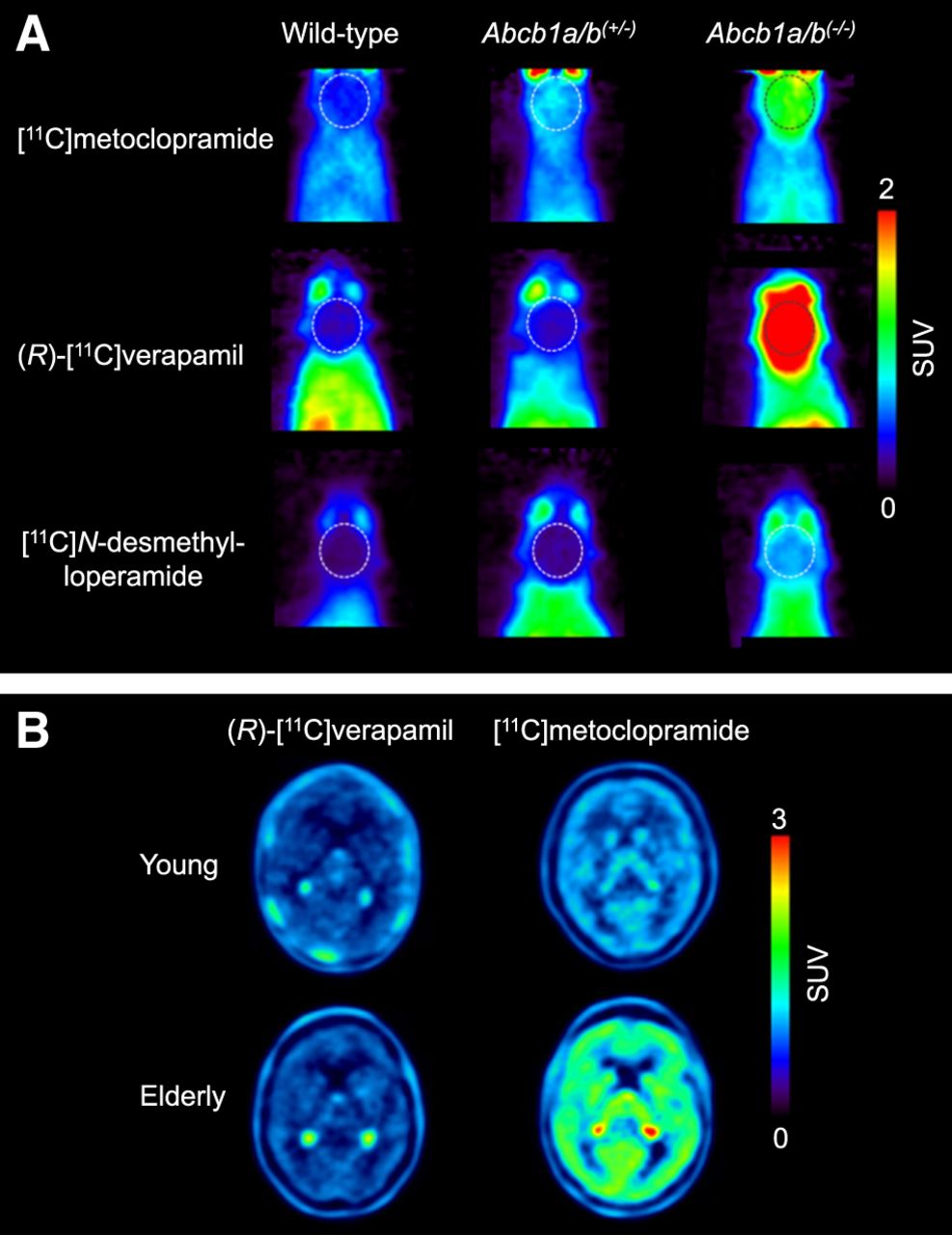
The ultimate goal is to use these radiotracers in CNS diseases in which moderate (<50%) changes in cerebral P-gp abundance are expected to occur (4). The initial assumption was that radiolabeled substrates, which show the greatest differences in brain uptake between P-gp knockout (Abcb1a/b(−/−)) and wild-type mice, would have the highest sensitivity for detecting small changes in P-gp expression, assuming a linear relationship between changes in brain distribution and the level of P-gp expression. Therefore, initial efforts focused on so-called avid substrates that are efficiently transported by P-gp, leading to low baseline brain uptake. Avid P-gp substrates include (R)-[11C]verapamil (10) and [11C]N-desmethyl-loperamide (Fig. 1) (11). Both radiotracers displayed a great increase in brain uptake in Abcb1a/b(−/−) relative to wild-type mice (Fig. 2A).

Sensitivity of [11C]metoclopramide to detect moderate decreases in cerebral P-gp abundance compared with (R)-[11C]verapamil and [11C]N-desmethyl-loperamide. (A) Coronal PET average images (0–30 min) of brain (highlighted with broken line) obtained after intravenous injection of [11C]metoclopramide, (R)-[11C]verapamil, or [11C]N-desmethyl-loperamide in wild-type, Abcb1a/b(+/−), and Abcb1a/b(−/−) mice (reprinted from (20)). (B) Horizontal PET average images (0–40 min) of brain of young and elderly healthy men imaged either with (R)-[11C]verapamil (reprinted from (19)) or [11C]metoclopramide (reprinted from (24)).
Regarding radiochemical purity, [11C]N-desmethyl-loperamide has the great advantage of being metabolically stable in humans, although it produces brain-penetrant radiolabeled metabolites in mice (11). (R)-[11C]verapamil, on the other hand, shows extensive peripheral metabolism both in rodents and in humans and generates, to a large extent, brain-penetrant radiolabeled metabolites (12). (R)-[11C]verapamil is metabolized mainly by cytochrome P450 3A enzymes. This confers vulnerability to drug–drug interactions with numerous cytochrome P450 3A inhibitors or inducers, potentially leading to significant changes in the peripheral metabolism of (R)-[11C]verapamil, which may confound the quantification of cerebral P-gp activity using kinetic modeling (13).
Neither verapamil nor N-desmethyl-loperamide is a substrate of human BCRP, thus rendering them selective for P-gp over BCRP (14,15). With respect to transporter selectivity, possible species differences in transporter specificity need to be considered. This is exemplified by the serotonin 5-hydroxytryptamine receptor 1A radioligand [18F]MPPF or the benzodiazepine receptor radioligand [11C]flumazenil, which are transported by rodent but not by human P-gp (16). Therefore, the assessment of transporter selectivity should be complemented with in vitro transport experiments in human transporter-overexpressing cell lines and not rely solely on experiments on wild-type and transporter knockout mice.
Clinical PET Studies with Avid P-gp Substrates
(R)-[11C]verapamil and [11C]N-desmethyl-loperamide were validated for their ability to assess P-gp activity at the human BBB, by performing PET scans after P-gp inhibition with tariquidar. Both radiotracers showed tariquidar dose-dependent increases in brain uptake in humans, with up to 5-fold increases at the maximum used tariquidar doses (17,18). Although [11C]N-desmethyl-loperamide was not further tested in humans beyond initial P-gp inhibition studies, (R)-[11C]verapamil or racemic [11C]verapamil were tested in several conditions, in which either an increase (drug-resistant epilepsy, depression) or a decrease (healthy aging, Alzheimer disease, Parkinson disease, carriers of single-nucleotide polymorphisms in the ABCB1 gene) of cerebral P-gp abundance was expected to occur. At best, moderate changes (∼30%) in the brain distribution of (R)-[11C]verapamil were observed, which were far from the pronounced effects seen in Abcb1a/b(−/−) mice or in inhibitor-treated rodents or humans.
Reasons for Limited Sensitivity of (R)-[11C]Verapamil
(R)-[11C]verapamil is efficiently transported by P-gp, and its brain uptake is maximally restricted by P-gp. Disease-induced changes in P-gp abundance are assumed to be small (4) relative to P-gp’s maximum transport capacity (i.e., Vmax in the classic Michaelis–Menten equation), leading to only small changes in the transport rate of (R)-[11C]verapamil. It was shown that the sensitivity of (R)-[11C]verapamil to detect moderate alterations in P-gp abundance can be improved after partial P-gp inhibition with tariquidar, which lowers the maximum transport capacity. (R)-[11C]verapamil in combination with partial P-gp inhibition was successfully applied to detect a decrease in cerebral P-gp activity in healthy aging (19) and a seizure-induced increase in P-gp activity in epilepsy patients (13). Because of the complexity and variability of the partial P-gp inhibition protocol and safety issues, a more sensitive P-gp substrate that does not require the administration of a P-gp inhibitor to measure P-gp activity is preferred.
Updated Selection Criteria for Effective P-gp Substrate Radiotracers
On the basis of the experience gained with previously developed radiolabeled P-gp substrates, there is a need for updated radiotracer selection criteria (Table 1). The most important lesson learned is related to the observation that a high imaging contrast between conditions of full and absent P-gp activity does not necessarily translate into high sensitivity to detect moderate alterations in P-gp abundance. It is therefore recommended to include an additional evaluation step, in which the ability of a radiotracer to detect a moderate reduction in P-gp abundance is assessed (Table 1). Instead of classic homozygous Abcb1a/b(−/−) mice that do no express P-gp at all, the use of heterozygous Abcb1a/b(+/−) mice is suggested, which are naturally generated in the breeding of Abcb1a/b(−/−) mice and have an approximately 50% decrease in P-gp abundance at the BBB (20). In an attempt to develop a radiotracer with better imaging properties than (R)-[11C]verapamil and [11C]N-desmethyl-loperamide, [11C]metoclopramide was developed (Fig. 1) (21). This tracer is less efficiently transported by P-gp at the BBB, resulting in an approximately 3-fold higher volume of distribution in the human brain than that of (R)-[11C]verapamil, when P-gp is fully functional (22). The choice of a weak P-gp substrate was motivated by the need to detect disease-induced increases in P-gp activity, which is difficult to achieve with avid P-gp substrates, given their low baseline brain uptake. When comparing the brain uptake of [11C]metoclopramide, (R)-[11C]verapamil, and [11C]N-desmethyl-loperamide between wild-type and Abcb1a/b(+/−) mice (20), it was discovered that [11C]metoclopramide has a better sensitivity than the other 2 radiotracers to detect a 50% decrease in P-gp abundance (Fig. 2A). Similarly, administration of a low dose of tariquidar (1 mg/kg) resulted in a higher increase in brain uptake of [11C]metoclopramide in rats than that of [11C]N-desmethyl-loperamide (23). The higher sensitivity of [11C]metoclopramide to detect moderate decreases in P-gp abundance, without the need to coadminister a P-gp inhibitor, was confirmed in a clinical PET study in which [11C]metoclopramide showed greater sensitivity than (R)-[11C]verapamil to detect an age-related decrease in cerebral P-gp activity (Fig. 2B) (19,24). Although the molecular determinants for the improved sensitivity of [11C]metoclopramide are not fully understood, this case clearly argues for the inclusion of Abcb1a/b(+/−) mice in the evaluation of novel radiotracers (Table 1).
Criteria for Selection of Effective P-gp Substrate Radiotracer
The ability of a radiotracer to detect an increase in P-gp abundance can be assessed by treating mice with a P-gp induction protocol (Table 1). Contrary to inhibition protocols, P-gp induction protocols at the BBB have been reported only in rodents. Chronic exposure to 5-pregnen-3β-ol-20-one-16α-carbonitrile, an activator of the rodent pregnane X receptor, can be used to upregulate the expression of P-gp at the mouse BBB (25). This led to a significantly increased brain efflux of [11C]metoclopramide, supporting the ability of [11C]metoclopramide to detect increased P-gp activity (25). This was confirmed in a clinical PET study in drug-resistant epilepsy patients, in whom seizure-induced increases in cerebral P-gp activity could be detected with [11C]metoclopramide (26). In this context, the radiolabeled antiseizure drug [11C]phenytoin (Fig. 1) was proposed to address the importance of efflux transporter activity in limiting its brain distribution in drug-resistant epilepsy. [11C]phenytoin was confirmed as a weak P-gp substrate at the rodent BBB (27) and was tested on humans (28). However, its sensitivity to detect changes in P-gp activity at the human BBB remains to be demonstrated.
In vitro transport studies showed that metoclopramide is not transported by human BCRP, and studies on rodents demonstrated lack of brain uptake of radiolabeled metabolites (21). Moreover, [11C]metoclopramide is metabolized primarily by cytochrome P450 2D6, which is a noninducible enzyme, as opposed to cytochrome P450 3A. Consequently, the potent cytochrome P450 inducers carbamazepine or St. John’s wort had no effect on the peripheral metabolism of [11C]metoclopramide in rats or humans (29,30). To assess the vulnerability of novel radiotracers to enzyme-mediated drug–drug interactions, it is suggested to assess radiotracer metabolism under conditions of cytochrome P450 inhibition (e.g., ritonavir) and induction (e.g., rifampicin or carbamazepine) (Table 1) (29).
11C is a short-lived PET radionuclide available only in centers equipped with a cyclotron. For broader applicability, a radionuclide with a longer half-life, such as 18F (half-life, 109.8 min), is preferred. Savolainen et al. developed an 18F-labeled weak P-gp substrate for PET, named [18F]MC225 (Fig. 1) (31). The first preclinical results looked encouraging in that [18F]MC225 is selective for P-gp over BCRP, has negligible brain uptake of radiolabeled metabolites, and has a higher baseline brain uptake than do avid P-gp substrates (31). Although [18F]MC225 has already been tested on healthy human subjects (32), it is not yet clear whether [18F]MC225 has better sensitivity than [11C]N-desmethyl-loperamide or (R)-[11C]verapamil to detect moderate changes in P-gp activity.
With respect to the clinical translation of novel radiotracers, using radiolabeled isotopologs of clinically applied drugs is preferred over new chemical entities, as the former do not require the conduct of preclinical toxicity studies. Moreover, this potentially allows for coinjecting the unlabeled drug with its radiolabeled analog in clinical PET studies. This could prove advantageous to saturate possible specific radiotracer-binding sites in the brain that may confound the measurement of P-gp activity. For instance, metoclopramide is known to bind to dopamine D2 and serotonin 5-hydroxytryptamine receptor 3 in the brain. To assess whether specific binding of [11C]metoclopramide contributes to the PET signal in the human brain, the brain distribution of [11C]metoclopramide was compared without and with coinjection of a therapeutic dose of unlabeled metoclopramide (21,24). The absence of displaceable binding of [11C]metoclopramide suggests that the brain PET signal is mainly nonspecific and unlikely to confound the assessment of P-gp activity. Similarly, extensive in vitro screening showed that N-desmethyl-loperamide interacts with several CNS targets (11). However, the brain PET signal associated with [11C]N-desmethyl-loperamide, when P-gp is inhibited, is driven mainly by irreversible lysosomal trapping, which provides a target-unrelated mechanism to amplify the brain PET signal (33). To assess the possible contribution of specific binding to the brain PET signal of new radiotracers, classic saturation or displacement studies using the unlabeled compound are recommended (Table 1) (21).
CONCLUSIONS AND PERSPECTIVES
Learning from the development and evaluation of PET radiotracers for efflux transporter activity at the BBB during the last 2 decades has called for specific criteria, different from those classically used for other CNS targets (e.g., neuroreceptors, enzymes, or reuptake transporters). This is because the target tissue (i.e., the BBB) is an interface rather than an anatomic organ, and radiotracers do not properly bind to their target but are actively transported by it. Initially developed P-gp substrate radiotracers (9) have limited clinical utility as they lack sensitivity to detect moderate and physiologically relevant alterations in cerebral P-gp activity in different brain diseases. The criteria summarized in Table 1 are recommended to be considered for developing and selecting an effective P-gp radiotracer.
As is the case for many other CNS targets, an ideal radiotracer that fulfills all criteria to study P-gp activity at the BBB from animals to humans has not been discovered yet. Nevertheless, because of its limitations, (R)-[11C]verapamil, which provided pioneering translational data, should now be abandoned. Today, the selection of one or another radiotracer should be driven mainly by the intended application framework, the expected level of P-gp modulation, and the clinical perspectives of the project.
Current radiotracers to assess P-gp activity are designed to enable accurate quantification of their transfer rate constants across the BBB, using dynamic PET acquisitions and kinetic modeling. The use of [11C]metoclopramide notably unveiled the role of P-gp in mediating the clearance of solutes from the brain, in addition to its well-established role in limiting the brain entry of substrates (22). State-of-the-art kinetic modeling requires invasive arterial blood sampling, and efforts should probably be made to find alternative and less invasive approaches to assess P-gp activity at the BBB (34).
The absolute and relative expression of P-gp and BCRP at the BBB is strikingly different between rodents and humans, with an approximately 2 times lower abundance of P-gp in humans and a BCRP/P-gp ratio of about 1 in humans versus about 0.3 in rodents (35). This suggests a limited human translatability of the importance of BCRP function at the rodent BBB. Efforts should probably be made to develop radiotracers to study BCRP activity, apart from that of P-gp. The functional interplay between P-gp and BCRP was unveiled at the human BBB using the dual P-gp/BCRP substrate [11C]tariquidar coinjected with unlabeled tariquidar at a dose that selectively inhibits P-gp (36). Such an approach is, however, difficult to implement in a clinical setting. Unfortunately, most known BCRP substrates are also transported by P-gp. To our knowledge, no BCRP-specific radiotracer has been validated yet.
Moreover, other efflux transporters than P-gp and BCRP may also play a role at the BBB. Targeted proteomics studies showed expression of multidrug resistance–associated protein 4 (MRP4/ABCC4) at the mouse and human BBB, although at much lower levels than those of P-gp and BCRP (35). Moreover, the abundance of MRP4 is about 8-fold lower in humans than in mice. It is therefore certainly of interest to develop MRP-specific radiotracers to assess the role of MRP4 at the BBB across species.
Efflux transporters may offer functional brain protection when BBB integrity is impaired, as observed in several neurologic diseases (1). Imaging of P-gp activity should thus be combined with markers of BBB permeability, such as contrast-enhanced MRI (37) or dedicated PET radiotracers, to address both the physical and the functional involvement of the BBB in the progression of neurologic diseases.
DISCLOSURE
This research was funded by the Austrian Science Fund (FWF) (grant I4470-B) and the Agence Nationale de la Recherche (ANR-19-CE17-0027). No other potential conflict of interest relevant to this article was reported.
ACKNOWLEDGMENTS
Matthias Jackwerth and Severin Mairinger helped prepare figures and the graphical abstract.
Footnotes
Published online Feb. 27, 2025.
- © 2025 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication December 11, 2024.
- Accepted for publication February 10, 2025.
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.