


Abstract
Chelated somatostatin agonists have been shown to be sensitive to N-terminal radiometal modifications, with Ga-DOTA agonists having significantly higher binding affinity than their Lu-, In-, and Y-DOTA correlates. Recently, somatostatin antagonists have been successfully developed as alternative tracers to agonists. The aim of this study was to evaluate whether chelated somatostatin antagonists are also sensitive to radiometal modifications and how. We have synthesized 3 different somatostatin antagonists, DOTA-p-NO2-Phe-c[d-Cys-Tyr-d-Aph(Cbm)-Lys-Thr-Cys]-d-Tyr-NH2, DOTA-Cpa-c[d-Cys-Aph(Hor)-d-Aph(Cbm)-Lys-Thr-Cys]-d-Tyr-NH2 (DOTA-JR11), and DOTA-p-Cl-Phe-c[d-Cys-Tyr-d-Aph(Cbm)-Lys-Thr-Cys]-d-Tyr-NH2, and added various radiometals including In(III), Y(III), Lu(III), Cu(II), and Ga(III). We also replaced DOTA with 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA) and added Ga(III). The binding affinity of somatostatin receptors 1 through 5 was evaluated in all cases. In all 3 resulting antagonists, the Ga-DOTA analogs were the lowest-affinity radioligands, with a somatostatin receptor 2 binding affinity up to 60 times lower than the respective Y-DOTA, Lu-DOTA, or In-DOTA compounds. Interestingly, however, substitution of DOTA by the NODAGA chelator was able to increase massively its binding affinity in contrast to the Ga-DOTA analog. The 3 NODAGA analogs are antagonists in functional tests. In vivo biodistribution studies comparing 68Ga-DOTATATE agonist with 68Ga-DOTA-JR11 and 68Ga-NODAGA-JR11 showed not only that the JR11 antagonist radioligands were superior to the agonist ligands but also that 68Ga-NODAGA-JR11 was the tracer of choice and preferable to 68Ga-DOTA-JR11 in transplantable HEK293-hsst2 tumors in mice. One may therefore generalize that somatostatin receptor 2 antagonists are sensitive to radiometal modifications and may preferably be coupled with a 68Ga-NODAGA chelator–radiometal complex.
- NODAGA chelator
- neuroendocrine tumor targeting
- radiometal modifications
- somatostatin receptor 2 antagonists
- somatostatin receptors
We demonstrated a decade ago (1) that somatostatin analogs may be sensitive to N-terminal radiometal modifications. For instance, Ga(III)-DOTA-OC (OC = octreotide) has a 3-fold higher affinity for somatostatin receptor 2 (sst2) than Y(III)-DOTA-OC, Ga(III)-DOTATOC has a 6-fold higher affinity than Y(III)-DOTATOC, and Ga(III)-DOTATATE has an 8-fold higher affinity than Y(III)-DOTATATE (1). These improved binding affinities also translated into improved internalization rates and concomitantly higher tumor uptake (2). In those studies, it appeared adequate to generalize that 67/68Ga is a radiometal that systematically improved the sst2 affinity of DOTA-conjugated somatostatin agonists and their pharmacokinetics. It was further confirmed in vivo in patients with neuroendocrine tumors that 68Ga-DOTANOC, 68Ga-DOTATOC, or 68Ga-DOTATATE was a better imaging agent than the 111In-DOTA congeners (2–8).
It has recently been shown that potent somatostatin receptor antagonists, known to poorly internalize into tumor cells, can visualize tumors in vivo as well as or even better than the corresponding agonists (9). This unexpected phenomenon was found both for sst2- and for somatostatin receptor 3 (sst3)-selective somatostatin analogs and may be due to the binding of the antagonist to a larger number of sites and to its slower dissociation rate. A pilot clinical trial with radiolabeled DOTA-linked sst2 antagonists recently confirmed the animal data (10). More potent sst2 antagonists aiming at that same purpose have been developed (11). To achieve this goal, several functional tests able to distinguish an agonist from an antagonist had to be developed (11,12).
In the light of these recent developments, it was therefore a logical consequence to investigate whether somatostatin antagonists were maintaining the same sensitivity to N-terminal modifications. We therefore selected the somatostatin antagonists with the best affinity and highest hydrophilicity that we have recently developed (11,13)—such as DOTA-p-NO2-Phe-c[d-Cys-Tyr-d-Aph(Cbm)-Lys-Thr-Cys]-d-Tyr-NH2 (DOTA-JR10), DOTA-Cpa-c[d-Cys-Aph(Hor)-d-Aph(Cbm)-Lys-Thr-Cys]-d-Tyr-NH2 (DOTA-JR11), and DOTA-p-Cl-Phe-c[d-Cys-Tyr-d-Aph(Cbm)-Lys-Thr-Cys]-d-Tyr-NH2 (DOTA-LM3) (Table 1) (11,13)—and coupled various metal ions to these DOTA conjugates such as Y(III), In(III), Lu(III), Cu(II), and Ga(III). In some analogs, we replaced DOTA with 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA), known to be a particularly good chelator for Ga(III) and Cu(II) (13,14). The sst1–sst5 binding affinities of these compounds were determined in vitro. Selected compounds with high sst2 affinity were tested for their biodistribution in vivo in animals bearing HEK293-hsst2–expressing tumors. As a control agonist, the pharmacokinetics of 68Ga-DOTATATE were compared with those of 68Ga-labeled somatostatin-based antagonists.
Chemical Structure and IC50 Values of sst2 Antagonists Based on JR10, JR11, and LM3 Family and Their Metallated Conjugates
MATERIALS AND METHODS
Reagents and Cell Lines
All reagents were of the best grade available and were purchased from common suppliers. The 68Ge/68Ga-generator IGG100 was available from Eckert & Ziegler. The sst2-specific antibody R2-88 was provided by Dr. Agnes Schonbrunn. The secondary antibody Alexa Fluor 488 goat antirabbit IgG (H+L) was from Molecular Probes, Inc. Tyr3-octreotide (TOC) was from Novartis.
The HEK293 cell line expressing the T7-epitope–tagged human sst2 (HEK-hsst2) was cultured at 37°C and 5% CO2 in Dulbecco modified Eagle medium containing 10% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 μg/mL), and G418 (500 μg/mL) (15). All culture reagents were from Gibco BRL, Life Technologies.
Synthesis of Analogs, Coupling to Chelators and Radiometals
The unnatural amino acids d-Aph(Cbm) (d-4-amino-Phe-carbamoyl) and Aph(Hor) (amino-Phe-hydroorotic acid) were synthesized as described earlier (13,16). The peptide analogs JR10, JR11, and LM3 and the corresponding DOTA and NODAGA conjugates (Table 1) were synthesized following standard solid-phase peptide synthesis on a methyl-benzhydrylamine resin as previously described (11,13).
The natIn, natY, natLu, natCu, and natGa complexes of the conjugates were prepared using an excess (2- up to 3.5-fold) of natInCl3, natYCl3 × xH2O, natLuCl3 × 6H2O, natCuCl2 × 2H2O, and natGa(NO3)3 × H2O, respectively, in ammonium acetate buffer, 0.2 M, pH 5, at 95°C (30 min) or at room temperature (natGa and natCu-NODAGA conjugates). Free metal ions were separated by preparative high-performance liquid chromatography (11), or they were eliminated by SepPak C-18 purification (Waters), in which the metallopeptides were eluted with ethanol. The fractions containing the metallopeptides were evaporated to dryness, redissolved in water, and lyophilized.
68Ga-labeled DOTATATE, DOTA-JR11, and NODAGA-JR11 were prepared using the Modular-Lab PharmTracer module from Eckert & Ziegler. Briefly, the 68Ge/68Ga-generator was eluted with 7 mL of HCl 0.1 N, and the eluate (∼900 MBq) was loaded onto a cation exchange column (Strata-XC; Phenomenex). 68Ga was eluted with 800 μL of a mixture of acetone/HCl (97.6%/0.02N) directly in a vial containing 2 mL of sodium acetate buffer (0.2 M, pH 4.0) and the minimum necessary amount of 10 μg of the conjugate. DOTATATE and DOTA-JR11 were labeled at 95°C within 8 min and NODAGA-JR11 at room temperature within 10 min, followed by SepPak C-18 purification to remove uncomplexed radiometal. The radiotracers were obtained in a radiochemical yield greater than 97% after purification and in specific activities ranging between 80 and 100 MBq/nmol. Quality control was performed by analytic high-performance liquid chromatography (13). The radiotracer solutions were prepared by dilution with 0.9% NaCl.
In Vitro sst1–sst5 Autoradiography
Receptor autoradiography was performed on 20-μm-thick cryostat (HM 500; Microm) sections of membrane pellets prepared from cell lines expressing the human sst1–sst5 as previously described (1,17). For each of the tested compounds, complete displacement experiments with the universal somatostatin radioligand 125I-[Leu8,d-Trp22,Tyr25]-somatostatin-28 (125I-LTT-SRIF-28) (74,000 MBq/mmol [2,000 Ci/mmol]; Anawa) using 15,000 cpm/100 μL and increasing concentrations of the unlabeled peptide ranging from 0.1 to 1,000 nM were performed. As a control, unlabeled SRIF-28 was run in parallel using the same increasing concentrations. The sections were incubated with 125I-LTT-SRIF-28 for 2 h at room temperature in Tris-HCl buffer (170 mmol/L, pH 8.2) containing 1% bovine serum albumin, bacitracin (40 mg/L), and MgCl2 (10 mmol/L) to inhibit endogenous proteases. The incubated sections were washed twice for 5 min in cold Tris-HCl (170 mmol/L, pH 8.2) containing 0.25% bovine serum albumin. After a brief dip in Tris-HCl (170 mmol/L, pH 8.2), the sections were dried quickly and exposed for 1 wk to Kodak BioMax MR film. Inhibitory concentration of 50% (IC50) values were calculated after quantification of the data using a computer-assisted image processing system as described previously (18). Tissue standards (autoradiographic 125I or 14C microscales; GE Healthcare) that contained known amounts of isotope, cross-calibrated to tissue-equivalent ligand concentrations, were used for quantification (11,19).
In Vitro Functional Assay for Agonism or Antagonism
An immunofluorescence microscopy-based internalization assay for sst2 was performed as previously described (15,17). HEK-hsst2 cells were grown on 35-mm 4-well plates (Cellstar; Greiner Bio-One GmbH) coated with poly-d-lysine (20 μg/mL) (Sigma-Aldrich). To distinguish whether the tested analogs are agonists or antagonists with respect to stimulating receptor internalization, HEK-hsst2 cells were treated for 30 min at 37°C in growth medium, either with vehicle alone (negative control) or with 10 nM TOC (positive control), 10 nM TOC in the presence of an excess (1 μM) of the somatostatin analogs, or 1 μM of the somatostatin analogs alone when the analogs were tested for agonism. The cells were processed for immunofluorescence microscopy and then imaged using a Leica DM RB immunofluorescence microscope and an Olympus DP10 camera.
Biodistribution in HEK-hsst2–Bearing Animals
All animal experiments were performed in accordance with the guidelines for the use of living animals in scientific studies and the German Law for the protection of animals. Female athymic nude mice, 4–6 wk old, were injected subcutaneously in the right shoulder with 107 HEK-hsst2 cells, freshly suspended in 100 μL of sterile phosphate-buffered saline. The tumors were allowed to grow for 14–18 d (tumor weight, 250–350 mg).
Mice were injected with 68Ga-DOTATATE, 68Ga-DOTA-JR11, or 68Ga-NODAGA-JR11 (100 pmol/100 μL/∼5–8 MBq) via the tail vein and were euthanized at 1 and 2 h after injection. Nonspecific uptake of all radiopeptides was determined with a coinjection of a 1,500-fold excess of the corresponding unlabeled conjugate. Organs of interest and blood were collected, rinsed of excess blood, blotted dry, weighed, and counted in a γ-counter. The results were expressed as percentage of injected activity per gram of tissue (%IA/g) and represent the mean ± SD of n = 3–5. The total counts injected per mouse were determined by extrapolation from counts of a known aliquot of the injected solution.
Small-Animal PET Studies
PET scans were obtained using a dedicated small-animal PET scanner (Focus 120 microPET scanner; Concorde Microsystems Inc.). 68Ga-DOTATATE, 68Ga-DOTA-JR11, and 68Ga-NODAGA-JR11 were administered to mice with HEK-hsst2 tumor xenografts, as described above. Animals were anesthetized with 1.5% isoflurane, and static scans were acquired at 1 and 2 h after injection, for 20–30 min. Blocking experiments were performed as described above, and static scans were obtained at 1 h after injection. PET images were reconstructed with filtered backprojection. No correction was applied for attenuation. Images were generated using AMIDE software. The color scale was set from 0% to 20% to allow for qualitative comparison among the images.
Data Analysis
Statistical analysis was performed by unpaired 2-tailed t testing using Prism software (GraphPad Software Inc.). P values of less than 0.05 were considered significant.
RESULTS
Affinity Studies and Agonist or Antagonist Properties
Table 1 summarizes the IC50 values for all compounds using 125I-LTT-SRIF-28. All analogs are highly specific for sst2.
In the JR11 family, yttrium or indium coupling to DOTA retains a high sst2 affinity comparable to the DOTA-JR11 compound without metal. Although indium diminishes the sst2 affinity 8-fold, copper and especially gallium induce a massive loss of the sst2 affinity by almost 80 times. Interestingly, when DOTA is replaced by NODAGA, the effect of gallium on sst2 affinity is completely restored, making Ga-NODAGA-JR11 a promising radiotracer (Table 1).
In the JR10 family, the same tendency of lowering the sst2 affinity by coupling gallium to DOTA is observed, when compared with indium or lutetium coupling. The sst2 affinity is, however, improved in the Ga-NODAGA-JR10 analog (Table 1).
In the LM3 family finally, a further impressive example of the loss of sst2 affinity of Ga-DOTA compared with In-DOTA is observed. Here again, replacing DOTA by NODAGA massively improves the affinity of the Ga-coupled analog and makes it adequate for clinical applications (Table 1).
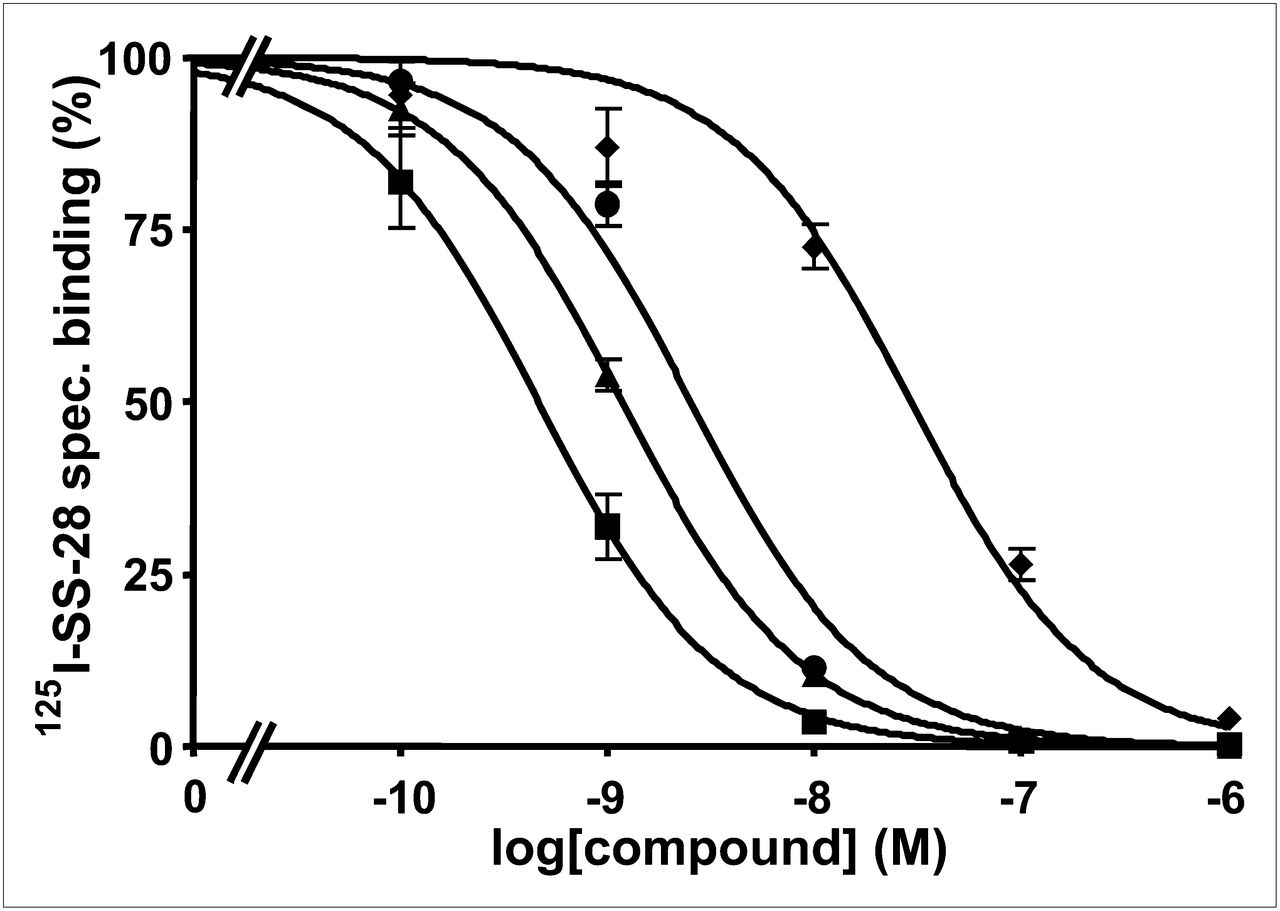
Two examples of competition experiments with JR11 (Fig. 1) and LM3 (Fig. 2) analogs (presently the most promising somatostatin antagonist radioligands) are presented to demonstrate the different binding affinities of gallium-coupled analogs with DOTA or NODAGA, as compared with reference compounds.
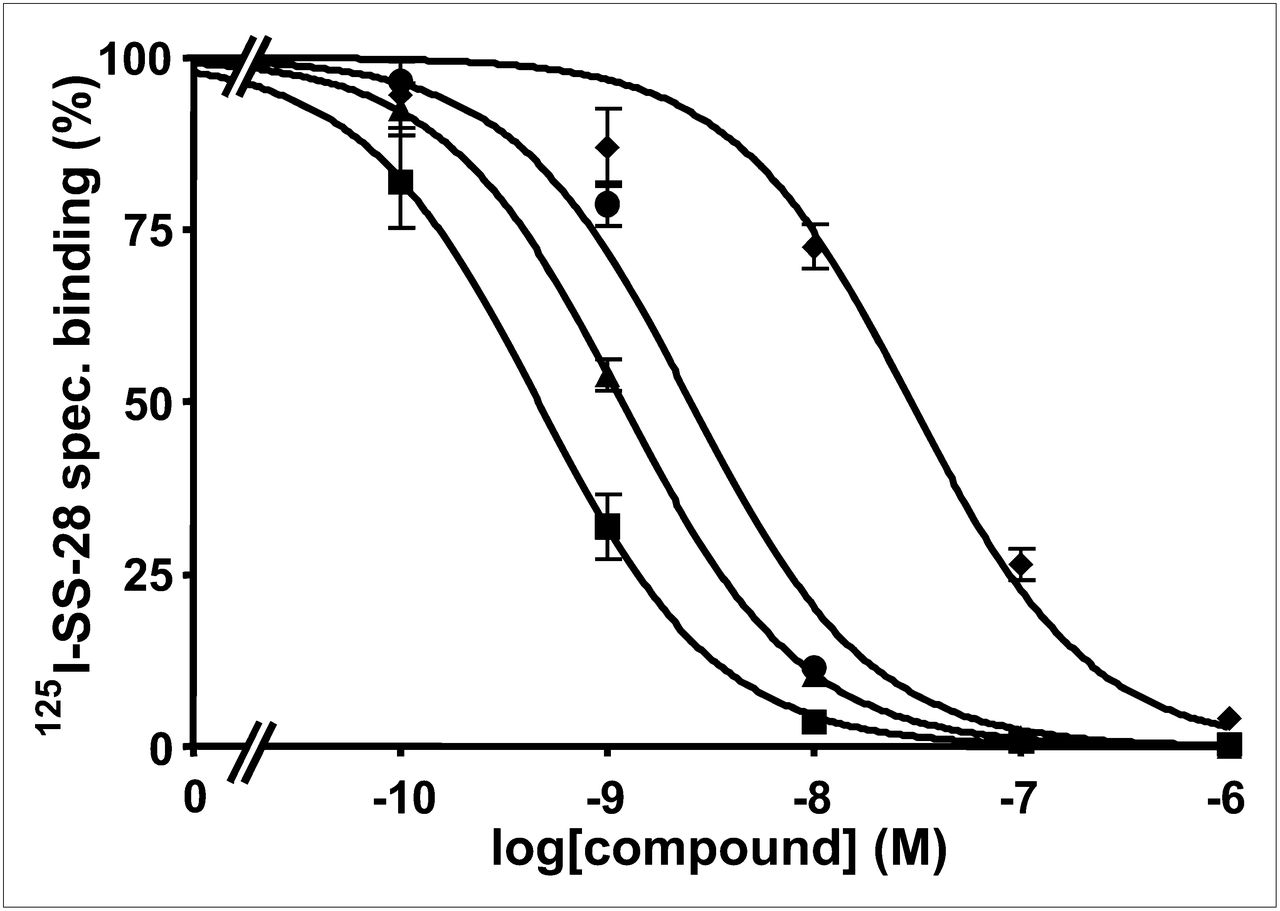
Competition experiments with cell membrane pellets of CCL39 cells expressing human sst2, using 125I-LTT-SRIF-28 as radioligand in presence of increasing concentrations of Ga-NODAGA-JR11 (▲), Y-DOTA-JR11 (■▪), Ga-DOTA-JR11 (♦), SRIF-28 (●) (as reference compound). spec = specific.
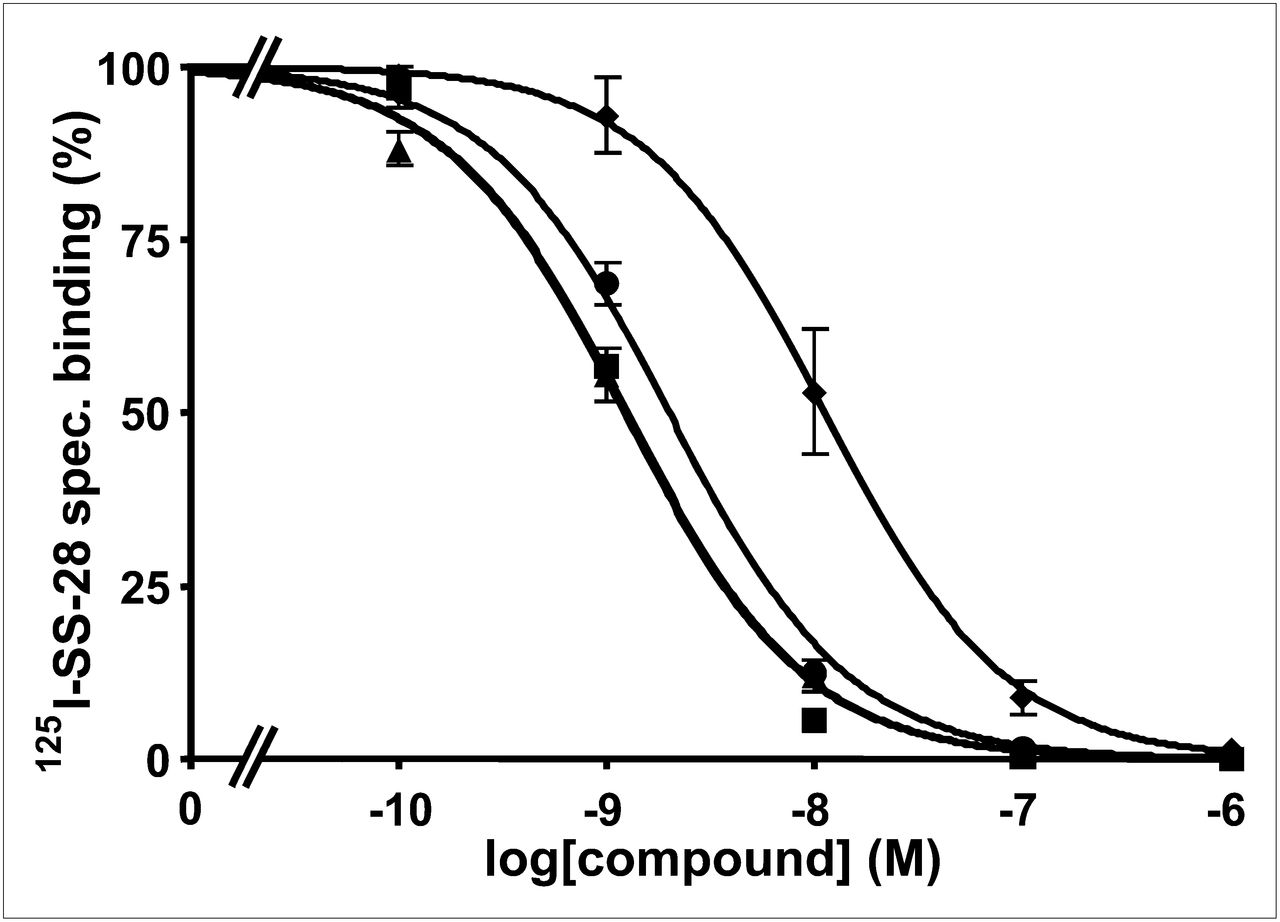
Competition experiments with cell membrane pellets of CCL39 cells expressing human sst2, using 125I-LTT-SRIF-28 as radioligand in presence of increasing concentrations of Ga-NODAGA-LM3 (▲), In-DOTA-LM3 (■▪), Ga-DOTA-LM3 (♦), and SRIF-28 (●) (as reference compound). spec = specific.
Receptor internalization assays are a suitable method to characterize receptor ligands for their functional behavior. Therefore, we have used an immunofluorescence microscopy–based internalization assay to analyze the Ga-NODAGA analogs for their ability to either stimulate sst2 internalization in HEK-hsst2 cells or antagonize the agonistic effect of TOC to stimulate sst2 internalization. Figure 3 shows that all 3 analogs are antagonists because alone at a concentration of 1 μM they are not able to stimulate sst2 internalization but they efficiently antagonize the agonistic TOC effect at a concentration of 1 μM.

Internalization assay based on sst2 immunofluorescence microscopy to determine whether Ga-NODAGA-JR10, Ga-NODAGA-LM3, and Ga-NODAGA-JR11 are agonists or antagonists. HEK-hsst2 cells were treated either with vehicle (no peptide) or with TOC (10 nM; positive control). To test for agonism or antagonism, cells were incubated either with 1 μM of the 3 Ga-NODAGA analogs alone (left column) or with 1 μM of the 3 Ga-NODAGA analogs in presence of TOC (10 nM). After incubation with peptides, cells were processed for immunocytochemistry as described in “Materials and Methods” section. All 3 Ga-NODAGA analogs act as antagonists because given alone they are not able to stimulate sst2 internalization, but they efficiently antagonize TOC-induced sst2 internalization.
In Vivo Biodistribution Results
68Ga-DOTATATE, 68Ga-DOTA-JR11, and 68Ga-NODAGA-JR11 biodistributions were compared at 1 and 2 h after injection. The data are reported in Tables 2–4. The radiotracers accumulated in the tumor, kidneys, and sst2-positive organs, such as the stomach and pancreas. The 2 antagonists, 68Ga-DOTA-JR11 and 68Ga-NODAGA-JR11, showed significantly higher tumor uptake (23.8 ± 3.7 and 30.7 ± 1.6 %IA/g, respectively, at 1 h after injection) than did the agonist 68Ga-DOTATATE (17.8 ± 2.2 %IA/g, P < 0.05). However, significantly higher accumulation of radioactivity in the kidneys was also found for 68Ga-DOTA-JR11 and 68Ga-NODAGA-JR11 (12.7 ± 3.5 and 10.4 ± 1.4 %IA/g, respectively) than for 68Ga-DOTATATE (5.2 ± 0.9 %IA/g, P < 0.05). The uptake in the sst2-positive organs was at the same level for 68Ga-DOTATATE and 68Ga-NODAGA-JR11 (e.g., stomach: 8.6 ± 1.9 and 9.9 ± 2.5 %IA/g, respectively, and pancreas: 10.8 ± 1.7 and 11.4 ± 3.6 %IA/g, respectively, P > 0.05), whereas significantly lower values were found for 68Ga-DOTA-JR11 (stomach: 0.5 ± 0.1 %IA/g, and pancreas: 0.5 ± 0.1%IA/g, respectively, P < 0.05). Blocking experiments confirm the receptor-mediated uptake in the tumor and sst2-positive organs of all 3 radiotracers. Tumor uptake remained essentially the same from 1 to 2 h after injection for all 3 radiotracers. However, the antagonist 68Ga-NODAGA-JR11 washed out from the sst2-positive organs more quickly than the agonist 68Ga-DOTATATE: two thirds of 68Ga-NODAGA-JR11 was washed out from the stomach and pancreas after 2 h, as compared with only one third of 68Ga-DOTATATE. Among all 3 radiotracers, tumor–to–non-tumor ratios were in favor of 68Ga-NODAGA-JR11.
Biodistribution Results of 68Ga-DOTATATE in Nude Mice Bearing HEK-hsst2 Tumor Xenografts
Biodistribution Results of 68Ga-DOTA-JR11 in Nude Mice Bearing HEK-hsst2 Tumor Xenografts
Biodistribution Results of 68Ga-NODAGA-JR11 in Nude Mice Bearing HEK-hsst2 Tumor Xenografts
Small-Animal PET Images
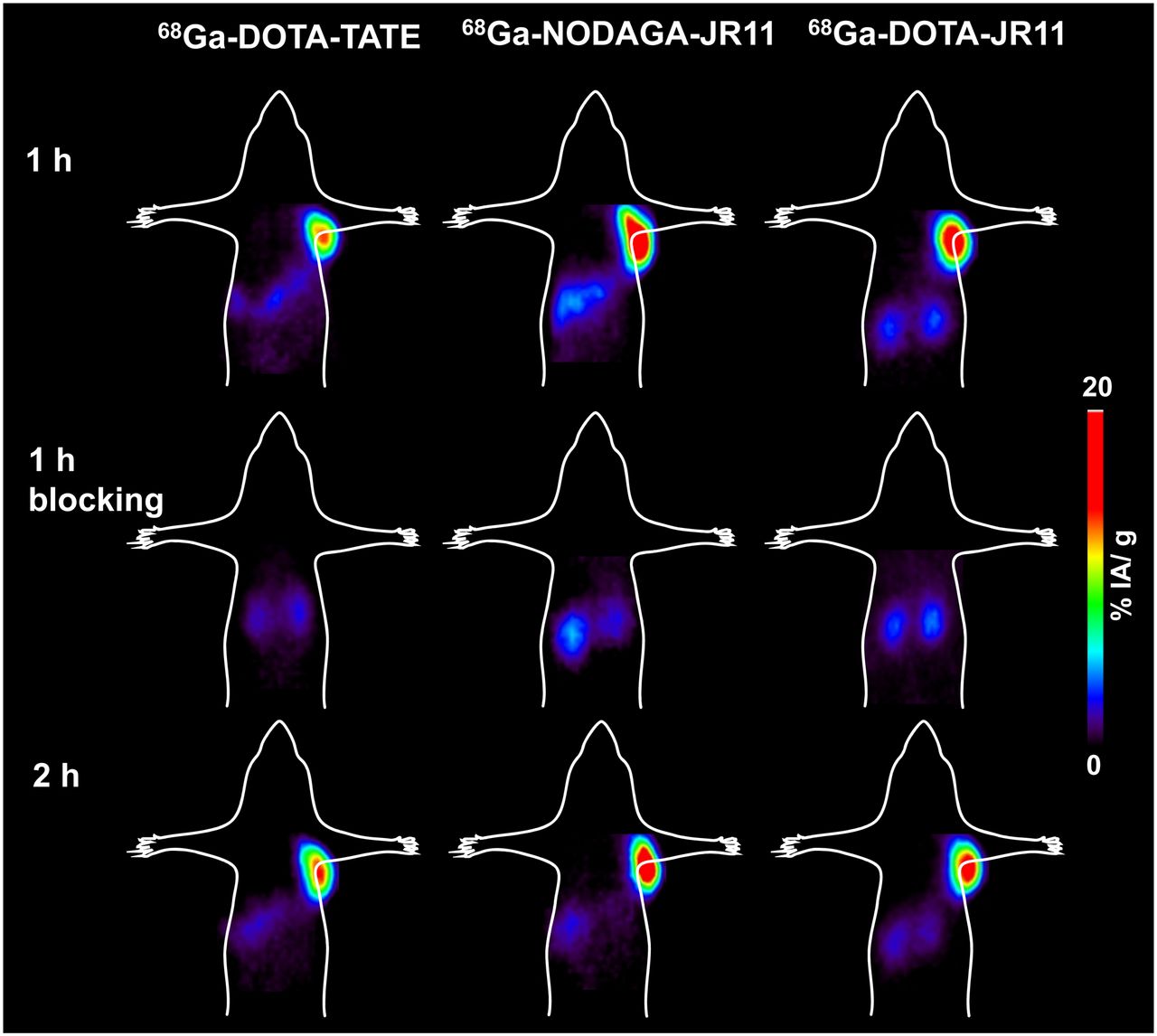
The PET images of 68Ga-DOTATATE, 68Ga-DOTA-JR11, and 68Ga-NODAGA-JR11 in Figure 4, reflecting the biodistribution results, illustrate not only the higher tumor uptake of the antagonists than the agonists but also the higher kidney uptake at 1 and 2 h after injection. Tumor-to-background contrast is better at 2 h after injection, especially for the antagonists. Because tumor blocking was demonstrated, the PET images of all radiotracers confirmed their specificity.
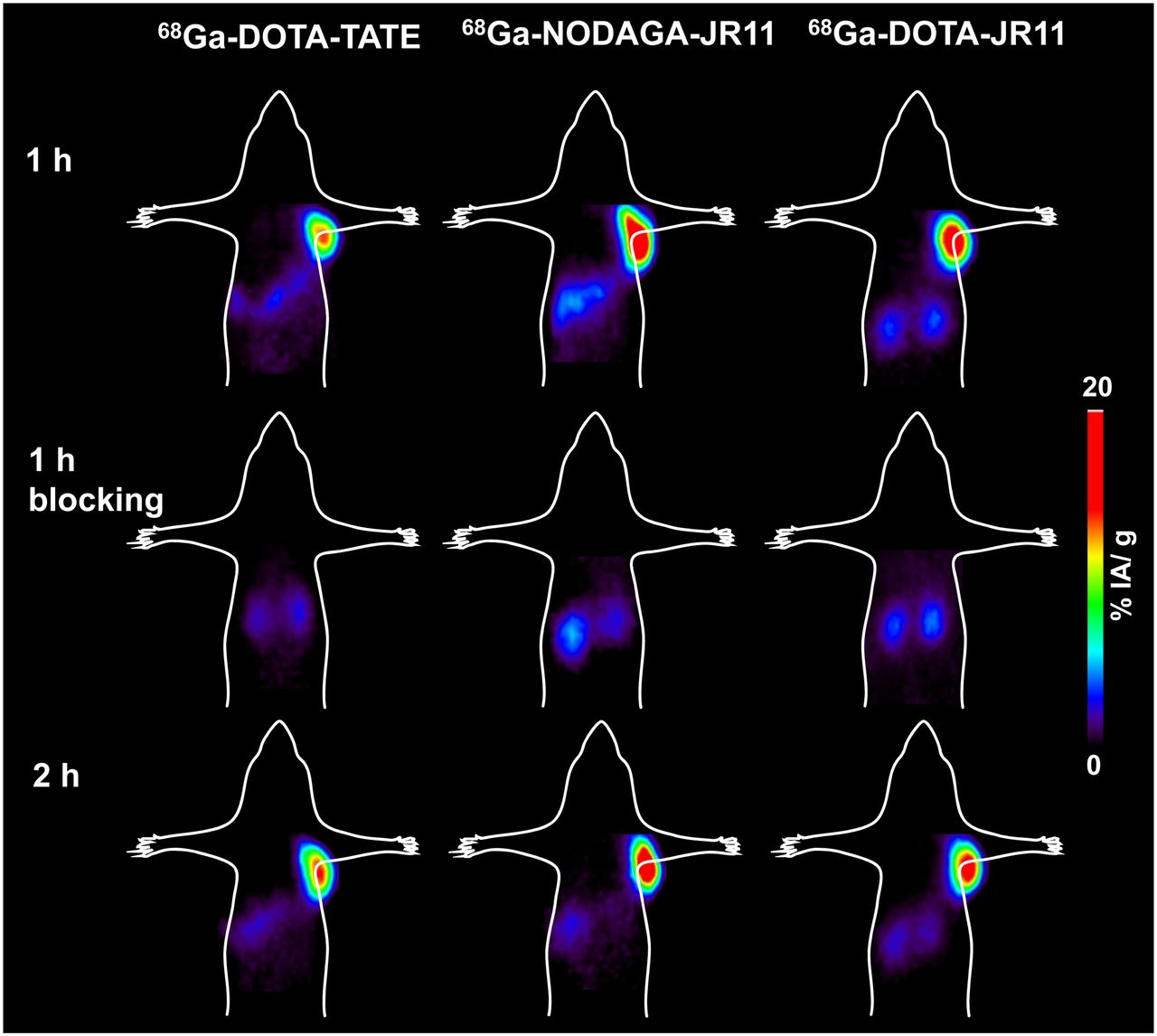
Small-animal PET images (coronal sections) of mice bearing HEK-hsst2 tumor injected with 68Ga-DOTATATE, 68Ga-DOTA-JR11, and 68Ga-NODAGA-JR11 (100 pmol/∼5–8 MBq) 1 and 2 h after injection show potentiality of radioantagonists to image sst2-expressing tumors in vivo. Images also illustrate higher tumor uptake of radioantagonists than agonists, and they confirm specificity of all radiotracers because no tumor is visualized in blocking experiments.
DISCUSSION
In the past decade, several reports have shown that adding a radiometal to a chelator–somatostatin–peptide complex had the potential to alter its receptor binding affinity or receptor subtype selectivity (1,2). Indeed, metallated M(III)-DOTA-octapeptide agonists targeting sst2 show pharmacologic differences for different M(III) radiometals of high relevance in nuclear oncology. In particular, in a broad family of different octapeptides the 67/68Ga-DOTA-octapeptides performed better than the 111In-, 90Y-, and 177Lu-DOTA-octapeptides in regard to sst2 affinity, rate of internalization, and tumor uptake (1,2). These earlier preclinical studies were translated into the clinic successfully, and several 68Ga-labeled peptides such as 68Ga-DOTATOC, 68Ga-DOTATATE, and 68Ga-DOTANOC are being used in the localization of neuroendocrine tumors worldwide. All these peptide structures have full agonistic properties.
We have recently shown that radiolabeled somatostatin-based antagonists targeting sst2 and sst3 are superior to the above-mentioned agonists if labeled with the γ-emitter 111In (9). The long-term goal is therefore to develop antagonist-based imaging agents for SPECT/CT and PET/CT and agents for targeted radionuclide therapy of neuroendocrine tumors.
In the in vitro part of the present study, all metallated DOTA- and NODAGA-conjugated somatostatin analogs of JR10, JR11, and LM3 (11,13) were shown to be of high affinity and high sst2 selectivity and were confirmed to have antagonistic properties. Several important conclusions can be drawn from these studies: first, a clear trend for a distinct affinity influence of the radiometal was observed using the sst2-selective DOTA-coupled antagonists of JR10, JR11, and LM3. Whereas DOTA agonists showed higher affinity when complexed to Ga(III) than to indium, yttrium, or lutetium, the inverse was found for the antagonists. Second, there is an 8-fold difference between In-DOTA-JR11 (IC50, 3.8 ± 0.7 nM) and Y-DOTA-JR11 (IC50, 0.47 ± 0.05 nM), indicating that 111In may not be a suitable surrogate of 90Y and therefore not reliable for dosimetric studies of the therapeutic radiopeptide. Third, another striking finding from these studies is the comparison between 68Ga-DOTATATE and 68Ga-DOTA-JR11 illustrating the great potential of the antagonists. 68Ga-DOTATATE (IC50, 0.2 ± 0.04 nM) has the highest binding affinity (lowest IC50 value) of all radiolabeled somatostatin-based agonists whereas 68Ga-DOTA-JR11 has 150-fold lower binding affinity (IC50, 29 ± 2.7 nM). Looking at pharmacokinetics and tumor uptake, in particular, the low-affinity antagonist is slightly superior, which can be explained by the higher number of binding sites for antagonists versus agonists, outweighing the affinity differences. The superiority of the antagonist was also impressively demonstrated with in vitro autoradiographic studies of human tumor specimens (20) and also confirmed by the PET images, which show high and specific tumor uptake and an excellent background clearance already at 1 and 2 h after injection.
We do not have a full explanation for the strong influence of the choice of radiometal. We explained the metal ion–dependent differences in the case of the agonists with differences in the coordination sphere as revealed by x-ray crystal structures of the model peptides Y-DOTA-d-Phe-NH2, Ga-DOTA-d-Phe-NH2 and Ga(NODASA) (21,22). X-ray structures are given in the supplemental information (supplemental materials are available online only at http://jnm.snmjournals.org). Y-DOTA-d-Phe-NH2 has an 8-fold coordination including the amide carboxy oxygen embedding the Y(III) inside the DOTA-monoamide cage. On the contrary, in Ga-DOTA-d-Phe-NH2, Ga(III) is hexacoordinated with a pseudooctahedral cis geometry, affording a free carboxymethyl group and a carboxymethylamide spacer. This keeps the pharmacophoric peptide at a distance from the chelate (21) and may allow higher flexibility of the cyclic peptide and therefore higher receptor binding affinity. Structural differences between Y-DOTATOC and Ga-DOTATOC were also confirmed by 1H and 13C NMR studies in solution (23).
Why the same metal coordination has a converse effect in the case of the chelate–antagonists is not clear. One can hypothesize that the spacer effect is also instrumental for the antagonists but toward lowering the affinity. This conclusion can be reached from the affinity of Cu-DOTA-JR11, which is also low (IC50, 16 ± 1.2 nM), and the coordination chemistry of Cu(II)-DOTA, which is similar to that of Ga(III)-DOTA. The geometry is pseudooctahedral and hexacoordinate, and one can strongly assume that in solution the carboxymethyl amide function is acting as a spacer (24–26).
The 8-fold affinity difference between In-DOTA-JR11 and Y-DOTA-JR11 is remarkable, but again an explanation is lacking. Both metal ions, In(III) and Y(III), have spheric symmetry and differ in the ionic radius (92 pm for In(III), 101.9 pm for Y(III), both for complexes of coordination number 8). We have recently also solved the x-ray crystal structure of In-DOTA-d-Phe-NH2 and found octacoordination. In-DOTA-d-Phe-NH2 crystallizes as the minor diastereoisomer, whereas Y-DOTA-d-Phe-NH2 adopts the major isomer conformation. More insight into differences is brought by 1H NMR studies in aqueous solution. They support octacoordination, but due to the smaller size of In(III) there is much more fluxionality within the In-DOTA-d-Phe-NH2 cage, which again may influence the binding affinity (Heppeler et al., unpublished data).
Because the relatively high IC50 values of 29, 8.9, and 12.5 nM for Ga-DOTA-JR11, Ga-DOTA-JR10, and Ga-DOTA-LM3, respectively, may affect the quality of images in vivo, we searched for alternative Ga(III) chelators such as NODAGA to determine whether a chelator–Ga complex would restore a higher binding affinity. Even though the NODAGA–peptide complex may have a decreased sst2 affinity, compared with the DOTA peptides, adding Ga3+ did indeed restore a high-affinity binding that is the basis of an improved in vivo tumor uptake. If we compare the sst2 affinity of Ga-DOTATOC (IC50, 2.5 ± 0.5 nmol/L) with Ga-NODAGA-TOC (IC50, 3.5 ± 1.6 nmol/L) (14), we find no significant difference between the 2 metallopeptides.
Moreover, the biodistribution experiments showed that the antagonists Ga-DOTA-JR11 and Ga-NODAGA-JR11 are better tracers than the agonist 68Ga-DOTATATE. 68Ga-DOTA-JR11, having a dramatically lower affinity for the sst2 (∼150-fold) than does 68Ga-DOTATATE, showed a 1.3-fold higher tumor uptake, whereas 68Ga-NODAGA-JR11, with a 6-fold lower affinity, showed a tumor uptake up to 1.7-fold higher. On the contrary, the tumor uptake in the sst2-positive organs was higher for 68Ga-DOTATATE and 68Ga-NODAGA-JR11 than for 68Ga-DOTA-JR11, reflecting somehow their higher affinities. The IC50 values, which reflect the affinity of the radiotracer for the corresponding receptors, are definitively a clear indication for its in vivo uptake in the sst2-positive tissues. However, this value is probably more predictive for the in vivo uptake in a well-organized, physiologic system expressing the receptors, such as the sst-positive normal tissues, rather than in a more disrupted system, such as the tumor environment. High target density, high association rate, and increased regional blood supply by pathologic tumor vessels may play an important role in the uptake and retention of the radiotracer in the tumor. On the other hand, a distinctly faster washout from the sst2-positive organs was found for 68Ga-NODAGA-JR11 than for 68Ga-DOTATATE, whereas tumor uptake remained essentially the same. Therefore, tumor-to-background ratio is higher for 68Ga-NODAGA-JR11 than for 68Ga-DOTATATE over time, with the exception of the tumor-to-kidney ratio. It is, however, worth mentioning that in the case of diagnostic radiotracers the kidney uptake is not as serious a drawback as in the case of therapeutic radiopharmaceuticals. Tumor-to-blood and tumor-to-muscle are 2 relevant ratios allowing increased image contrast—ratios that are persistently much higher for the 68Ga-NODAGA-JR11 over time. Additionally, an improvement on the tumor-to-liver ratio can be achieved with 68Ga-NODAGA-JR11 versus 68Ga-DOTATATE at 2 h. This ratio is important for sensitivity and accuracy in the diagnosis and staging of neuroendocrine tumors because often the liver is the primary site of metastasis for most of these tumors.
CONCLUSION
In this study, we demonstrated that complexation with distinct radiometals or replacing the chelator or chelate of a given peptide antagonist may dramatically affect the affinity and the in vivo profile of the radiotracer. Additionally, we showed that the radiolabeled antagonists, in particular if the chelator is NODAGA as in 68Ga-NODAGA-JR11, have higher tumor uptake than the agonists, even when the affinity of the antagonist is lower by orders of magnitude. Even though 68Ga-DOTATATE is a clinically accepted tracer to localize neuroendocrine tumors and is the preferred PET radiopharmaceutical in most laboratories with interest in this tumor entity, we argue that in the future 68Ga-NODAGA-JR11 or equally improved antagonists should be selected for this purpose. Whether antagonists labeled with therapeutic radionuclides, for example, 177Lu-DOTA-JR11, may represent a targeted vector for internal radiotherapy needs to be shown in clinical studies that we have started recently.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jul. 31, 2012.
- © 2012 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication January 5, 2012.
- Accepted for publication April 30, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Limitations of the radiotheranostic concept in neuroendocrine tumors due to lineage-dependent somatostatin receptor expression on hematopoietic stem and progenitor cells
- Optimized Methods for the Production of High-Purity 203Pb Using Electroplated Thallium Targets
- Head-to-Head Comparison of 68Ga-NODAGA-JR11 and 68Ga-DOTATATE PET/CT in Patients with Metastatic, Well-Differentiated Neuroendocrine Tumors: Interim Analysis of a Prospective Bicenter Study
- 225Ac-MACROPATATE: A Novel {alpha}-Particle Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors
- A Randomized, Factorial Phase II Study to Determine the Optimal Dosing Regimen for 68Ga-Satoreotide Trizoxetan as an Imaging Agent in Patients with Gastroenteropancreatic Neuroendocrine Tumors
- First-in-Humans Study of the SSTR Antagonist 177Lu-DOTA-LM3 for Peptide Receptor Radionuclide Therapy in Patients with Metastatic Neuroendocrine Neoplasms: Dosimetry, Safety, and Efficacy
- Multimodal Imaging of 2-Cycle PRRT with 177Lu-DOTA-JR11 and 177Lu-DOTATOC in an Orthotopic Neuroendocrine Xenograft Tumor Mouse Model
- Head-to-Head Comparison of 68Ga-DOTA-JR11 and 68Ga-DOTATATE PET/CT in Patients with Metastatic, Well-Differentiated Neuroendocrine Tumors: A Prospective Study
- Phase I Trial of Well-Differentiated Neuroendocrine Tumors (NETs) with Radiolabeled Somatostatin Antagonist 177Lu-Satoreotide Tetraxetan
- International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature
- Sensitivity Comparison of 68Ga-OPS202 and 68Ga-DOTATOC PET/CT in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase II Imaging Study
- Safety, Biodistribution, and Radiation Dosimetry of 68Ga-OPS202 in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase I Imaging Study
- Somatostatin Receptor Imaging of Neuroendocrine Tumors: From Agonists to Antagonists
- SSTR-Mediated Imaging in Breast Cancer: Is There a Role for Radiolabeled Somatostatin Receptor Antagonists?
- Biodistribution, Pharmacokinetics, and Dosimetry of 177Lu-, 90Y-, and 111In-Labeled Somatostatin Receptor Antagonist OPS201 in Comparison to the Agonist 177Lu-DOTATATE: The Mass Effect
- Somatostatin Receptor Antagonists for Imaging and Therapy
- Significance of a Single-Time-Point Somatostatin Receptor SPECT/Multiphase CT Protocol in the Diagnostic Work-up of Gastroenteropancreatic Neuroendocrine Neoplasms
- Comparison of the Therapeutic Response to Treatment with a 177Lu-Labeled Somatostatin Receptor Agonist and Antagonist in Preclinical Models
- Development of 68Ga- and 89Zr-Labeled Exendin-4 as Potential Radiotracers for the Imaging of Insulinomas by PET
- In Vivo Radioimaging of Bradykinin Receptor B1, a Widely Overexpressed Molecule in Human Cancer
- Preclinical Evaluation of a High-Affinity 18F-Trifluoroborate Octreotate Derivative for Somatostatin Receptor Imaging
- Comparison of Somatostatin Receptor Agonist and Antagonist for Peptide Receptor Radionuclide Therapy: A Pilot Study