


Abstract
Clinical studies have demonstrated the potential of radiometallated exendin-4 derivatives for the imaging of glucagonlike peptide-1 receptor–overexpressing insulinomas. Recently investigated exendin-4 derivatives were radiolabeled with the SPECT isotopes 99mTc or 111In. Despite promising results, the low spatial resolution associated with SPECT and the occasional need to perform imaging several days after injection for the demarcation of insulinomas from the kidneys represent current limitations. The aim of this work was the development of exendin-4 derivatives for the imaging of insulinomas by high-resolution PET at early or late time points after injection of the radiotracer. Methods: An exendin-4 derivative conjugated to desferrioxamine (DFO) was used for radiolabeling with the PET isotopes 68Ga and 89Zr. Both radiotracers were evaluated in vitro with RIN-m5F cells for their cell internalization properties as well as affinities and specificities toward the glucagonlike peptide-1 receptor. Serum stabilities of the radiopeptides were assessed in blood serum, and their distribution coefficient was determined by the shake-flask method. Biodistribution experiments were performed with nude mice bearing RIN-m5F xenografts. For all experiments, clinically evaluated [Lys40-(AHX-DTPA-111In)NH2]exendin-4 was used as a reference compound. Results: [Lys40-(AHX-DFO)NH2]exendin-4 was labeled with 89Zr and 68Ga in high radiochemical yield and purity. In vitro experiments showed favorable cell uptake and receptor affinity for [Lys40-(AHX-DFO-68Ga)NH2]exendin-4, and [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 and [Lys40-(AHX-DTPA-111In)NH2]exendin-4 performed similarly well. In biodistribution experiments, [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 exhibited a significantly enhanced tumor uptake 1 h after injection in comparison to the other 2 radiotracers. Tumor uptake of [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 was comparable to that of [Lys40-(AHX-DTPA-111In)NH2]exendin-4 at 1–48 h after injection. All compounds showed a fast blood clearance and low accumulation in receptor-negative organs and tissue with the exception of the kidneys, a known characteristic for exendin-4–based radiotracers. Conclusion: 68Ga- and 89Zr-radiolabeled [Lys40-(AHX-DFO)NH2]exendin-4 exhibit characteristics comparable or superior to the clinically tested reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 and, thus, represent potential new tracers for the imaging of insulinomas by PET.
Insulinomas are rare, β cell–derived neuroendocrine tumors of pancreatic origin. In most cases, they are benign and overexpress the glucagonlike peptide-1 receptor (GLP-1r) with a high incidence and density (1). Surgery is the preferred therapy, but the detection of small-sized insulinomas (usually <2 cm) (2) for exact preoperative localization remains challenging with conventional methods (e.g., CT, MR imaging, ultrasound). As a consequence, alternatives to the noninvasive detection of insulinomas are needed.
Glucagonlike peptide-1 (GLP-1) is a potent incretin hormone and the endogenous ligand for the GLP-1r. GLP-1 is released from intestinal L cells in response to nutrient ingestion (3). In humans, multiple forms of GLP-1 are secreted but most GLP-1 in circulation is GLP-1(7-36)NH2 (4). Exendin-3 and exendin-4 are analogous reptilian agonists of GLP-1r and share an amino acid homology to GLP-1 of 50% and 53%, respectively. Unlike GLP-1, exendin-4 it is not degraded by dipeptidylpeptidase 4, a major protease involved in the metabolism of the hormones. In addition, exendin-4 exhibits a 10-fold-increased affinity toward human GLP-1r (5). Early preclinical studies using 125I-labeled GLP-1(7-36) amide and exendin-3 have demonstrated the general feasibility of GLP-1r targeting with radiolabeled peptides but were hampered by either insufficient stability of the radiopeptide, challenging radiolabeling procedures, or a fast washout of radioactivity from cancer cells and tumors (6). Radiometallation of stable GLP-1 analogs such as exendin-4 has since helped to overcome some of these initial limitations. This is in part due to the higher degree of flexibility and convenient labeling procedures offered by metallic radionuclides in comparison to radiohalogens.
Several radiolabeled exendin-4 (and -3) have been reported in the past years. These derivatives were functionalized with different chelators (diethylenetriaminepentaacetic acid [DTPA], hydrazinonicotinic acid, DOTA, and 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid) suitable for radiolabeling with 111In (2,7,8), 99mTc (8,9), 68Ga (7,8,10–12), and 64Cu (10,12,13), respectively. More recently, examples of 18F-labeled exendins have also been described (14–17). Despite intensive research efforts made toward the development of GLP-1r imaging agents, only a few examples have so far been evaluated clinically (9,18,19). Encouraging results were obtained with 111In-labeled derivatives, namely [Lys40-(AHX-DTPA-111In)NH2]-exendin-4 (AHX = 6-amino-hexanoic acid) and [Lys40-(AHX-DOTA-111In)NH2]-exendin-4 (18,19). With both radiotracers, insulinomas that were hardly detectable by conventional methods could be clearly visualized with SPECT. In addition, application of exendin-4 radiolabeled with the longer-lived 111In (half-life [t1/2] = 2.8 d) enabled the subsequent resection of the tumor mass by radioguided surgery (18). These studies also revealed that the inherent low spatial resolution associated with SPECT was a limitation. In several patients, it was not possible at early time points to demarcate tumors of the pancreas from the kidneys, organs in close anatomic proximity that display a high and persistent uptake of radioactivity. In these cases, a second SPECT/CT scan 3–7 d after injection after sufficient clearance of radioactivity from the kidneys was required (18). Obviously, application of exendin-4 derivatives labeled with positron-emitting radionuclides for higher-resolution PET could provide a solution. First preclinical investigations with 68Ga-labeled [Lys40-(AHX-DOTA)NH2]exendin-4 indicated favorable pharmacokinetics and a fast and high accumulation of the radiotracer in tumors (and in the kidneys) (8). During the preparation of this article, the detection of occult insulinoma by 68Ga-NOTA-exendin-4 PET/CT in a single patient was reported (20). However, whether PET imaging with 68Ga-labeled exendin-4 enables the reliable differentiation of small tumors of the pancreas from the kidneys at early time points after injection remains to be shown in a larger cohort. Alternatively, application of longer-lived positron-emitting radiometals may allow deploying the high resolution of PET for imaging of insulinomas at later time points, ideally earlier than those reported for 111In-labeled exendin-4.
Here, we report an exendin-4 derivative, [Lys40-(AHX-DFO)NH2]exendin-4, functionalized with the siderophore desferrioxamine (DFO), a chelator enabling radiolabeling with both 68Ga (t1/2 = 67.7 min) and 89Zr (t1/2 = 78.4 h) for potential imaging of insulinomas by PET at early and late points, respectively. The radiotracers [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 and [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 were fully evaluated in vitro and in vivo and compared side by side with the clinically tested reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4.
MATERIALS AND METHODS
Detailed description of materials and methods including those used for radiolabeling reactions, cell experiments, serum stability studies, distribution coefficient determination, and biodistributions in mice can be found in the supplemental data (available at http://jnm.snmjournals.org).
RESULTS
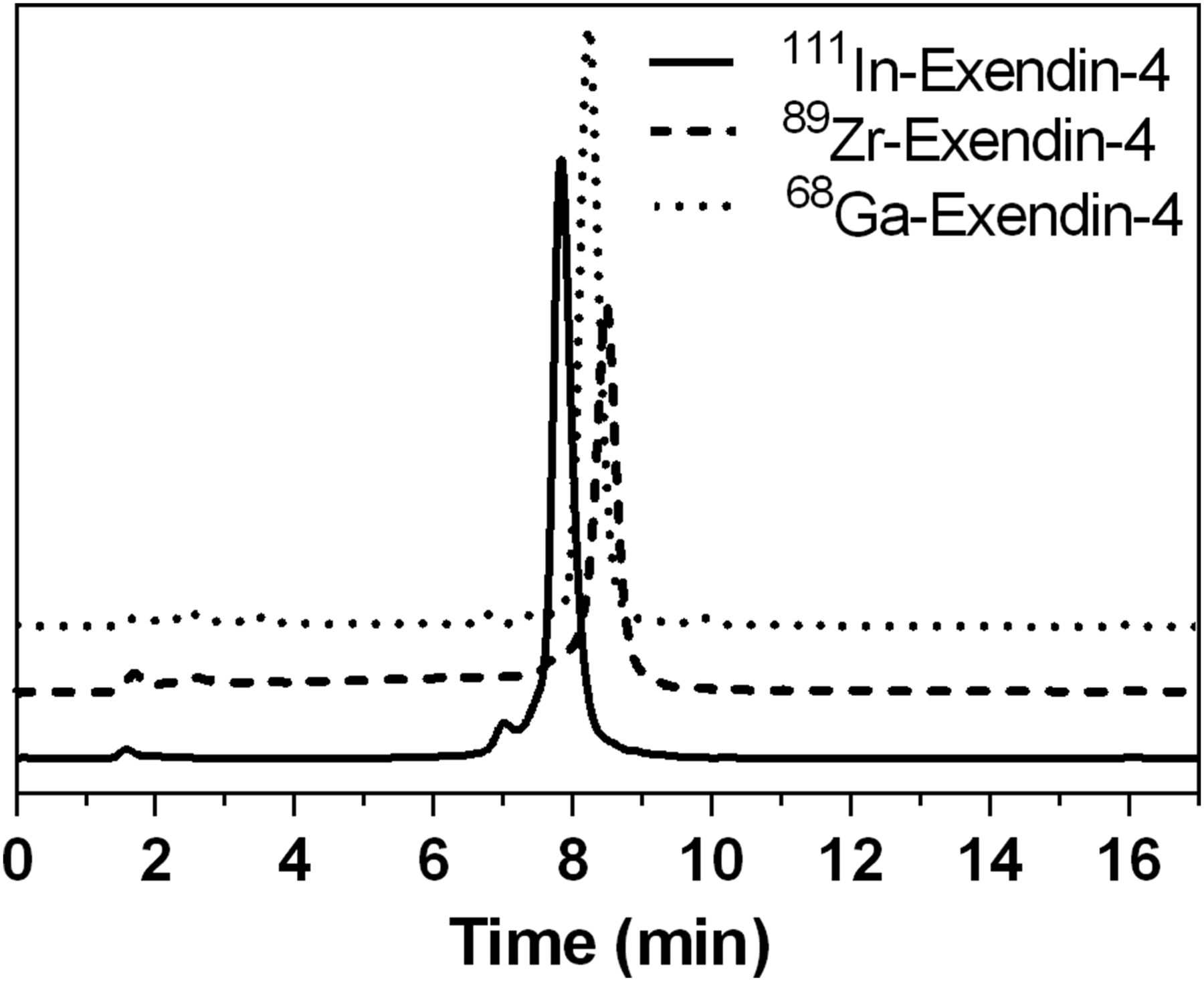
Radiolabeling of the peptide conjugates with 68Ga, 89Zr, and 111In provided the corresponding radiometallated peptides in excellent radiochemical yield and radiochemical purity and sufficient specific activities (As; Table 1). Representative γ–high-performance liquid chromatograms of the radiolabeled products obtained are shown in Figure 1.
Results of Radiolabeling Experiments

Radio–high-performance liquid chromatograms of [Lys40-(AHX-DFO-89Zr)NH2]exendin-4, [Lys40-(AHX-DFO-68Ga)NH2]exendin-4, and reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4.
The results of cell experiments (n = 2–3 in triplicate) are summarized in Table 2 and shown in Figures 2 and 3. Binding of all 3 compounds to, and internalization into RIN-m5F cells, was GLP-1r–specific, as shown by the blocking experiment. In all cases, the presence of excess natural exendin-4 decreased the cellular binding of the radiotracers to, and internalization into RIN-m5F cells, to less than 0.5% of the applied radioactivity, thus demonstrating receptor specificity. For internalization experiments, the time–activity curve did not reach a plateau after 4 h of incubation at 37°C. This observation is in agreement with published in vitro data for related, radiometal-labeled exendin-4 (and -3) derivatives (2,7).
Physicochemical Properties of Radiolabeled Exendin-4 Derivatives

Time-dependent receptor specific internalization of [Lys40-(AHX-DFO-89Zr)NH2]exendin-4, [Lys40-(AHX-DFO-68Ga)NH2]exendin-4, and reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 of at least 2 independent experiments (triplicates in each), expressed as percentage of added radioactivity/106 cells ± SD. Overlapping lines near 0% activity/106 cells represent results of blocking experiments (main text).

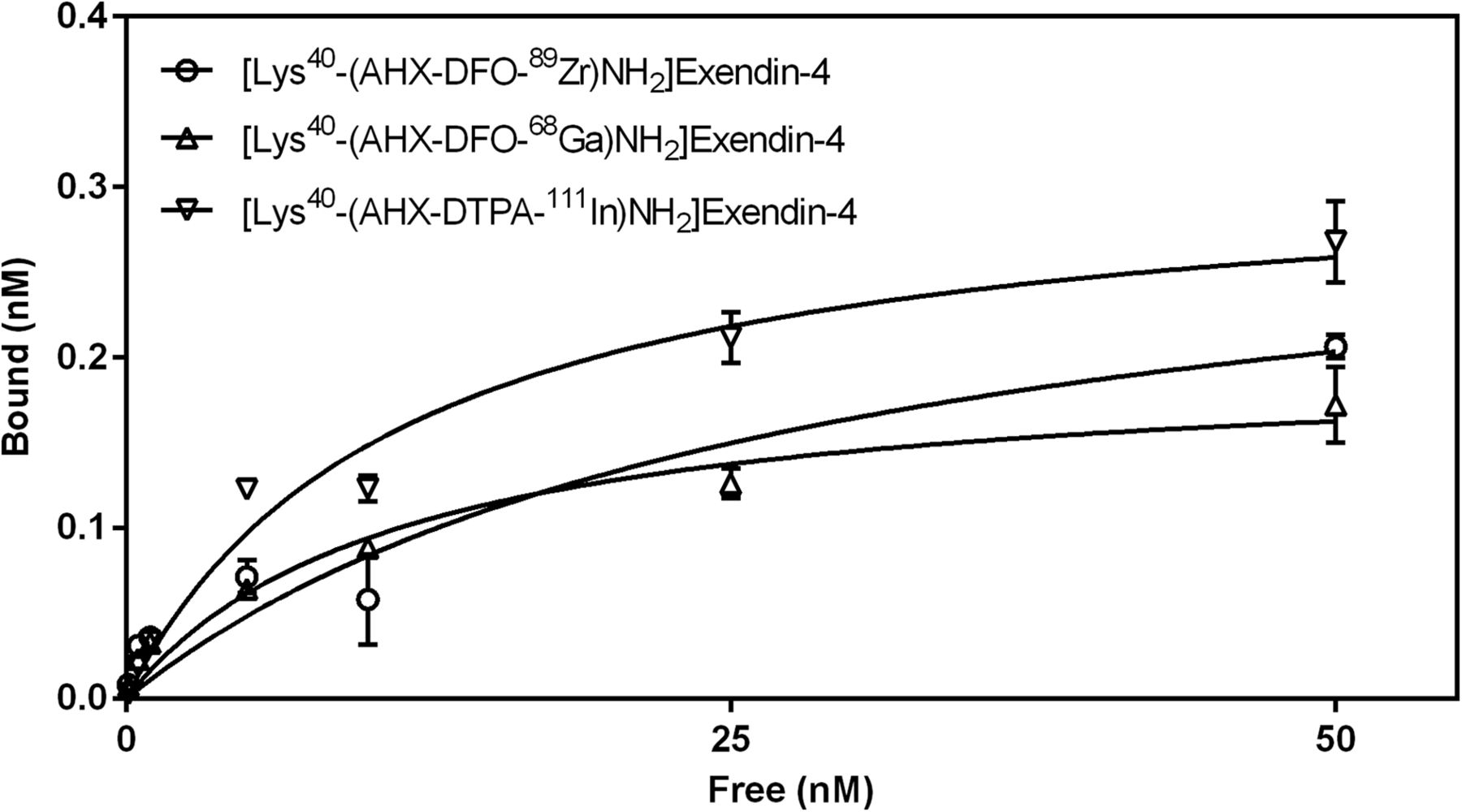
Receptor saturation binding curves of [Lys40-(AHX-DFO-89Zr)NH2]exendin-4, [Lys40-(AHX-DFO-68Ga)NH2]exendin-4, and reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 of 2 independent experiments (triplicates in each).
[Lys40-(AHX-DFO-68Ga)NH2]exendin-4 and [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 were stable for the time periods investigated, 4 and 24 h, respectively (Table 2).
The distribution coefficient of all 3 compounds was found to be in the same range (−1.7 to −2.0; Table 2), indicating a hydrophilic character of the radiotracers at physiologic pH.
Biodistribution experiments for the assessment of the uptake of radiolabeled exendin-4 derivatives in GLP-1r–positive and –negative organs were performed in RIN-m5F–xenografted Foxn1NU nude mice at 1, 4, 24, and 48 h after injection for the 89Zr- and 111In-labeled peptides and 1 h after injection for the 68Ga-labeled peptide. The results are shown in Figures 4–6 and supplemental data.
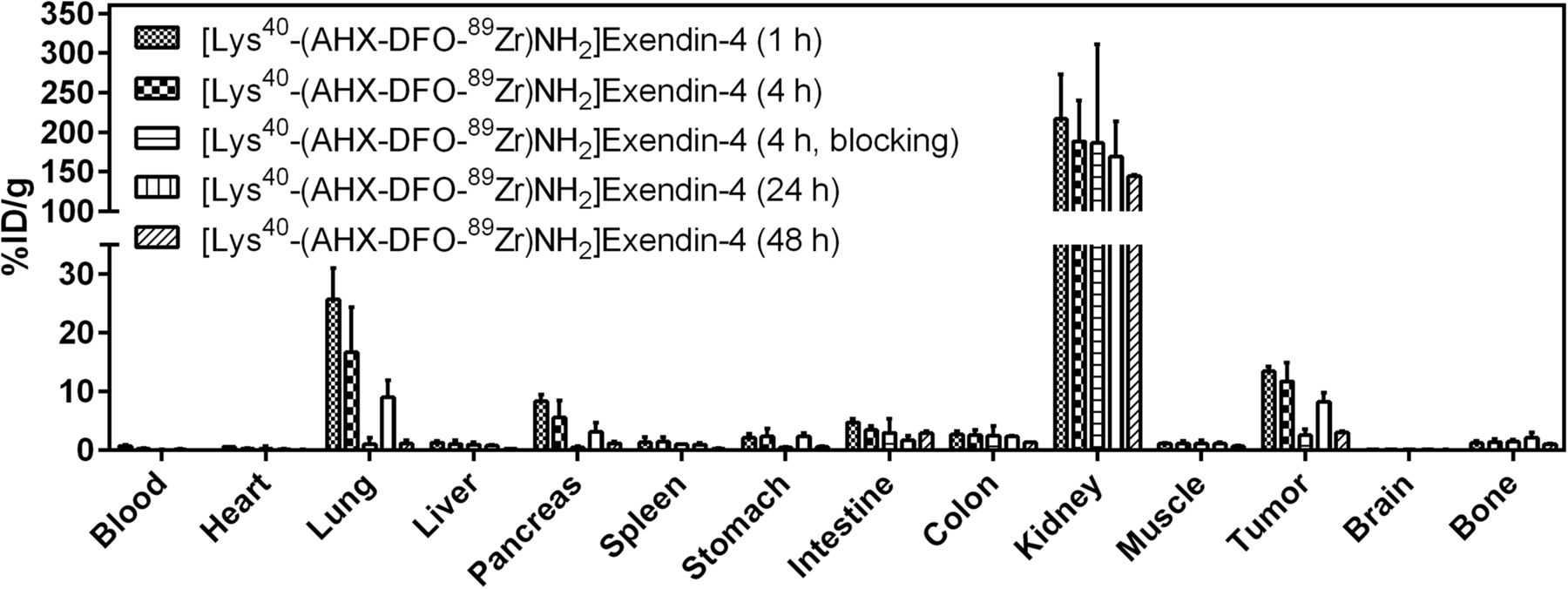
Biodistribution of [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 in Foxn1NU mice (n = 5). Values (%ID/g ± SD) are expressed as mean %ID/g of tissue. Blocking was done by preinjection of 1,000-fold excess of exendin-4.
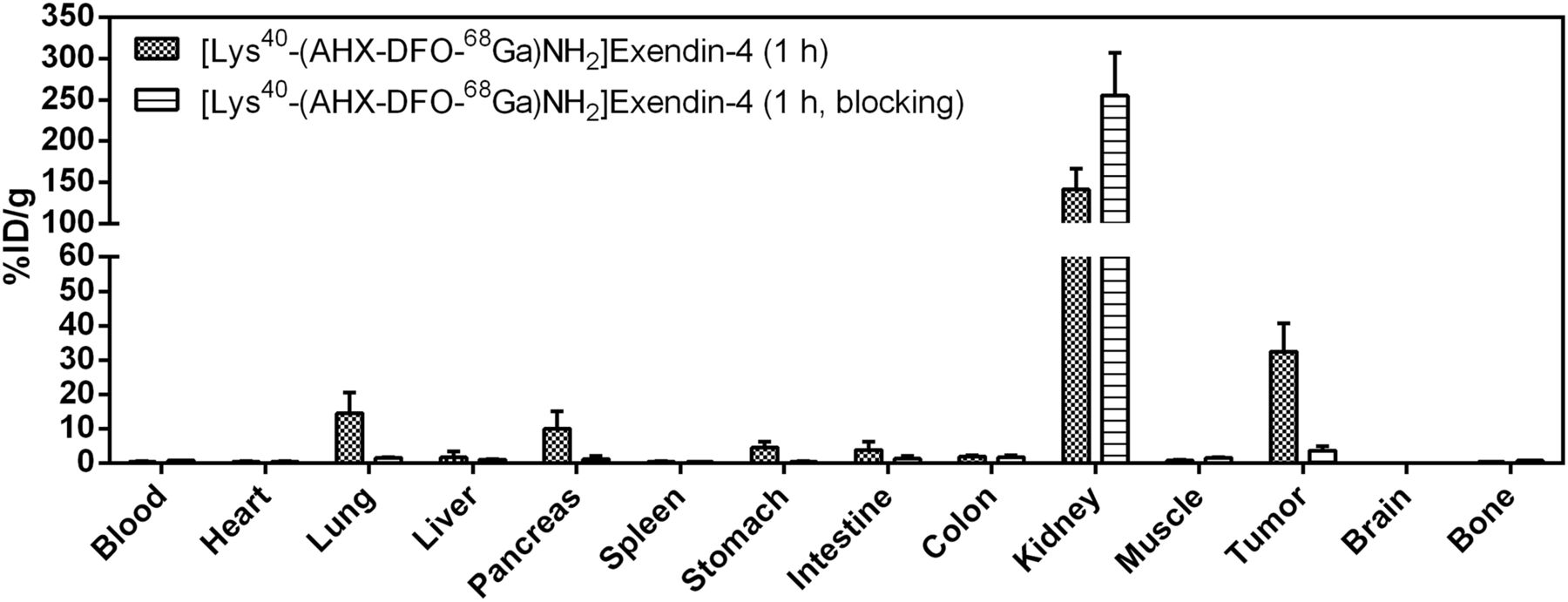
Biodistribution of [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 in Foxn1NU mice (n = 5). Values (%ID/g ± SD) are expressed as mean %ID/g of tissue. Blocking was done by preinjection of 1,000-fold excess of exendin-4.
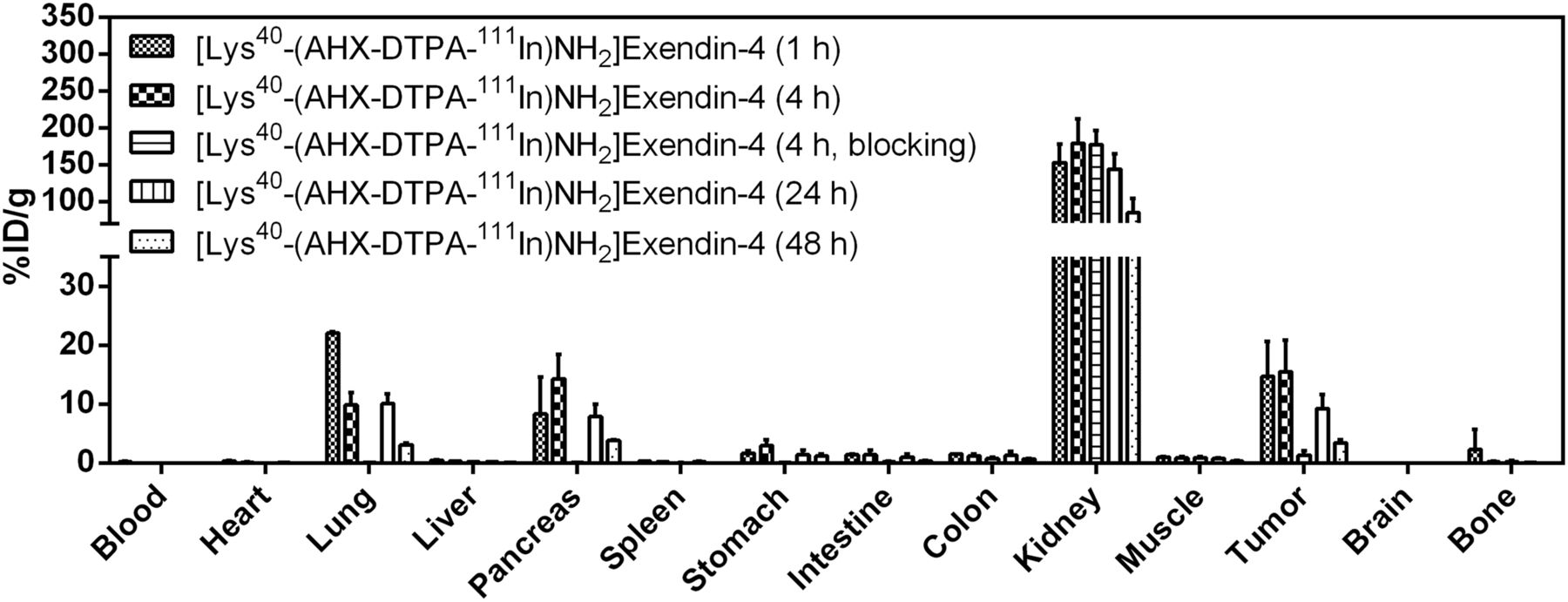
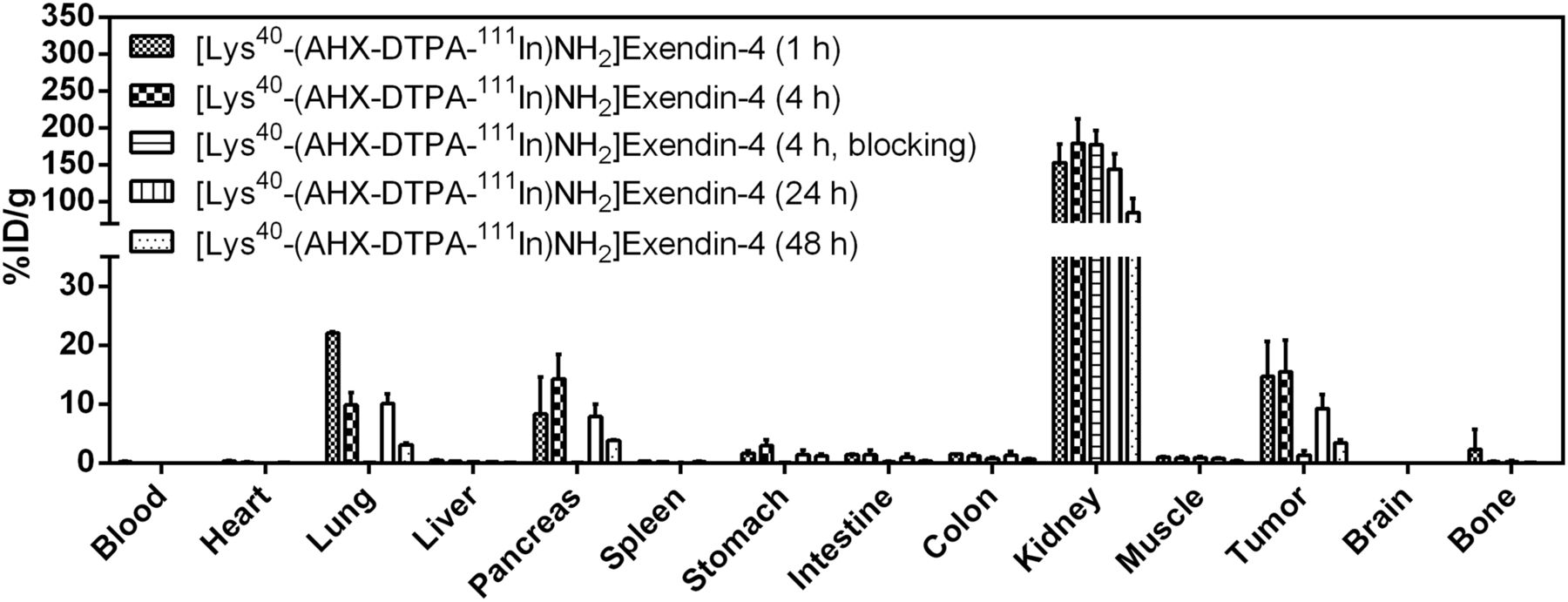
Biodistribution of reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 in Foxn1NU mice (n = 5). Values (%ID/g ± SD) are expressed as mean %ID/g of tissue. Blocking was done by preinjection of 1,000-fold excess exendin-4
All 3 radiopeptides showed receptor-specific accumulation in the tumors and GLP-1r–positive organs (lung, pancreas, intestine, stomach). Tumor uptake of the reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 was 14.7 ± 5.9 percentage injected dose per gram (%ID/g) at 1 h after injection and remained constant until 4 h (15.6 ± 5.3 %ID/g), after which time a slow washout was observed (9.3 ± 2.3 %ID/g [24 h] and 3.5 ± 0.5 %ID/g [48 h]). A similar behavior was observed for [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 but with a slightly lower tumor uptake of 13.5 ± 0.8 %ID/g at 1 h, 11.7 ± 3.2 %ID/g at 4 h, 8.2 ± 1.6 %ID/g at 24 h, and 3.1 ± 0.2 %ID/g at 48 h. In contrast, [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 exhibited a significantly higher tumor uptake of 32.5 ± 8.3 %ID/g at 1 h after injection. Receptor-specific uptake of the radiopeptides was confirmed by coadministration of an excess of exendin-4 (1-39) in a separate experiment at 4 h after injection (89Zr and 111In) and 1 h after injection (68Ga), resulting in a tumor blockade of more than 91% for [Lys40-(AHX-DTPA-111In)NH2]exendin-4, more than 81% for [Lys40-(AHX-DFO-89Zr)NH2]exendin-4, and more than 88% for [Lys40-(AHX-DFO-68Ga)NH2]exendin-4, respectively. High and persistent unspecific uptake of radioactivity in the kidneys was observed for all 3 derivatives, with the highest value for [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 at 1 h after injection (216.9 ± 56.2 %ID/g). For all compounds investigated, a rapid clearance of radioactivity from receptor-negative tissues was observed, whereas the washout from receptor-positive organs and the kidneys was slow.
DISCUSSION
GLP-1r targeting with radiolabeled exendin derivatives has been intensively investigated in recent years. Indeed these efforts are justified by the fact that current conventional detection methods often lack the required sensitivity to localize reliably small insulinoma lesions before surgery. In addition, GLP-1r targeting with radiotracers holds great promise for the noninvasive determination of pancreatic β-cell mass during the development of type 1 diabetes (21) and for monitoring islet cell grafts that are transplanted as a potential new therapeutic means to achieve glycemic control of the disease (22).
First clinical studies with 111In-labeled exendin-4 have clearly demonstrated the benefits of GLP-1r imaging of insulinomas using nuclear imaging modalities (18,19); however, these studies have also revealed some limitations. In particular, the low spatial resolution associated with SPECT impeded in several cases the demarcation of pancreatic tumors from the kidneys, which made a second scan after 3–7 d necessary. To address these issues, we set out to investigate the utility of exendin-4 derivatives radiolabeled with PET radionuclides, in particular radiometals. The higher resolution of PET than SPECT may allow for the detection of insulinomas despite the high unspecific uptake of radioactivity in the kidneys.
Considering the possibility that image acquisition at later time points might still be necessary, we planned to include not only short-lived but also longer-lived PET radiometals. Specifically, we envisioned the employment of 68Ga (t1/2 = 67.7 min) and 89Zr (t1/2 = 78.4 h). Even though the radiolabeling of peptides and small molecules with 89Zr has been reported (23,24), one has to acknowledge that the physical half-life of 89Zr is not a good match for the short biologic half-life and fast excretion rate of a peptide carrier (usually minutes to a few hours), unless specific reasons require late-time-point imaging as in the example described herein.
Ideally, radiotracers based on 2 different radiometals should be accessible from the same radiolabeling precursor and display similar physicochemical properties. To achieve this goal, we chose an exendin-4 derivative functionalized with the siderophore DFO for our investigations (supplemental data). DFO is a hydroxamic acid–based, acyclic chelator, which has been safely used in the clinic for the treatment of acute iron poisoning. It has also been shown invaluable for the development of 89Zr radiopharmaceuticals, in particular in the context of immuno-PET as demonstrated in various preclinical and clinical studies (25). Because of the similarities of iron and gallium with regards to charge and ionic radii, DFO has also been reported for the chelation of 67/68Ga (26,27). Thus, the chelator DFO provides a unique opportunity to prepare, study, and compare structurally similar 68Ga- and 89Zr-labeled exendin-4 derivatives starting from the same precursor. For a direct comparison of novel 68Ga- and 89Zr-labeled exendin-4 derivatives, all experiments described henceforth were run in parallel with the clinically evaluated reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 (2).
[Lys40-(AHX-DFO)NH2]exendin-4 and [Lys40-(AHX-DTPA)NH2]exendin-4 were radiolabeled with 68Ga/89Zr and 111In, respectively, by modified procedures described in the literature (Table 1). With 89Zr, the As of [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 up to 10 GBq/μmol could be achieved within 2 h at room temperature; however, to reach an As of greater than 20 GBq/μmol it was necessary to extend the reaction time to 14–16 h for quantitative complexation of the radiometal. 68Ga labeling was conducted with prepurified 68Ga-GaCl3 generator eluates. To our surprise, no satisfying radiochemical yields were observed within an appropriate time by incubation of [Lys40-(AHX-DFO)NH2]exendin-4 at room temperature or at elevated temperatures using a heating block. Finally, reaction in a microwave reactor for 1 min at 95°C yielded [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 in high radiochemical yield and purity at an As of 55 GBq/μmol. Reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 was prepared at an As of 52 GBq/μmol as previously described (2). The As achieved for the individual exendin-4 radiotracers described herein are in the range of those described in the literature (13–700 GBq/μmol) for preclinical (2,7,8,11,28) and clinical investigations (9,18,19,29).
High-performance liquid chromatography analysis of 89Zr-labeled exendin-4 derivatives showed no radiolysis on storage up to 16 h, which might have been an issue with regards to the long reaction times required to achieve an As sufficient for clinical applications. Serum stability studies also indicated no release of the radioactive metal from the complex during 4 h of incubation for [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 and 24 h for [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 (Table 2).
Several cell lines and animal models have been described in the literature for the preclinical evaluation of radiolabeled exendins. Murine INS-1, RIN-m5F (6), isolated primary β cells (2,8), and stably transfected CHL (10) cells have been used most frequently. Accordingly, animal models based on xenografts of these different cell lines have been described. As an alternative, a transgenic Rip1Tag2 mouse model, which develops endogenous insulinomas spontaneously, has also been described. However, the latter animal model is not generally accessible, and the handling of primary β cells derived therefrom is not trivial (30). Thus, we conducted our studies with commercial RIN-m5F cells. To enable a comparison of our results with published data for radiometallated exendin-4 derivatives, we used the clinically established radiotracer [Lys40-(AHX-DTPA-111In)NH2]exendin-4 as an internal reference compound (Table 2). GLP-1r–specific cell binding internalization was confirmed for all 3 compounds in vitro with a linear trend in uptake during 4 h of incubation (Fig. 2). For the reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 and [Lys40-(AHX-DFO-89Zr)NH2]exendin-4, the rate of cell internalization was similar (4.3%–5.5%) whereas [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 exhibited an increased uptake by a factor of 2 (9.4%). Dissociation constants, a measure of affinity of the radiolabeled peptides toward the GLP-1r, were in the low 2-digit nanomolar range for all compounds investigated (Fig. 3); however, a 2-fold-lower receptor affinity was found for [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 than for the other compounds (∼28 vs. 11 nM).
In vivo experiments confirmed the in vitro results. Biodistribution data (Figs. 4–6) of the 2 new investigative compounds and the reference compound showed a clean organ distribution pattern for all 3 radiotracers, with rapid clearance from the blood pool already 1 h after injection. Receptor-specific uptake of the radiotracers in the tumor xenografts and in GLP-1r–positive organs (lung, pancreas, intestine, stomach) was confirmed by blocking experiments with an excess of native exendin-4 (1-39), which significantly decreased the radiotracer uptake in these organs. Values of tumor uptake were in the same order of magnitude for [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 and the reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 at all time points whereas that of [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 was significantly higher at the 1 h time point (∼30% vs. 13–15 %ID/g). The different in vitro and in vivo characteristics of the radiolabeled exendin-4 derivatives were not surprising. In fact, this phenomenon has previously been described for other radiolabeled peptide conjugates, which are identical in all regards but differ in terms of the radiometal used (31–34). Thus, the effect of different metallic radionuclides on the biologic properties of a radiotracer should be investigated carefully in each case.
Unspecific uptake of radioactivity in the kidneys was high for all 3 radiolabeled exendin-4 derivatives, with values ranging from 153 to 217 %ID/g 1 h after injection, with the highest uptake for [Lys40-(AHX-DFO-89Zr)NH2]exendin-4. The retention of radioactivity in the kidneys remained high over the time of the experiments. Even after 48 h after injection, only about 30%–40% of the initially accumulated radioactivity was cleared from the organs. Uptake of radioactivity in the kidneys is commonly observed for radiolabeled peptides because of renal excretion; however, persistent retention is a particular feature of radiometallated exendin derivatives (35). Although this phenomenon is yet not fully understood in detail, it has been suggested that binding of the radiopeptide to the multiligand receptor megalin on glomerular filtration may lead to internalization and lysosomal entrapment of radioactive metabolites (36). Because high uptake of radioactivity in radiation-sensitive kidneys could be a limitation for clinical applications, different strategies are currently being pursued to overcome this issue (8,37).
CONCLUSION
We report herein the radiolabeling of a novel DFO-functionalized exendin-4 derivative, [Lys40-(AHX-DFO)NH2]exendin-4, with 68Ga and 89Zr and full preclinical evaluation in vitro and in vivo of the corresponding products [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 and [Lys40-(AHX-DFO-89Zr)NH2]exendin-4, respectively. A direct, side-by-side comparison with the clinically evaluated reference compound [Lys40-(AHX-DTPA-111In)NH2]exendin-4 showed that the novel radiotracers exhibit characteristics comparable or superior to GLP-1r–targeted imaging. [Lys40-(AHX-DFO-68Ga)NH2]exendin-4 is a promising candidate for clinical translation in terms of PET imaging at early time points after injection of the radiotracer. On the other hand, [Lys40-(AHX-DFO-89Zr)NH2]exendin-4 may represent a potential PET tracer for image acquisition at late time points provided that the kidney uptake of the radiotracer is not a limitation. Whether early- or late-time-point imaging of insulinomas by PET is advantageous for patient care is currently being investigated at our institution with related exendin-4 derivatives.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the L. & Th. La Roche Foundation (Switzerland). All animal experiments were conducted in compliance with the Swiss animal protection laws and with the ethical principles and guidelines for scientific animal trials established by the Swiss Academy of Medical Sciences and the Swiss Academy of Natural Sciences. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Dr. Danielle Vugts (VU University Medical Center, The Netherlands) for valuable advice on 89Zr-radiolabelling procedures and Dr. Ole Maas, Lisa McDougall, Caroline Ziegler, and Svenja Scheiwiler (University of Basel Hospital, Division of Nuclear Medicine) for technical assistance.
Footnotes
Published online Aug. 6, 2015.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication April 10, 2015.
- Accepted for publication July 27, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}