


Abstract
Interleukin-2 (IL2) binds with high affinity to the IL2 receptors overexpressed on activated T lymphocytes in various pathologic conditions. Radiolabeling of IL2 with a positron-emitting isotope could provide a tool for noninvasive PET of activated T cells in immune-mediated diseases. We report the labeling of IL2 with N-succinimidyl 4-18F-fluorobenzoate (18F-SFB) for the synthesis of N-(4-18F-fluorobenzoyl)interleukin-2 (18F-FB-IL2) and the in vitro and in vivo evaluation of this novel radiopharmaceutical for the detection of IL2 receptor–positive cells by PET. Methods: 18F-SFB was efficiently prepared using a 3-step radiochemical pathway. Purified 18F-SFB was incubated with IL2 in borate buffer (pH 8.5) and ethanol at 50°C for 10 min. 18F-FB-IL2 was purified by reversed-phase high-performance liquid chromatography. As in vitro quality controls, a biologic binding assay, sodium dodecyl sulfate polyacrylamide gel electrophoresis, mass spectrometry, and 3-chloroacetic acid precipitation stability tests were performed. Biodistribution studies were performed in BALB/c mice to evaluate the distribution of the tracer in healthy animals. PET experiments were performed in severe combined immunodeficiency disease mice inoculated with phytohemoagglutinin-activated human peripheral blood mononuclear cells (hPBMc). Whole-body images were acquired 30 min after injection of 5–15 MBq of 18F-FB-IL2. Results: 18F-SFB was produced with a 34%–38% radiochemical yield. The radiochemical purity after solid-phase extraction purification ranged from 93% to 96%. Conjugation of 18F-SFB to IL2 yielded 18F-FB-IL2 as the major product. The radiochemical yield of 18F-FB-IL2 after high-performance liquid chromatography purification was 25%–35% based on 18F-SFB. 18F-FB-IL2 was stable in plasma at 37°C and capable of stimulating T cells to an extent similar to native IL2. A biodistribution study showed highest tracer uptake in the kidneys and bladder due to rapid renal clearance of the tracer. Small-animal PET images showed binding of 18F-FB-IL2 to activated hPBMc proportional to the number of injected cells. Conclusion: We report the successful labeling of IL2 with 18F for PET of activated T lymphocytes. 18F-FB-IL2 is stable, is biologically active, and allows in vivo detection of activated T lymphocytes.
Interleukin 2 (IL2) is a small single-chain glycoprotein (15.5 kDa) of 133 amino acids that is synthesized and secreted by activated T lymphocytes (1). Originally it was known as T cell growth factor because of its growth-stimulating proprieties. IL2 binds with high affinity to the cell membrane IL2 receptor, which is mainly expressed on the cell surface of activated T lymphocytes. IL2 plays a regulatory role during the inflammation, because it induces lymphokine secretion and expression of class II major histocompatibility complex molecules, by T lymphocytes. Activated T lymphocytes are involved in chronic inflammation, which is associated with inflammatory degenerative diseases, viral infections, graft rejection, host response to tumors, and organ-specific autoimmune diseases. Thus, the lymphocytic infiltration of the target tissue or organ by activated T lymphocytes may represent the histologic hallmark of many chronic inflammatory diseases. The ability to detect the immune-cell infiltration in vivo in tissues could, therefore, be a useful diagnostic tool and may provide a rationale for immune intervention and to follow up the efficacy of antiinflammatory drugs.
In the past, our group and others have synthesized several radiopharmaceuticals for SPECT of activated T lymphocytes (2,3). In particular, scintigraphy using radiolabeled IL2 has already demonstrated its clinical utility and potential for diagnostic and prognostic purposes and for therapy follow-up. 123I-IL2 and 99mTc-IL2 were used mostly for the diagnosis of chronic autoimmune diseases, such as insulitis in type 1 diabetes (4), celiac disease (5), Hasimoto thyroiditis (6), Crohn disease (7), and vulnerable atherosclerotic plaques (8). Despite the good clinical results, routine application of this technique was limited because of complex labeling procedures and the low spatial resolution of SPECT.
PET, on the other hand, intrinsically is more sensitive, has better spatial resolution, and allows quantitative measurements. These PET properties are essential to visualize small inflammatory lesions during the early stages of immune-mediated diseases.
N-succinimidyl 4-18F-fluorobenzoate (18F-SFB) was shown to be a suitable synthon for radiolabeling of peptides and proteins (9,10). 18F-SFB is an activated ester that can be linked to lysine residues in the protein via an acylation reaction. In this article, we describe the synthesis of the PET radiopharmaceutical N-(4-18F-fluorobenzoyl)interleukin-2 (18F-FB-IL2) as a new probe for the noninvasive detection of activated T lymphocytes, its in vitro and in vivo quality controls, and its validation in an animal model of inflammation.
MATERIALS AND METHODS
All chemicals were obtained from commercial suppliers Sigma-Aldrich, Fluka, and Merck and used without further purification. Solid-phase extraction cartridges were obtained from Waters Chromatography Division, Millipore Corp.
Synthesis of Ethyl 4-[Trimethylammonium]Benzoate (SFB Precursor)
Ethyl-4-[dimethylamino]benzoate (0.64 g, 3.3 mmol) was dissolved in 12 mL of dry benzene under an argon atmosphere. Methyl trifluoromethanesulfonate (0.4 mL) was added, and the mixture was refluxed for 5.5 h. Then, the mixture was cooled to room temperature.
The solvent was removed under vacuum, and a white precipitate was formed.
Synthesis of 18F-SFB
18F-SFB was prepared according the methods described by Wester et al. (10).
Aqueous 18F-fluoride was produced by irradiation of 18O-water with a Scanditronix MC-17 cyclotron via the 18O(p,n)18F nuclear reaction. The 18F-fluoride solution was passed through a Sep-Pak Light Accell Plus QMA anion exchange cartridge (Waters) to recover the 18O-enriched water.
18F-fluoride was eluted from the QMA anion exchange cartridge with 1 mL of K2CO3 (1 mg/mL) and collected in a vial with 5 mg of Kryptofix 222 (Merck). To this solution, 1 mL of acetonitrile (MeCN) was added, and the solvents were evaporated at 130°C. The radioactive residue (18F-KF and Kryptofix complex) was carefully dried 3 times by addition and evaporation of anhydrous MeCN (0.5 mL at 130°C). After the addition of 10 mg of ethyl 4-[trimethylammonium]benzoate in dimethylformamide (0.25 mL), the mixture was heated at 90°C for 12 min. Then, 1 M HCl (0.5 mL) was added. The reaction mixture was heated at 100°C for 5 min. After being cooled, the reaction mixture was passed through a C18 Sep-Pak cartridge for solid-phase extraction. After the cartridge was washed with water, purified 18F-fluorobenzoic acid was eluted from the cartridge with 2 mL of MeCN into a vial containing 10 mg of Kryptofix 222 and 5 mg of K2CO3.
The eluate was dried under an argon stream at 130°C. Complete drying was ensured by the addition and evaporation of anhydrous MeCN (3 × 0.5 mL).
Then, the solution of O-(N-succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (20 mg) in anhydrous MeCN (0.5 mL) was added, and the mixture was heated at 90°C for 5 min. The mixture was cooled and diluted with 0.03 M HCl.
18F-SFB was diluted with 15 mL of water before being passed through an Oasis HLB cartridge (30 mg, 1 mL) for solid-phase extraction. The cartridge was washed with 5 mL of water and eluted with EtOH (0.5 mL) to obtain the final pure 18F-SFB solution.
The labeling procedure was fully automated using a Zymark robotic system.
Conjugation of 18F-SFB to IL2
Human recombinant IL2 (Proleukin; Novartis) was reconstituted at 2 mg/mL in nitrogen-purged water for injection and stored in 100-μL aliquots at −80°C for further use.
For the conjugation reaction, 100 μL of IL2 solution were incubated with the solution of 18F-SFB in 0.5 mL of ethanol in the presence of 0.5 mL of borate buffer, pH 8.3. To optimize reaction conditions, reaction time and temperature were varied. The product was purified by high-performance liquid chromatography (HPLC) using an Elite LaChrom Hitachi L-7100 pump system with an Econosphere C18-column (10 μm, 250 mm × 10 mm) equipped with both ultraviolet detection (Elite LaChrom VWR L-2400 UV detector set at 254 nm; Hitachi) and a Bicron radioactivity monitor. Gradient elution was performed using a mixture of 0.1% aqueous trifluoroacetic acid (solvent A) and 0.1% trifluoroacetic acid in EtOH (solvent B). The following gradient profile was used: 0–5 min 0% B, 5–10 min 40% B, 10–35 min 65% B, 35–45 min 100% B, and 45–47 min 0% B at a flow rate of 1 mL/min. Retention times were 26 min for 18F-FB-IL2, 28 min for 18F-fluorobenzoic acid, and 32 min for 18F-SFB. The product was collected from HPLC in approximately 55% ethanol in water. For the animal studies, the product was diluted 1:10 with saline, to ensure that the ethanol content was below 10%.
In Vitro Quality Controls
HPLC.
The purified product was analyzed by reversed-phase HPLC analysis as described above. Retention times were 24 min for 18F-FB-IL2, 26 min for 4-18F-fluorobenzoic acid, and 30 min for 18F-SFB.
Thin-Layer Chromatography (TLC).
TLC was performed with Merck F-254 silica gel strips. Strips were eluted with ethyl acetate:n-hexane (3:1). Free 18F-SFB migrated with the solvent front (Rf = 1) whereas 18F-FB-IL2 remained at the origin (Rf = 0).
The compounds on the TLC plates were detected by ultraviolet light (254 nm).
For radiolabeled compounds, the detection on the TLC plates was performed by phosphor storage imaging (multisensitive screens; Packard). These screens were exposed to the TLC strips for a few seconds and subsequently read out using a Cyclone phosphor storage imager (PerkinElmer) and analyzed with OptiQuant software (PerkinElmer).
Stability Tests.
To assess the stability of 18F-FB-IL2 over time, 3-chloroacetic acid precipitation was performed. 18F-FB-IL2 was dissolved in human plasma at 37°C. Samples were taken after 0, 15, 30, 60, 90, and 120 min. The proteins in these samples were precipitated with 20% ice-cold 3-chloroacetic acid and centrifuged for 5 min at 3,000g. Low-molecular-weight 18F-labeled fragments were in the supernatant, and the protein bound radioactivity was in the pellet. The supernatant and pellet were separated, and radioactivity in each fraction was counted in the automatic γ-counter (LKB Wallac).
Protein Determination.
Protein levels in each sample were determined using 2 methods: via a Micro BCA Protein Assay kit (Pierce) with bovine serum albumin as a calibration standard and via a photospectrometric analysis using a cuvette-free spectrophotometer NanoDrop (N-1000 Spectrophotometer; Thermo Fisher Scientific Inc.). Gel electrophoresis was performed by loading 10 μg of native and 1 μg of radiolabeled IL2 on a 12.5% polyacrylamide gel. Gels were run on a Mini Gel electrophoresis apparatus, with a standard molecular-weight mixture, and then stained with Coomassie Brilliant Blue (Thermo Fisher Scientific Inc.) as described by the manufacturer. The electrophoretic band of the radiolabeled sample was compared with native IL2 (12.5–15 kDa).
Mass Spectrometry (MS).
For identification of labeled IL2, matrix-assisted laser desorption and ionization time-of-flight MS (MALDI-TOF-MS) was used. Decayed 18F-FB-IL2 and 3,5-dimethoxy-4-hydroxycinnamic acid (sinapic acid) were dissolved in 70% acetonitrile in 0.1% trifluoroacetic acid. This matrix solution was mixed in equal volumes with the sample solution. One microliter of each mixture was spotted by the dried-droplet method on the MALDI plate (MTP; Applied Biosystem) and then dried for analysis (11). The MTP was introduced into the high-vacuum chamber of the mass spectrometer MALDI-TOF (Voyager DE Applied Biosystem). Spectra were analyzed by Data Explorer (version 4.0.0.0.; Applied Biosystems). MALDI-TOF operation conditions were set as follows: mode of operation was linear, polarity was positive, the acceleration voltage was 25,000 V, delayed extraction time was 1050 ns, and the acquisition mass range was 5,000–20,000 Da.
Degree of Conjugation.
To calculate the number of 4-fluorobenzoic residues conjugated to each IL2 molecule, the amount of radioactivity incorporated in the protein (in MBq) and the specific activity of 18F-SFB (in MBq/nmol) were measured. Both values were corrected for decay to the end of the synthesis. In addition, the concentration of the protein recovered after reaction was determined using NanoDrop (in mg/mL) and converted into the total amount of protein (in nmol). The substitution ratio is equal to the amount of 4-fluorobenzoyl residues divided by the amount of IL2 molecules, which can be calculated by the following formula: substitution ratio = (radioactivity [MBq]/specific activity [MBq/nmol])/(amount of protein [nmol]).
Cell Proliferation Assay.
Human peripheral blood mononuclear cells (hPBMc) were isolated from healthy volunteers by density-gradient centrifugation (Lymphoprep; Axis-Shield) as described by Bøyum (12). These cells were kept in RPMI 1640 supplemented with l-glutamine, 10% fetal calf serum, penicillin (100 IU/mL), and streptomycin (100 μg/mL) (Gibco). Isolated hPBMCs were incubated for 48 h with phytohemoagglutinin (PHA-P) (5 μg/mL; Sigma-Aldrich) at 37°C and 5% CO2 to activate them. Subsequently, fluorescence-activated cell sorting was performed after the cells were stained with an anti-CD25 antibody (e-Bioscience) to verify the percentage of cell activation.
Because IL2 stimulates proliferation of PHA-P–activated T lymphocytes, the biologic activity of radiolabeled IL2 and native IL2 can be measured by 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay. Activated hPBMc (50–70 × 104 per well) was plated in a 96-well plate. Cells were incubated for 24 h with different concentrations (0.1, 1, 10, 100, and 1,000 units/mL) of either native IL2 or decayed 18F-FB-IL2. Then, 0.01 mg of MTT was added to each well, and cells were incubated for 3–4 h at 37°C and 5% CO2. MTT is reduced by viable cells in an insoluble, colored (dark purple) formazan product. The cells were solubilized with an organic solvent (isopropanol), and absorbance was read by an enzyme-linked immunosorbent assay reader at 490 nm. The cellular proliferation of decayed 18F-FB-IL2–treated cells was compared with the proliferation of untreated cells and of cells treated with native IL2. Dose–response curves were calculated by the following equation: % cell proliferation (λ = 490 nm) = (absorbance of cells treated with radiopharmaceutical/absorbance of untreated cells) × 100. All assays were run in triplicate.
Animal Experiments
Targeting and Blocking Studies.
Animal experiments were performed according to Dutch Regulations for Animal Welfare. The protocol was approved by the Ethical Committee of the University of Groningen.
Biodistribution studies of 18F-FB-IL2 were performed in normal BALB/c mice (control, n = 12).
For targeting experiments, Fox-chase severe combined immunodeficiency disease (SCID) beige mutant mice were used (Harlan). Eleven mice were divided into 4 groups and subcutaneously implanted with an increasing number of activated T lymphocytes in 150 μL of Matrigel (Becton Dickinson) in the right shoulder (5 × 106, 10 × 106, 13 × 106, and 20 × 106 cells, respectively) 60–90 min before tracer administration. In the left shoulder, the same amount of Matrigel was inoculated as negative control.
For blocking experiment, 4 mice were inoculated with 10 × 106 phytohemoagglutinin-activated T lymphocytes in 150 μL of Matrigel in the right shoulder. Animals were pretreated with a 100-fold excess of IL2 in 0.1 mL of water 30 min before intravenous injection of 18F-FB-IL2.
Mice were anesthetized with 2% isoflurane (Pharmachemie BV) in medical air and intravenously injected with 9.86 ± 4.22 MBq in 45% EtOH in water. After 15, 60, or 90 min, animals were terminated. Major organs were dissected and weighed. Radioactivity in these organs was measured with an automatic γ-counter (LKB; Wallac). Radiopharmaceutical uptake was expressed as a percentage of injected dose per gram (%ID/g) using the following formula: ([activity in the target organ/grams of tissue]/injected dose) × 100%.
PET and Data Reconstruction.
For PET studies (Focus 220; Siemens Medical Solution USA, Inc.), 2 mice were scanned simultaneously in each scan session, anesthetized with 2% isoflurane in medical air, and injected trough the penile vein with approximately 1.5 μg of radiolabeled IL2. Images were acquired 30 min after injection, under anesthesia. A static PET scan was acquired for 30 min, followed by a transmission scan using a 57Co point source.
A list-mode protocol was used, and the dataset was fully corrected for random coincidences, scatter, and attenuation. Three-dimensional regions of interest were manually drawn around the site of cell inoculation and the contralateral site. %ID/g values were calculated.
Histology
To confirm the presence of lymphocytic infiltration, we histologically evaluated 5-μm hematoxylin- and eosin-stained skin sections of SCID mice. The skin was dissected, immediately frozen in liquid nitrogen, and stored at −80°C.
RESULTS
Synthesis of 18F-SFB
For radiolabeling of 18F-SFB, the most critical step was the conversion of 4-18F-fluorobenzoic acid into the corresponding activated ester, because traces of moisture severely affected the yield of the reaction. Best results were obtained when 4-18F-fluorobenzoic acid was purified by solid-phase extraction using a C18 cartridge and eluted in a vial with 5 mg of K2CO3 and 15 mg of Kryptofix 222. Residual water was subsequently removed by azeotropic distillation with acetonitrile. The dried potassium 4-18F-fluorobenzoate-kryptofix complex then readily reacted with O-(N-succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate to give the desired product. The automated system produced 18F-SFB with a good radiochemical yield (34%–38%). The radiochemical purity after solid-phase extraction ranged from 93% to 96%, as determined by HPLC.
Conjugation of 18F-SFB with IL2
The best efficiency for conjugation reaction of 18F-SFB with IL2 was obtained using 100 μL of IL2 (2 mg/mL) in borate buffer (pH 8.5):ethanol (1:1).
For optimization of the conjugation condition, the labeling temperature and the incubation time were varied. The conjugation reaction was tested at 20°C, 37°C, 50°C, and 60°C. The highest labeling yield was obtained when the conjugation reaction was performed at 50°C. If the temperature of the conjugation reaction was further increased to 60°C, the yield decreased, probably because of the instability of the protein at high temperatures. In addition, 18F-SFB is more rapidly hydrolyzed to 18F-fluorobenzoic acid (degradation product) at elevated temperatures.
The influence of the incubation time on the radiochemical yield of the conjugation process was also tested. The conversion of the conjugation of IL2 with 18F-SFB was monitored by silica gel TLC analysis using ethyl acetate:hexane (3:1) as the mobile phase. The reaction was followed from 0 to 30 min, and the reaction mixture samples were taken every 5 min. The conjugation reaction is fast, and the reaction is complete within 10 min. Longer reaction times did not significantly increase the labeling yield. In fact, longer reaction times will likely result in increased protein denaturation. Thus, optimal conditions for the conjugation of 18F-SFB to IL2 were a reaction temperature of 50°C and an incubation time of 10 min. Under these conditions, the radiochemical yield of 18F-FB-IL2 after HPLC purification was 25%–35% (on the basis of 18F-SFB). Practical yields were over 400–500 MBq, because only short cyclone irradiation times were used (<15 min).
The specific activity of the product was about 117 ± 6 GBq/μmol, which was sufficient for animal experiments.
Quality control by reversed-phase HPLC revealed that 18F-FB-IL2 is eluted slightly faster than native IL2, as a result of the introduction of 18F-fluorobenzoate residues. Radiochemical purity of the final product was greater than 95%.
Protein Determination and Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
The Micro BCA Protein Assay is one of the most widely used methods to determine the protein concentration, with a detection level of 5 μg/mL. Therefore, NanoDrop photospectrometry was also used. The average concentration of purified 18F-FB-IL2 after the elution by semipreparative HPLC was 0.047 ± 0.009 mg/mL using these methods. SDS-PAGE showed a single band for 18F-FB-IL2, running at the same height as native IL2 (15 kDa), thus indicating the absence of covalent aggregates or fragments of 18F-FB-IL2 and proving the integrity of the protein during the labeling procedure.
MS
MALDI-TOF analysis, because it is more accurate than SDS-PAGE in determining the molecular mass of the protein, was also used to confirm the identity of 18F-FB-IL2. The mass spectra demonstrated that the mass of fluorinated IL2 was in the same range as native IL2, which has a mass of 15,636 Da, as determined by MALDI-TOF (data not shown). MALDI-TOF data suggest that after labeling, each labeled IL2 molecule was coupled with 1 molecule of SFB (Fig. 1).

MALDI-TOF profile of 18F-FB-IL2.
Degree of Conjugation
Because the accuracy of MALDI-TOF is insufficient to determine the number of FB groups per protein molecule, we also calculated the level of the conjugation on the basis of radioactivity incorporated in the protein, the specific activity of 18F-SFB, and the amount of the protein recovered after reaction. With this calculation, we determined that on average 1.5–1.8 4-fluorobenzoic residues were bound to each molecule of labeled IL2.
Cell Proliferation Experiments
PHA-P could strongly stimulate the activation of hPBMC, resulting in a maximal expression of CD25 on the cell surface of lymphocytes after 48 h (60% of positive cells by fluorescence-activated cell sorting analysis with anti-CD25 staining).
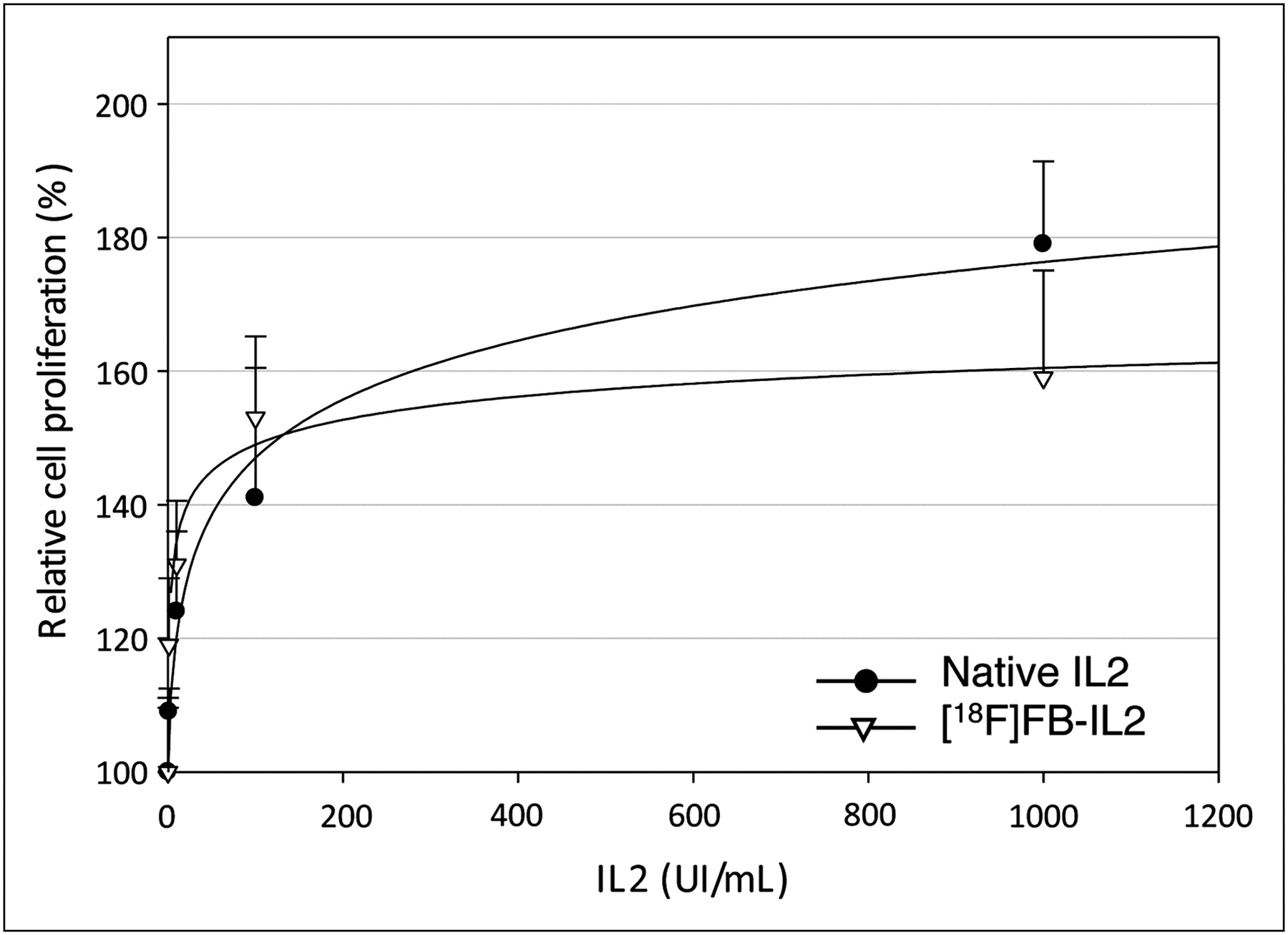
As shown in Figure 2, stimulation of cell proliferation by decayed 18F-FB-IL2 was not significantly different from stimulation by native IL2 (P = 0.612, 0.389, 0.575, 0.230, and 0.339 for 0.1, 1, 10, 100, and 1,000 IU/mL, respectively).

MTT cell proliferation assay of phytohemoagglutinin-activated hPBMc stimulated by native IL2 or 18F-FB-IL2. Results are expressed as percentage of increase in cellular proliferation as compared with untreated cells (mean ± SD of 3 independent experiments, each performed in triplicate). There is no significant difference between labeled and unlabeled IL2 at any tested concentration.
Stability Test
As shown in Figure 3, 95.1% ± 2.6% and 95.1% ± 3.0% of the labeled product is still intact after 1 and 2 h of incubation in phosphate-buffered saline. In human plasma, no degradation was observed. Thus, the stability of the tracer appears sufficient for imaging purposes.
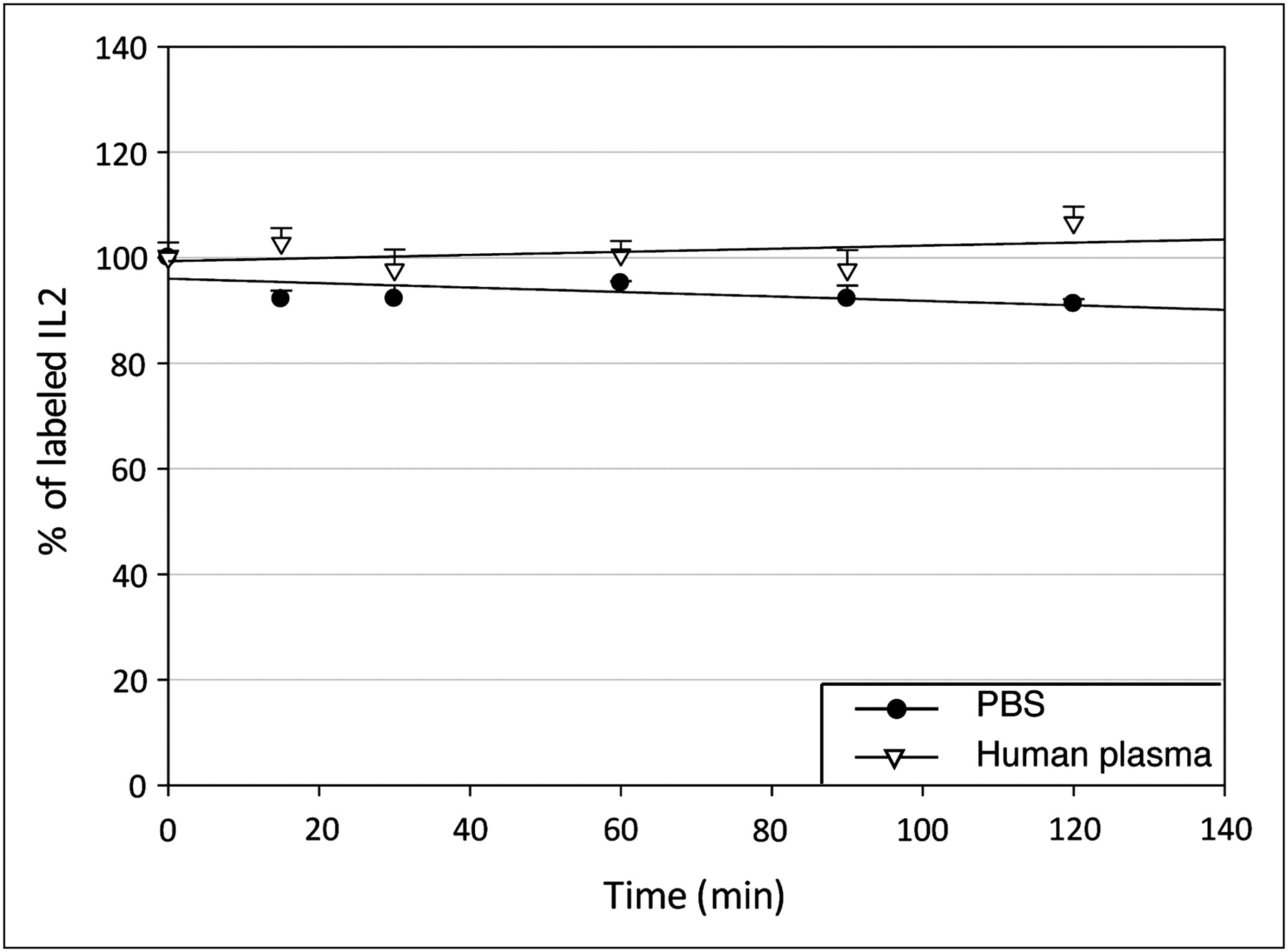
3-chloroacetic acid precipitation assay as percentage of labeled IL2 at different times in phosphate-buffered saline and human plasma. PBS = phosphate-buffered saline.
Ex Vivo Biodistribution
Because human IL2 binds also to murine lymphocytes, biodistribution studies were performed in healthy BALB/c mice with a normal immune system, possibly predicting the behavior of the radiopharmaceutical in humans. Results of biodistribution studies are presented in Table 1. We observed a low uptake in all organs (SUV < 1), with the kidneys being the organs with highest uptake. There was negligible uptake in the spleen, stomach, and liver. Significant amounts of radioactivity were excreted in the bladder, indicating that this radiopharmaceutical is mainly cleared via the renal pathway, a characteristic that is similar to the native IL2 (1,13). 18F-FB-IL2 showed low bone uptake, which did not significantly increase over time, indicating that defluorination in vivo is negligible.
Biodistribution of Radiotracer 18F-FB-IL2 in 12 BALB/c Mice at Different Time Points
PET
Animal studies were performed in immune-incompetent SCID mice rather than wild-type animals, to avoid any host immune response to the exogenous human T lymphocytes. As soon as 30 min after intravenous injection of 18F-FB-IL2, activated T lymphocytes could be clearly visualized in the right shoulder of all animals. The kidneys were the organs with highest tracer uptake.
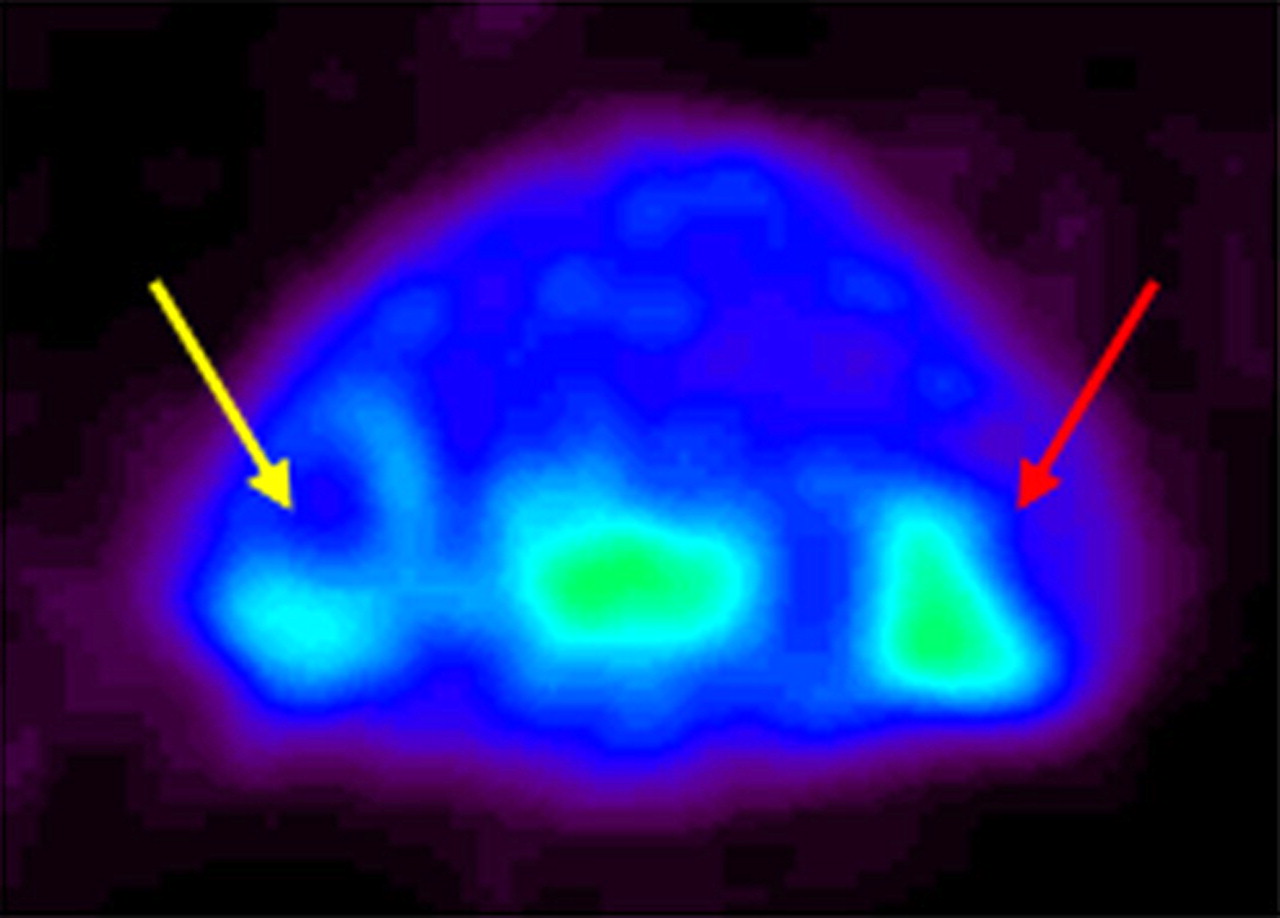
Unexpectedly, the small-animal PET images also showed radiopharmaceutical uptake at the border of the Matrigel injected in the left shoulder (control shoulder) of all mice (Fig. 4).

Small-animal PET images of SCID mice inoculated with phytohemoagglutinin-activated T lymphocytes. Small-animal PET image (transaxial section of mouse shoulders) shows 18F-FB-IL2 uptake in right shoulder (red arrow) and, to lesser extent, in contralateral, control shoulder (yellow arrow) due to migration of lymphocytes from injection site to contralateral site.
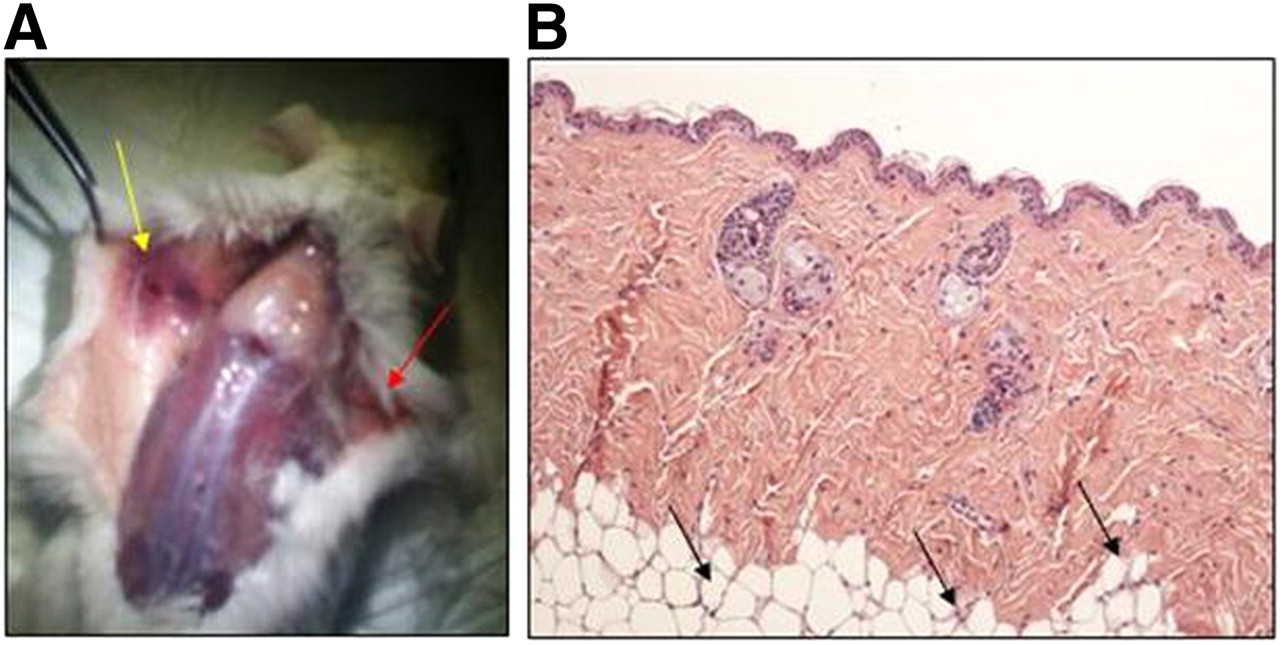
Visual inspection of the left shoulder of the terminated animals showed a strong inflammatory reaction at the site of Matrigel injection in the control shoulder. The typical signs of inflammation were observed, such as redness of the inflamed tissue characterized by vasodilatation and tissue damage (Fig. 5A). The inflammation was confirmed by the histologic examination of the skin, and lymphocytic infiltration was observed as in the right shoulder as well (Fig. 5B). Because SCID mice do not have endogenous mouse lymphocytes, this finding indicates that some lymphocytes have migrated from the right to the left shoulder, probably because of the Matrigel-induced inflammation in the control shoulder. These infiltrating lymphocytes are most likely responsible for the radiopharmaceutical uptake observed in the left shoulder.

(A) Visual inspection of derma of inoculated mouse, of which PET image is presented in Figure 4. Red arrow highlights site at which lymphocytes were inoculated with Matrigel in right shoulder, and yellow arrow indicates inflammatory reaction after inoculation of Matrigel only in left shoulder. (B) Hematoxylin and eosin staining of skin from left shoulder showing presence of lymphocytes at border of Matrigel (black arrows).
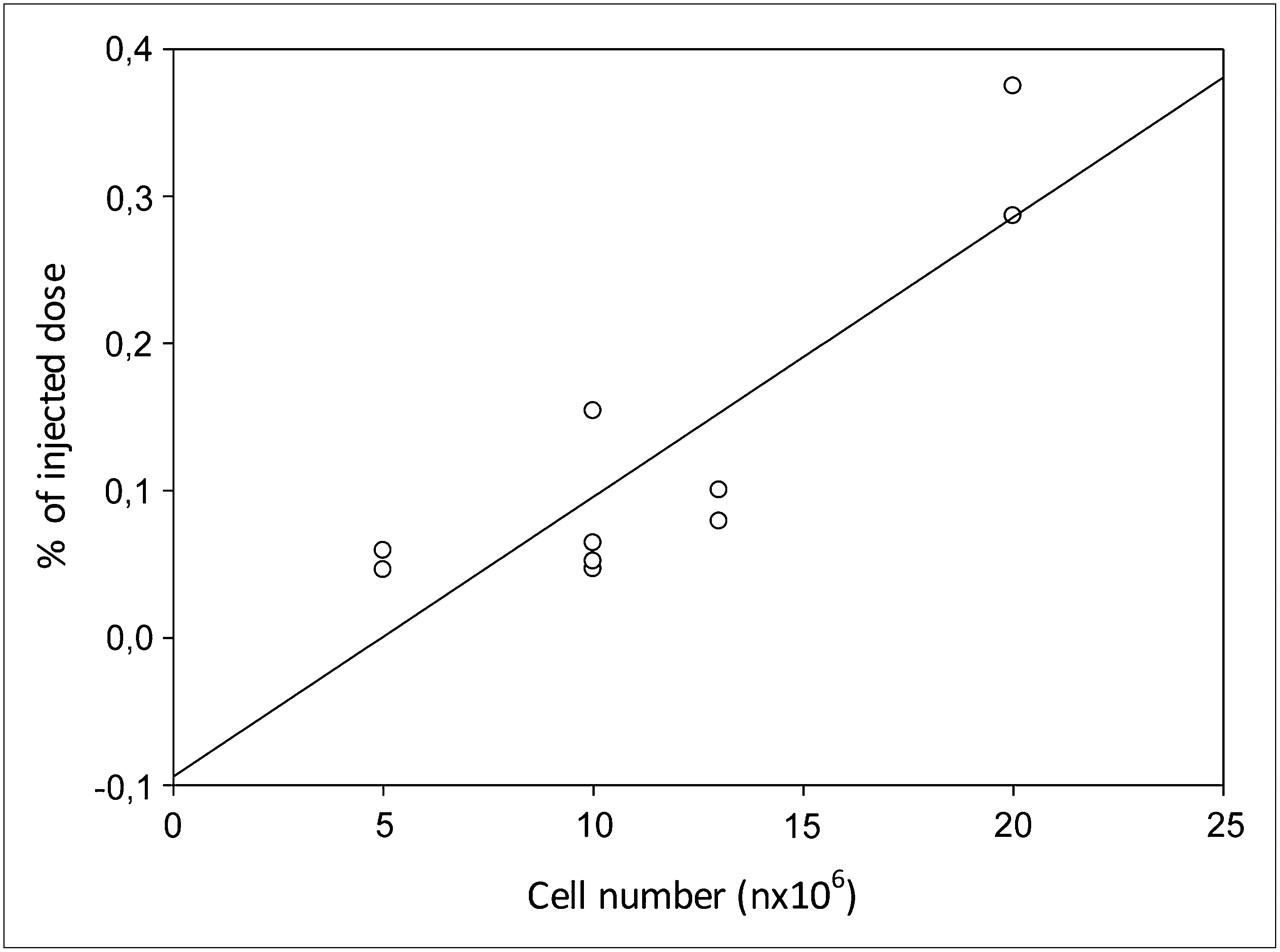
We found no correlation between 18F-FB-IL2 uptake in the right shoulder and the number of inoculated activated T lymphocytes (R2 = 0.099, P = 0.375), likely because of the variable cell migration to the contralateral shoulder. Indeed, when summing the uptake of both shoulders (the inoculation site and the migration site), we found a significant correlation (R2 = 0.768, P = 0.0012) (Fig. 6). 18F-FB-IL2 uptake, as %ID, was 0.052 ± 0.009, 0.084 ± 0.046, 0.093 ± 0.016, and 0.328 ± 0.062 for 5 × 106, 10 × 106, 13 × 106, and 20 × 106 cells, respectively.
The %ID in the inner core of the Matrigel in the left shoulder was negligible, with a value of 0.0016 ± 0.0014, and because no lymphocytes were found there at histology we considered it as a control tissue.
In vivo blocking studies in mice injected with 10 × 106 activated T lymphocytes are shown in Figure 7. The uptake of 18F-FB-IL2 decreased after animal treatment, with an excess of native IL2. Quantitative analysis showed a 76% ± 20% decrease in uptake in mice pretreated versus untreated ones (%ID/g, 0.0084 ± 0.046 vs. 0.024 ± 0.012, P = 0.048).

Correlation between number of inoculated lymphocytes and 18F-FB-IL2 uptake in both shoulders (R2 = 0.768, P = 0.0012).

In vivo binding of 18F-FB-IL2 to 10 × 106 phytohemoagglutinin-activated T lymphocytes inoculated in shoulder of SCID mice (n = 4) (left bar) and in mice (n = 4) pretreated with 100-fold excess of unlabeled IL2 (right bar). Data are mean ± SD of standardized uptake value calculated 30 min after radiopharmaceutical injection.
DISCUSSION
In vivo detection of lymphocytes using nuclear imaging techniques is an important diagnostic tool developed more than 30 y ago with the introduction of labeled autologous cells (14,15). Later, and because of the toxicity of direct radiolabeling on lymphocytes, several peptides and proteins were radiolabeled with SPECT isotopes, including IL1, FAS ligand peptides, and, more recently, antibodies against CD25, CD3, CD20, HLA-DR, and others (3,16–20). Until now, however, none of these peptides and antibodies had been used in daily clinical practice.
After the introduction of PET and the possibility of using this sensitive technique for the localization of small inflammatory lesions, only 18F-FDG was tested for imaging lymphocytes because of their high metabolic rate during the inflammation process. Despite its poor specificity, good results have been obtained in vasculitis (21,22), sarcoidosis (23,24), rheumatoid arthritis (25–27), inflammatory bowel diseases (28–31), and other chronic inflammatory conditions (32–37).
A specific radiopharmaceutical, such as radiolabeled IL2, that directly shows the presence of tissue-infiltrating activated T lymphocytes in diseases may allow the low specificity of 18F-FDG to be overcome. Indeed, studies performed with either 99mTc-labeled or 123I-labeled IL2 in over 800 patients clearly showed the clinical potential and utility of this radiopharmaceutical without any adverse side effect (5,7,8,38).
Therefore, the aim of our study was to develop a new PET tracer, 18F-FB-IL2, for imaging of activated T lymphocytes.
18F-fluoroacylation via 18F-SFB was the method chosen for labeling IL2. This agent is well described in the literature and is widely used for labeling proteins and peptides. The binding between the Lys residues of the protein–peptide and the labeled precursor (18F-SFB) is stable, compared with other 18F-fluorination agents, such as 4-nitrophenyl 18F-fluoropropionate. The protocol for the radiosynthesis of 18F-SFB is totally automated in a robotic system present in our laboratory, providing us reliable results in terms of radiochemical yield and purity. 18F-FB-IL2 was obtained in a good radiochemical yield (25%–30%, on the basis of 18F-SFB), with high radiochemical purity (>95%) and high specific activity (120 GBq/μmol).
The introduction of the prosthetic group slightly modifies the lipophilicity of 18F-FB-IL2. This difference is observed by analytic HPLC as a modification of the elution profile of the labeled protein. The retention time shifts from 26.5 to 24 min. This small modification, however, did not change the biologic properties of IL2 as proved by a biologic analysis (MTT assay). The radiopharmaceutical proved stable in vitro in human plasma and in vivo in animals.
In BALB/c mice 18F-FB-IL2 was rapidly washed out from all organs, except for the kidneys, suggesting that the major excretion route occurs by renal clearance, as is the case for native IL2.
Compared with 99mTc-labeled IL2, 18F-FB-IL2 showed a higher uptake in the kidneys and a lower uptake in the liver. This difference could be due to the additional positive charge in the technetium complex or some release of technetium from the complex. Furthermore, scavenger receptors on liver endothelial cells and Kupffer cells can filter the positively charged 99mTc-labeled protein from the blood pool. Because the 18F label in 18F-FB-IL2 is covalently bound to the protein, release of the radiolabel from the stable prosthetic group is highly unlikely. In addition, the 18F-labeled protein is neutral and therefore less prone to filtration from the blood by the liver scavenger receptors.
As an experimental model, immune-depressed SCID mice inoculated with different amounts of phytohemoagglutinin-activated T lymphocytes were used. We found a significant 76% decrease of the tracer uptake after injection of the cold protein, proving the specificity of the binding between labeled IL2 and IL2 receptors expressed on activated T lymphocytes.
When comparing the number of inoculated T lymphocytes with the accumulation of radioactivity at the injection site, only a poor correlation was observed. However, we found a significant linear correlation when radiopharmaceutical uptake at the injection site and at the control xenograft were combined (R2 = 0.768). Further investigation of the skin of the control shoulder provided a logical explanation of these results. We observed unexpected migration of part of the inoculated T lymphocytes to the contralateral shoulder, where only Matrigel was inoculated under the skin as control. The strong inflammatory reaction in the control shoulder cannot be explained by an immune response of the host immune system, because SCID mice lack an efficient immune system. A likely hypothesis for the migration of T lymphocytes from the inoculation site to the contralateral site, as we found in the literature, is that the mechanical trauma induced by the needle puncture and by the injection of Matrigel can induce attraction and migration of active immune cells (39). Endothelial cells and keratinocytes can also produce proinflammatory cytokines and chemokines that further stimulate chemotaxis of lymphocytes (39). This process is similar to atopic dermatitis. The 18F-FB-IL2 uptake found by small-animal PET in the control shoulder was not as homogeneous as the uptake in the right shoulder but was mainly detected on the periphery of the jellified Matrigel. Indeed, at histology, the core of the Matrigel did not show lymphocytes and did not show any 18F-FB-IL2 uptake.
CONCLUSION
The results of this study show that IL2 can be efficiently and reproducibly labeled with 18F. 18F-FB-IL2 is able to specifically target human activated T lymphocytes and to detect cell migration in vivo. This PET radiopharmaceutical appears a promising new probe for detecting activated T lymphocytes in pathologic conditions, such as autoimmune diseases and graft rejection.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Gaurav Malviya and Filippo Galli for help in performing some experiments. A portion of this study has been supported with a JDRF innovation grant (5-2006-943). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Apr. 12, 2012.
- © 2012 by the Society of Nuclear Medicine, Inc.
REFERENCES
- Received for publication April 4, 2011.
- Accepted for publication October 17, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Imaging of Activated T Cells
- Imaging Calreticulin for Early Detection of Immunogenic Cell Death During Anticancer Treatment
- Molecular Imaging of Chimeric Antigen Receptor T Cells by ICOS-ImmunoPET
- Development and Evaluation of Interleukin-2-Derived Radiotracers for PET Imaging of T Cells in Mice
- Combination treatment with hypofractionated radiotherapy plus IL-2/anti-IL-2 complexes and its theranostic evaluation
- A PET Imaging Strategy to Visualize Activated T Cells in Acute Graft-versus-Host Disease Elicited by Allogenic Hematopoietic Cell Transplant
- Synthesis and Characterization of 18F-Interleukin-8 Using a Cell-Free Translation System and 4-18F-Fluoro-L-Proline
- PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments
- Detection of Insulitis by Pancreatic Scintigraphy With 99mTc-Labeled IL-2 and MRI in Patients With LADA (Action LADA 10)