


Abstract
We analyzed the pattern of 11C-acetate and 18F-FDG uptake on PET/CT in patients with hepatocellular carcinoma (HCC). We also assessed the expression of important regulatory enzymes related to glycolysis and lipid synthesis in relation to 18F-FDG and 11C-acetate uptake in human HCC cell lines. The significance of 11C-acetate uptake regulation was further evaluated with regard to cell viability. Methods: 18F-FDG and 11C-acetate uptake patterns in HCC in 11 patients and in 5 HCC cell lines were assessed. We evaluated the gene expression of metabolic enzymes related to glycolysis and lipid synthesis in a cell line with the highest 18F-FDG uptake and another cell line with the highest 11C-acetate uptake. They included hexokinase II, adenosine triphosphate citrate lyase, acetyl coenzyme A (CoA) synthetase 1 (ACSS1), acetyl CoA synthetase 2 (ACSS2), acetyl CoA carboxylase, and fatty acid synthase. In a cell line with high 11C-acetate uptake, the enzymatic activities of ACSS1 and ACSS2 were blocked using respective small, interfering RNAs (siRNAs), and the impact on 11C-acetate uptake and cell viability was assessed. Results: In all 11 patients and 4 of the 5 cell lines, the uptake patterns of the 2 radiotracers were complementary. ACSS1 and ACSS2 were highly expressed in a cell line with low 18F-FDG uptake and high 11C-acetate uptake, whereas only ACSS2 was expressed in a cell line with high 18F-FDG uptake and low 11C-acetate uptake. Fatty acid synthase expression was seen in cells with high 18F-FDG or 11C-acetate uptake. These findings indicate the possibility that both glucose and acetate can be a compensatory carbon source for lipid synthesis in cancer. Transient transfection with ACSS1 or ACSS2 siRNA in cells with high 11C-acetate uptake decreased 11C-acetate uptake and cell viability. Conclusion: The patterns of 18F-FDG and 11C-acetate uptake seemed to complement each other in both human HCC and HCC cell lines. Fatty acid synthase expression was seen in cells with high 18F-FDG or 11C-acetate uptake, suggesting glucose- or acetate-dependent lipid synthesis. Acetyl CoA synthetase appears to be important in 11C-acetate uptake and acetate-dependent lipid synthesis for the growth of cancer cells with a low-glycolysis phenotype. Inhibition of acetyl CoA synthetase in these cells may be promising for anticancer treatment.
A glycolytic phenotype even in the presence of available oxygen, the so-called Warburg effect, is considered to be a result of the low respiration rate in cancer cells (1). 18F-FDG, a glucose analog, is the most commonly used radiotracer in combination with PET for the detection of various malignancies based on this phenomenon. Previous studies have found, however, that some tumors do not show increased 18F-FDG uptake. For example, primary hepatocellular carcinomas (HCCs) exhibit a wide spectrum of 18F-FDG uptake, considerably reducing its sensitivity for tumor detection. The false-negative rate of 18F-FDG PET in HCCs is high—up to about 40%−50% (2). Other than HCCs, many solid tumors such as prostate cancers, renal cell carcinomas, low-grade sarcomas, low-grade lymphomas, or some tumors with well-differentiated histology also display low glucose metabolism (3–5). Consequently, development of new radiotracers is of the utmost clinical importance to increase the accuracy of diagnosis in tumors that do not show increased 18F-FDG uptake on PET.
11C-acetate was originally used in imaging cardiac oxidative metabolism. In the myocardium, 11C-acetate is rapidly converted into acetyl coenzyme A (CoA) after cellular uptake and dominantly enters the tricarboxylic acid cycle for CO2 generation (6). Shreve et al. reported a reduced rate of clearance of 11C-acetate in renal cell carcinoma, allowing differentiation of cancerous cells from normal and diseased nonneoplastic renal tissue (7). Since then, 11C-acetate has been proposed as an alternative radiotracer for detecting tumors not seen on 18F-FDG studies (2,8). The metabolic fate of 11C-acetate is not well understood in tumor cells. In a study using 14C-acetate, tumor cells incorporated 14C activity into the lipid-soluble fraction rather than the water-soluble or CO2 fraction. Considering the positive correlation between accumulation of the lipid-soluble fraction of 14C and growth activity, the increased 14C-acetate uptake in tumor cells was attributed mainly to the increased lipid synthesis caused by the high level of proliferative activity in the tumors (9).
The extensive involvement of lipid synthesis in 11C-acetate uptake has also been confirmed in prostate tumor cells in vitro and in tumor models using small-animal PET (10). Treatment with inhibitors for fatty acid synthesis blocks 11C-acetate uptake in prostate cancer cell lines. In addition, the expression level of fatty acid synthase (FAS) for lipid synthesis correlates well with 11C-acetate uptake in tumor-bearing mouse models. However, we have observed the expression of FAS even in cancer cells with high 18F-FDG uptake and low 11C-acetate uptake. Additional regulatory enzymes appear to affect 11C-acetate uptake in cancer cells. In this study, we analyzed the pattern of 11C-acetate and 18F-FDG uptake on PET/CT in patients with HCC. We also assessed the expression of important regulatory enzymes related to glycolysis and lipid synthesis in relation to 18F-FDG and 11C-acetate uptake in human HCC cell lines. The significance of 11C-acetate uptake regulation was further evaluated with regard to cell viability.
MATERIALS AND METHODS
Patients, Imaging Procedures, and Image Interpretation
Eleven patients (10 men and 1 woman; mean age, 54.2 y; range, 31–68 y) with newly diagnosed HCC were enrolled from December 2007 to October 2008. Our institutional review board approved the protocol for this prospective study, and written informed consent was obtained from all patients. The patients were instructed to fast for more than 6 h, and 370–555 MBq of 18F-FDG were injected intravenously. One hour later, scans were acquired on an integrated PET/CT system (Biograph 40; Siemens Medical Solutions) at 3 min per bed position. Transmission scans from low-dose CT were used for attenuation correction. The attenuation-corrected PET data were iteratively reconstructed. On a separate day, a PET/CT scan was also obtained on the same scanner 20 min after intravenous injection of 370–555 MBq of 11C-acetate at 2 min per bed position. Attenuation correction and image reconstruction were done using the same protocol as for the 18F-FDG data. Both PET/CT scans were visually analyzed by 2 experienced nuclear medicine specialists. A consensus was reached for each scan. HCCs with radiotracer uptake higher than in the surrounding liver were classified as high uptake, and those showing uptake similar to that in the liver were classified as low uptake. Mixed uptake was defined as areas of high and low uptake in the same tumor. For semiquantitative analysis, a region of interest was drawn over the tumors and the peak standardized uptake value was recorded.
Cell Lines and Culture
Human HCC cell lines from the Korean Cell Line Bank (SNU 387, 398, 449, and 475) and American Type Culture Collection (Hep3B) were used in this study. All cell lines from the Korean Bank were derived from HCC patients with positive hepatitis B virus infection. The major Edmonson–Steiner grades for each cell line were as follows: grades II–III for SNU 449, grades III–IV for SNU 387 and 475, and SNU 398 for anaplastic HCC. The cells were maintained in RPMI-1640 (BioWhittaker) or Dulbecco modified Eagle medium containing penicillin-streptomycin (100 U/mL), gentamicin sulfate (50 mg/mL), and 10% fetal bovine serum in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Plate Imaging of 18F-FDG and 11C-Acetate and Cell Growth Curve
All HCC cell lines were plated on 12-well plates with 3 × 105 cells per well for 24 h. Six hours before the uptake experiment, the medium was changed to a glucose-free medium (RPMI-1640). Approximately 0.037 MBq (1 μCi) or 0.111 MBq (3 μCi) of 18F-FDG and 0.37 MBq (10 μCi) or 1.11 MBq (30 μCi) of 11C-acetate were added to the cells, followed by incubation for 20 min. The cells were washed with phosphate-buffered saline 3 times and then imaged for 5 min on a PET scanner (Allegro; Philips-ADAC Medical Systems). The images were reconstructed using an iterative reconstruction algorithm (low-action maximal likelihood). After imaging, the cells were washed twice with phosphate-buffered saline, and 20 μL of reporter lysis buffer (Promega) were added to each well. The lysed cells were then harvested to measure the amount of radioactivity by γ-counter (Wallac 148 Wizard 3; Perkin Elmer). The radioactivity measured was divided by the radioactivity added and normalized to protein content obtained by bicinchoninic acid assay (Pierce Biotechnology) with bovine serum albumin as the protein standard. Cell growth rates were determined by seeding 1 × 105 cells in 6-well plates in an appropriate medium for 24–96 h. The cells were trypsinized after 24, 48, 72, and 96 h and counted with trypan blue stain, 0.4% (GIBCO). All experiments were performed in triplicate.
Reverse-Transcriptase Polymerase Chain Reaction of Enzymes for Glycolysis and Lipid Synthesis
Hexokinase II, as a key enzyme for glycolysis, regulates the first step of the glycolytic pathway and is known to maintain the malignant phenotype of cancers. Adenosine triphosphate citrate lyase (ACL) is an enzyme of glucose-dependent lipid synthesis. Acetyl CoA synthetase 1 (ACSS1, mitochondrial) and 2 (ACSS2, cytosolic) were selected as enzymes catalyzing the conversion of acetate to acetyl CoA. Acetyl CoA carboxylase and FAS were chosen as enzymes of lipid synthesis. Total RNA was prepared from a cell line with high 18F-FDG uptake (SNU 387) and a cell line with high 11C-acetate uptake (SNU 449) using the RNeasy kit (Qiagen) following the manufacturer's instructions. The sequences of primers used for analysis were as follows: For hexokinase II, the forward sequence was 5′-TCTATGCCATCCCTGAGGAC-3′ and the reverse sequence was 5′-TCTCTGCCTTCCACTCCACT-3′; for ACSS1, forward was 5′-ACCAAGATCGCCAAATATGC-3′ and reverse was 5′-TGCTTGTCCTTGCACTTCTG-3′; for ACSS2, forward was 5′-GGATTCCAGCTGCAGTCTTC-3′ and reverse was 5′-CATGCCACCACAAGTCAATC-3′; for acetyl CoA carboxylase, forward was 5′-CACGCTCAAGTCACCAAGAA-3′ and reverse was 5′-CTGCAGGTTCTCAATGCAAA-3′; and for FAS, forward was 5′-GAAGCTCGTGTTGACTTCTC-3′ and reverse was 5′-AGAAGACCACAAAGTAGTCC-3′. Complementary DNA was synthesized from 1 μg of RNA and Oligo dT in a reverse-transcriptase premix (Bioneer). Each polymerase chain reaction mixture contained 10 pmol of forward and reverse primers, deoxyribonucleoside triphosphate mixture, 2× GC buffer I (LA Taq; TaKaRa), and complementary DNA. Amplification was performed for 30 cycles for hexokinase II, ACL, ACSS1, ACSS2, acetyl CoA carboxylase, and FAS, with denaturation at 94°C for 45 s, annealing at 53°C for 45 s, and extension at 72°C for 45 s. Polymerase chain reaction mixture products were analyzed by 1.5% agarose gel electrophoresis.
Small, Interfering RNA (siRNA) Transfections for the Inhibition of Acetyl CoA Synthetase (ACS)
siRNA oligonucleotides targeting ACSS1 and ACSS2 and control siRNA were obtained from Invitrogen. The ACSS1 siRNA sequence was 5′-UCACCGUAUUUCAGCAACAGCCGG-3′. The ACSS2 siRNA sequence was 5′-UAUGCUUGGUGACAGGCUCAUCUCC-3′. SNU 449 cells expressing both ACSS1 and ACSS2 were plated in 12-well plates with 2 × 105 cells per well. The siRNA was added to 50% confluent cells with Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions and incubated for 24 h. PET and the measurement of percentage uptake after transfections were performed as described above.
Cell Viability After the Inhibition of ACS Using siRNA
Cell viability after the transfection of ACSS1, ACSS2, and control siRNAs in SNU 449 cells was determined by MTT assay (Sigma), which measures the conversion of the tetrazolium salt 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) to formazan. The cells were seeded at 3 × 104 cells per well on 24-well plates and incubated overnight at 37°C. The next day, the cells were transiently transfected with siRNAs, as described above. MTT assays were performed daily for 5 d to assess cell viability changes over time. In brief, 200 μL of MTT in phosphate-buffered saline (1 mg/mL) were added to each well. After incubation for 4 h at 37°C, the supernatant was discarded and 200 μL of dimethyl sulfoxide were added to dissolve MTT formazan crystals. Plates were then read on a microplate reader at 540 nm.
RESULTS
Patterns of 18F-FDG and 11C-Acetate Uptake in Human HCC
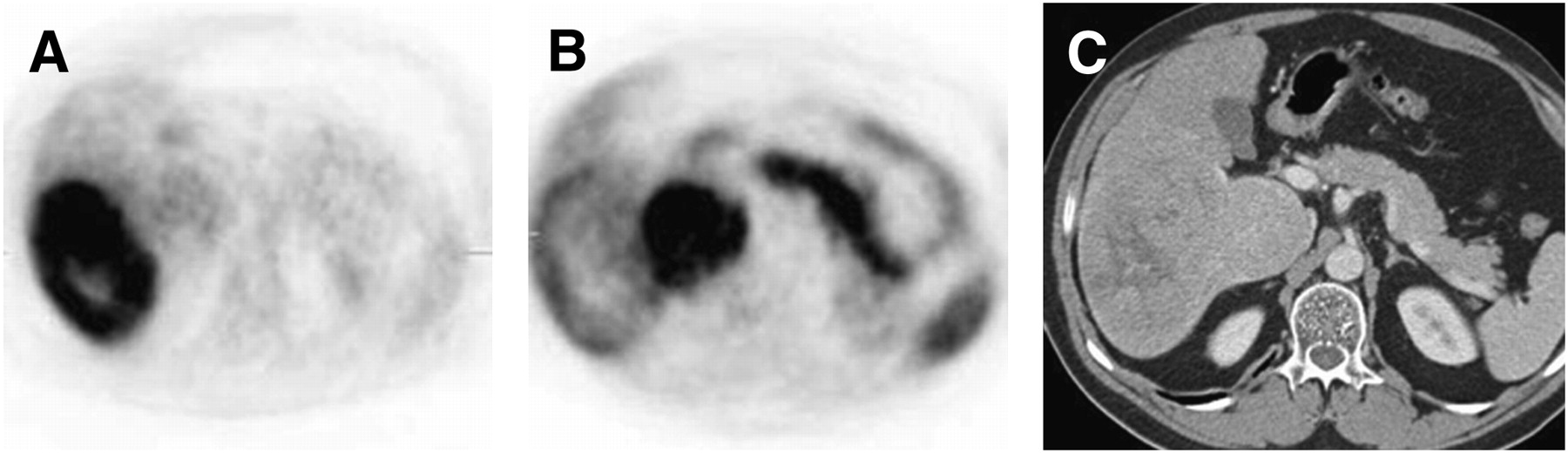
Six patients had low 18F-FDG uptake and high 11C-acetate uptake in their tumors (Table 1). One patient had high 18F-FDG uptake and low 11C-acetate uptake in the tumor. Four patients had mixed uptake within the tumor: The areas with high 18F-FDG uptake had low 11C-acetate uptake, whereas the regions with low 18F-FDG uptake had high 11C-acetate uptake (Fig. 1).

HCC with mixed uptake of 18F-FDG and 11C-acetate. (A) 18F-FDG uptake was higher in lateral portion of mass. (B) 11C-acetate uptake was noted mainly in medial portion of mass. (C) Axial view of CT scan showed large, multinodular mass in right lobe of liver.
Uptake Patterns of 18F-FDG and 11C-Acetate in Human HCC
PET of 18F-FDG and 11C-Acetate Uptake Versus Growth Rates in Cancer Cell Lines
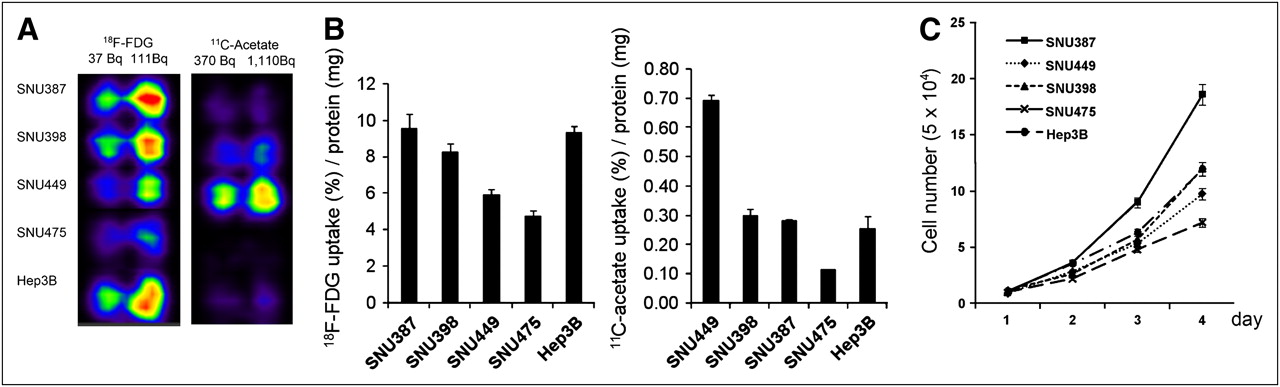
HCC cell lines significantly varied in 18F-FDG and 11C-acetate uptake (Figs. 2A and 2B). For 18F-FDG, high uptake was seen in SNU 387, SNU 398, and Hep3B cells, and low uptake was seen in SNU 449 and SNU 475 cells. In contrast, high 11C-acetate uptake was noted in SNU 449 cells, whereas the rest of the cell lines had low 11C-acetate uptake. Except for SNU 475, which showed low uptake for both 18F-FDG and 11C-acetate, the 2 radiotracer uptake patterns seemed to complement each other in HCC cell lines. Higher 18F-FDG uptake was found to correlate well with more rapid cell growth (Fig. 2C); in contrast, 11C-acetate uptake did not predict cell growth well.

PET and γ-counting of 18F-FDG and 11C-acetate uptake vs. growth rates in human HCC cell lines. (A and B) Patterns of 18F-FDG and 11C-acetate uptake were complementary to each other in all HCC cell lines except SNU 475, which showed low uptake for both 18F-FDG and 11C-acetate. (C) Higher 18F-FDG uptake correlated well with more rapid cell growth.
Expression of Enzymes Related to Glycolysis and Lipid Synthesis
SNU 387 cells with high 18F-FDG uptake showed increased expression of hexokinase II, whereas the expression of hexokinase II was relatively low in SNU 449 cells with low 18F-FDG uptake (Fig. 3). ACL, an enzyme for glucose-dependent lipid synthesis, was expressed in both cell lines. Of the 2 enzymes related to cellular activation of acetate, ACSS2 for the production of cytosolic acetyl CoA was expressed in both cell lines. ACSS1 for mitochondrial acetyl CoA was expressed only in SNU 449 cells, suggesting some use of mitochondrial acetyl CoA for lipid synthesis in the cytosol. Expression of acetyl CoA carboxylase and FAS was seen in both cell lines. These patterns of gene expression indicated glucose-dependent lipid synthesis in SNU 387 cells and acetate-dependent lipid synthesis in SNU 449 cells.
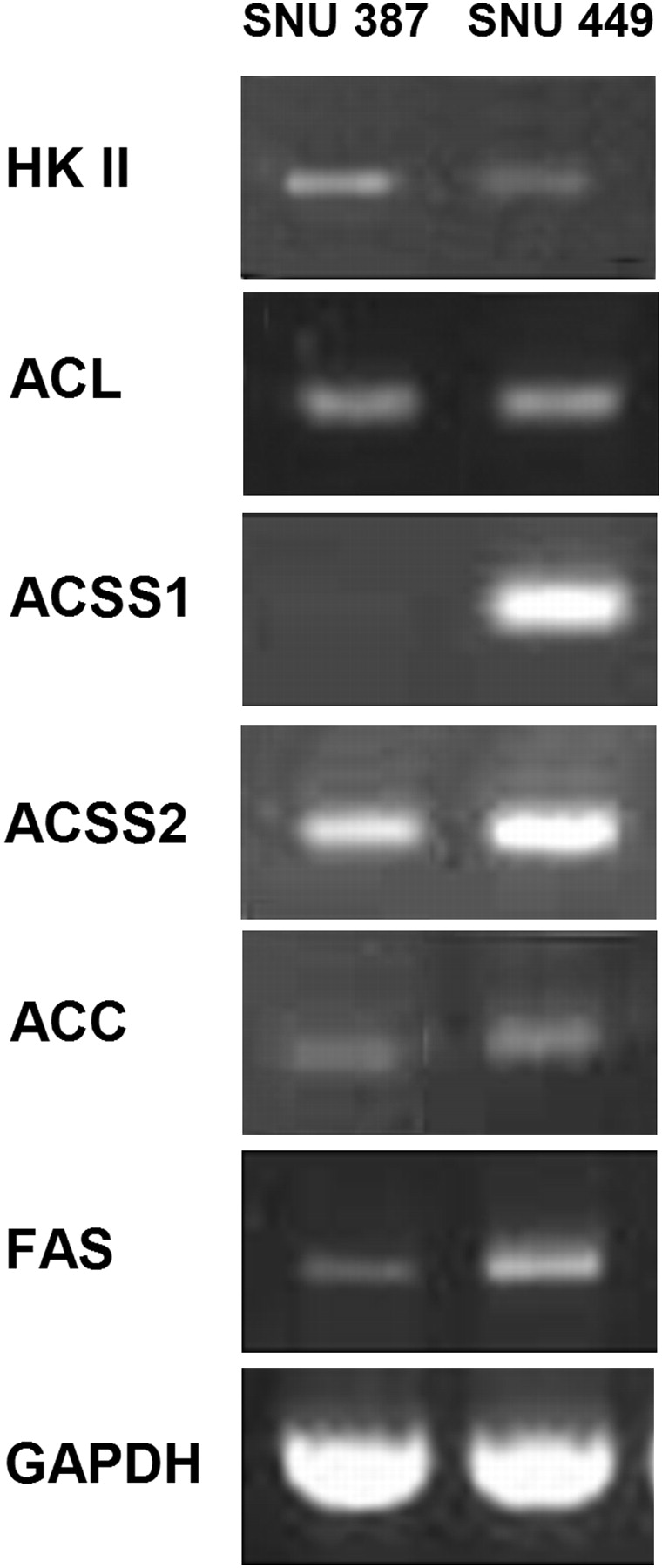
Gene expression patterns of enzymes related to glycolysis and lipid synthesis. SNU 387 cells with high 18F-FDG uptake showed increased expression of hexokinase II. In SNU 449 cells with high 11C-acetate uptake, both ACSS1 and ACSS2 were highly expressed. Acetyl CoA carboxylase and FAS expression in both cell lines suggested that glucose and acetate act as complementary carbon sources for de novo lipid synthesis in cancer cells.
Inhibition of ACSS1 or ACSS2 and Effects on 11C-Acetate Uptake and Cell Growth
Transient inhibition of ACSS1 or ACSS2 using the respective siRNAs was highly effective in reducing ACS gene expression in SNU 449 cells, as seen by reverse-transcriptase polymerase chain reaction mixture (Fig. 4A). On plate imaging and γ-counting, the amount of 11C-acetate uptake was also lower in wells with ACSS1 or ACSS2 siRNA than in the well with a control siRNA (Figs. 4B and 4C). Both ACSS1 and ACSS2 clearly play an important role in 11C-acetate uptake in SNU 449 cells. Transient transfection of SNU449 cells with ACSS1 or ACSS2 siRNAs resulted in reduced cell viability (Figs. 4D and 4E).

Inhibition of ACSS1 or ACSS2 and effects on 11C-acetate uptake and cell viability. (A) Transient inhibition of ACSS1 or ACSS2 using respective siRNAs was highly effective in reducing ACS gene expression, as seen in reverse-transcriptase polymerase chain reaction. (B and C) On plate imaging and γ-counting, degree of 11C-acetate uptake was reduced with ACSS1 or ACSS2 siRNA. (D and E) Transient transfection of SNU449 cells with ACSS1 or ACSS2 siRNA decreased cell viability.
DISCUSSION
De novo lipid synthesis is extremely low in nonproliferating cells, since fat-containing diets usually provide enough lipids for their needs and FAS activity remains low (11,12). Rapidly growing tumors also take a large proportion of their lipid requirements from the host. However, de novo lipid synthesis independent of physiologic hormone regulation occurs as a significant source of lipids for proliferation of cancer cells (13,14). High expression of FAS, one of the main synthetic enzymes responsible for the fatty acid synthesis pathway, has been identified as a potential selective target for the diagnosis and treatment of many malignant tumors (15–17). FAS seems to be involved in many cellular processes during carcinogenesis, such as membrane biogenesis, tumor cell proliferation, DNA replication, apoptosis, energy storage, redox balance in hypoxic conditions, or signal transduction by lipid modification (18). The exact mechanisms by which increased lipid synthesis and FAS activity connect to a malignant phenotype remain the subject of active investigation.
Studies on lipogenesis in neoplastic tissues have shown that both glucose and acetate carbon can be used for lipid synthesis (13,19,20). In fact, increases in glucose uptake in highly glycolytic cancer cells are attributed to glucose-dependent de novo lipid synthesis to enhance cell proliferation (21). In this pathway, pyruvate from glycolysis enters a tricarboxylic acid cycle to become citrate. Citrate is then preferentially exported to the cytosol and cleaved into acetyl CoA and oxaloacetate by ACL (22). The acetyl CoA becomes a metabolic intermediate for lipid synthesis. In glucose-dependent cancer cells, ACL is considered a key enzyme linking glucose metabolism to lipid synthesis. In this study, we also found that glucose-dependent cancer cells expressed ACL and FAS, supporting the use of glucose carbons for de novo lipid synthesis in rapidly growing cancer cells.
Acetate can be another source of carbons for lipid synthesis, and radiolabeled acetate has been used to measure lipid synthesis in cultured fibroblasts, cancer cells, and human cancers (9,10,23,24). The relative contributions of glucose and acetate carbons have been measured in some studies. A sparing effect for glucose carbons has been reported in which acetate carbons account for a significant source of lipid synthesis at high extracellular concentrations relative to glucose (24). In the human intestine, acetate is produced by anaerobes, and acetate is present at 0.2–0.3 mM in the blood (25). Although acetate rises after alcohol consumption and is released from the liver under ketogenic conditions, acetate is unlikely to be abundant enough to show a glucose-sparing effect for lipid synthesis in vivo (26). Alternatively, in cells with low rates of glycolysis such as cytochrome c null cells, Hatzivassiliou et al. suggested the possibility that acetate-dependent de novo lipid synthesis compensates for the low rate of glucose-dependent lipid synthesis (21).
This study found that human HCCs or cancer cells with low glycolysis showed remarkable 11C-acetate uptake, supporting the use of 11C-acetate as an alternative radiotracer for tumors not seen on 18F-FDG studies (2,8). Although 11C-acetate can be used for many metabolic pathways, increased ACL and FAS expression implies an association with acetate-dependent lipid synthesis in cells with low glycolysis. Taken together, the results suggest that 18F-FDG and 11C-acetate probably complement each other in the biosynthesis of macromolecules, particularly lipids in cancer cells. However, further studies using enzymatic assays are needed to better verify the role of ACL in glucose- or acetate-dependent lipid synthesis.
Cancer cells with high 18F-FDG uptake experienced rapid cellular proliferation in our study. In human HCCs, higher 18F-FDG uptake is predictive of larger tumor size, poorer histologic differentiation, shorter survival, and higher rates of recurrence (27–30). The metabolic differences between normal liver and HCC may explain our findings to some extent. Little glycolysis occurs in normal liver tissue, as glycogen is synthesized from blood glucose and resecreted as glucose (19). A series of metabolic changes, including increased glycolysis, happens as the cancer progresses, until HCC ultimately becomes an extrahepatic tissue metabolically. HCCs with low 18F-FDG seem metabolically more similar to normal liver than do HCCs with high 18F-FDG uptake. Accordingly, HCCs with low 18F-FDG uptake could conceivably have less aggressive features than HCCs with high 18F-FDG uptake.
For cellular metabolism, acetate must be activated into acetyl CoA by ACS. Of the 2 isoforms, ACSS1 is a mitochondrial enzyme that produces acetyl CoA for oxidation in the tricarboxylic acid cycle (31). Mitochondrial acetyl CoA produced by ACSS1 becomes citrate, which is transported into the cytosol for lipid synthesis (32). ACSS2 is a cytosolic enzyme that converts acetate to acetyl CoA for the synthesis of fatty acids and cholesterol (33). We found ACSS2 expression in both 18F-FDG–avid and 11C-acetate–avid cells. The results suggest no major role of ACSS2 in lipid synthesis in glucose-dependent cells, likely because of low acetate uptake in these cells. In contrast, both ACSS1 and ACSS2 were highly expressed in cells with low glycolysis, and both enzymes correlated well with 11C-acetate uptake. The expression of ACSS1 in acetate-dependent SNU 449 cells suggested the use of mitochondrial acetyl CoA for lipid synthesis in the cytosol. We also observed that the inhibition of ACSS1 and ACSS2 had a significant impact on cell survival. Our findings prove that ACS is one of the key enzymes in 11C-acetate uptake and acetate-dependent lipid synthesis for the growth of cancer cells with a low-glycolysis phenotype.
This study had some limitations. First, we assessed the expression of important regulatory enzymes related to glycolysis and lipid synthesis at the transcriptional instead of translational level. Enzymatic activities of only HK II and ACS were assessed using 18F-FDG and 11C-acetate, respectively. Second, we did not directly measure 11C activity incorporated in the lipid fraction of the cell lines we used in this study. Third, these enzymes were not evaluated in tissue samples of the patients. Further studies are currently being performed to investigate the expression of these enzymes in HCCs from patients.
CONCLUSION
The patterns of 18F-FDG and 11C-acetate uptake seemed to complement each other in both human HCCs and HCC cell lines. Human HCCs or cancer cells with low glycolysis showed remarkable 11C-acetate uptake, supporting the use of 11C-acetate as an alternative radiotracer for the detection of tumors with low 18F-FDG uptake. ACL and FAS expression was seen in cells with high 18F-FDG or 11C-acetate uptake, suggesting glucose- or acetate-dependent lipid synthesis. ACS appears to be important in 11C-acetate uptake and cell survival in cancer cells with a low-glycolysis phenotype. Inhibition of ACS in these cells may be promising for anticancer treatment.
Acknowledgments
This work was supported by a Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (M20702010003-08N0201-00314) and by a grant from the 2007 Korean National Cancer Control Program, Ministry of Health and Welfare, Republic of Korea. The production of 11C-acetate was supported by the KIRAMS QURI project of MEST and KOSEF.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication January 28, 2009.
- Accepted for publication April 28, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mitochondrial ACSS1 regulates the oncometabolite 2-hydroxyglutarate and De Novo Pyrimidine biosynthesis under nutrient-deprived conditions in lymphoma
- Visualizing cancer-originated acetate uptake through MCT1 in reactive astrocytes demarcates tumor border and extends survival in glioblastoma patients
- Visualization of reactive astrocytes in living brain of Alzheimers disease patient
- 68Ga-FAPI PET/CT: Biodistribution and Preliminary Dosimetry Estimate of 2 DOTA-Containing FAP-Targeting Agents in Patients with Various Cancers
- Harnessing Preclinical Molecular Imaging to Inform Advances in Personalized Cancer Medicine
- Glucose-independent Acetate Metabolism Promotes Melanoma Cell Survival and Tumor Growth
- Late Imaging with [1-11C]Acetate Improves Detection of Tumor Fatty Acid Synthesis with PET
- Interrogating Tumor Metabolism and Tumor Microenvironments Using Molecular Positron Emission Tomography Imaging. Theranostic Approaches to Improve Therapeutics
- ATP Citrate Lyase Knockdown Induces Growth Arrest and Apoptosis through Different Cell- and Environment-Dependent Mechanisms
- ATP-Citrate Lyase: A Key Player in Cancer Metabolism
- Kruppel-like Factor 4 (KLF4) Activates the Transcription of the Gene for the Platelet Isoform of Phosphofructokinase (PFKP) in Breast Cancer