


The availability of plastic microspheres labeled with different γ-emitting isotopes has allowed the quantification of blood flow in different organs, including the heart (1). In the past 3 decades this has made it possible to study the changes in myocardial blood flow (MBF) under different physiologic and pathophysiologic conditions in experimental animals.
In a similar fashion, the development of PET has allowed the noninvasive quantification of regional MBF in healthy humans and patients with different cardiovascular diseases. This article summarizes how PET measurement of MBF has contributed to advance our understanding of cardiac physiology and pathophysiology.
METHODOLOGIC AND TECHNICAL CONSIDERATIONS
Several techniques, including Doppler catheterization and coronary sinus thermodilution, are available for measuring coronary blood flow (CBF) in humans. These techniques, which are fundamentally variations of indicator dilution methods, are invasive and affected by serious limitations (2). Although CBF, for which the symbol F is often used, has units of volume per time (i.e., mL/min), Doppler measurements usually allow assessment of flow velocity (cm/s) and only few techniques provide volumetric flow (3,4). Measurement of inert tracer clearance, which can be invasive (based on arteriovenous sampling) or semiinvasive (e.g., based on external detection of radionuclide washout, after intracoronary injection 133Xe) provides estimates of regional perfusion, although the mass of tissue subtended by the artery under study is unknown (2). SPECT allows the noninvasive assessment of directional changes in regional tissue perfusion, but its physical limitations do not permit quantification of MBF (5). PET has been shown to allow noninvasive and accurate quantification of regional MBF if suitable tracers are used and appropriate mathematic models are applied. These PET measurements of MBF, for which the symbol F/W is also used, have units of volume per time per unit weight of myocardium (i.e., mL/min/g).
To achieve the accurate quantification of tracer uptake, which is a characteristic of PET, correction for photon attenuation is crucial. At present, a rotating 68Ge (half-life = 287 d) source is commonly used for photon attenuation correction in oncology but also in cardiac viability and perfusion scanning. 68Ge sources have only a low photon flux, which necessitates long acquisition times of up to 20 min (6,7), although with current methods transmission times much shorter than 20 min can be used. This results in increased chance of motion artifacts and poor anatomic image quality as well as patient discomfort and economic loss because of lower patient throughput (8). Introduction of combined PET/CT systems, where the CT scan can be used for attenuation correction, is expected to allow improvement of these limitations since a high-end CT scanner is able to acquire images with far higher spatial resolution in a much shorter time (<4 s) compared with a conventional 68Ge transmission scan (68Ge attenuation correction). The feasibility of adequate photon attenuation correction with the CT scan of a hybrid PET/CT scanner has been demonstrated for static PET scans using 18F-FDG (9,10) and, more recently, also for dynamic scanning with 13N-labeled ammonia (13NH3) (11).
TRACERS AND CAMERAS
Various tracers have been used for measuring MBF by PET, including 15O-labeled water (H215O) (12–17), 13NH3 (18–23), the cationic potassium analog 82Rb (24,25), 62Cu-pyruvaldehyde bis(N4-methylthio-semicarbazone (62Cu-PTSM) (26–30) and 11C as well as 68Ga-labeled albumin microspheres (31,32), 94mTc-teboroxime (33), and 38K (34). Early PET studies used 13NH3 (18) and 82Rb (35) for qualitative assessments of regional MBF. Currently, 13NH3, H215O, and 82Rb are the most widely used PET perfusion tracers. 13NH3 and 82Rb are given intravenously as boluses. In the case of H215O, the tracer can be administered as an intravenous bolus injection (6,12,15,36), an intravenous slow infusion (6,37), or by inhalation of 15O-labeled carbon dioxide (C15O2), which is then converted to H215O by carbonic anhydrase in the lungs (14). Generator-produced 82Rb is a very appealing MBF tracer because it does not require a cyclotron on site and has a very short half-life (78 s). Although several models have been proposed for quantification of regional MBF using 82Rb (25,38–40), they are limited by the heavy dependence of the myocardial extraction of this tracer on the prevailing flow rate and myocardial metabolic state. Therefore, quantification of regional MBF with 82Rb may be inaccurate, particularly during hyperemia or in metabolically impaired myocardium. In addition, the high positron energy (3.15 MeV) of this radionuclide results in relatively poor image quality and in a reduced spatial resolution due to its relatively long positron track.
Several tracer kinetic models for quantification of MBF have been successfully validated in animals against the radiolabeled microsphere gold standard over a wide flow range for both H215O and 13NH3. The latter has also been validated against the argon gas technique in humans (41). Single-compartment models, based on Kety’s model for an inert freely diffusible tracer (42), are used for estimation of MBF using H215O (12,14,15,43). A 3-compartment model describing the kinetics of the myocardial metabolic trapping and whole-body metabolism of 13NH3 has been used for calculation of MBF using this tracer (21,23,44–46). The models have to include corrections for underestimation of radiotracer concentration due to the partial-volume effect and spillover from the left chamber onto the ventricular myocardium (44,47), which result from the limited spatial resolution of the PET camera and the motion of the heart. Additional corrections have been developed to account for the impact of flow (20) on myocardial extraction of 13NH3 and for the radiolabeled metabolites (48) of 13NH3, which accumulate in blood.
The equivalence of H215O and 13NH3 as perfusion tracers has been demonstrated in experimental animals (49) and in humans (50), but the proof of congruence of the tracers in ischemic and infarcted segments requires further investigation. The use of H215O as a perfusion tracer is potentially superior to 13NH3 because H215O is metabolically inert and freely diffusible across capillary and sarcolemmal membranes. Thus, it equilibrates rapidly between the vascular and extravascular spaces and its uptake by the heart does not vary despite wide variations in flow rate. The short half-life (123 s) of 15O allows repetitive MBF measurements at short intervals (10 min, equivalent to 5 half lives of 15O). Previously, an important shortcoming of the H215O technique was its need for additional 15O-carbon monoxide (C15O) blood-pool scanning to define the regions of interest and correct for the high 15O activity in the blood pool. An advanced technique has been proposed for generating myocardial images directly from dynamic H215O scans, eliminating the need for additional C15O blood-pool scans (51). This technique has recently been validated against microspheres in experimental animals (52,53), and its repeatability has been documented in humans (16,17).
The differences between H215O and 13NH3 are of little relevance when the measurements of MBF are performed in normal myocardium as proven by the comparable flow estimates obtained with the 2 tracers in healthy human subjects. However, in a highly heterogeneous tissue (e.g., jeopardized myocardium in patients with previous infarction), the diffusion/extraction and final uptake of H215O and 13NH3 are determined by the flow rates in each tissue compartment—that is, higher in viable tissue and lower in scar tissue. 13NH3 uptake (on which the model for the computation of MBF is based) in a given region of interest will reflect the average uptake and, hence, average flow in this mixture of viable and fibrotic tissue. On the other hand, since the uptake of H215O in scar tissue is negligible, washout of H215O (on which the model for the computation of MBF is based) will mainly reflect activity in better-perfused segments and the resulting flow can therefore be higher than that obtained with 13NH3 in the same region (54).
The PET cameras used for the quantification of MBF, as well as for other cardiac PET applications, work in 2-dimensional (2D) mode with collimating septa between the detector rings to reduce the number of interplane scattered photons (55). A new generation of 3-dimensional (3D)-only tomograms has become available with potential benefits over the 2D systems, particularly a higher efficiency. Recently, absolute quantification of MBF with H215O and 3D PET has been validated in experimental animals (53).
APPLICATIONS OF PET TO STUDY CARDIAC PHYSIOLOGY AND PATHOPHYSIOLOGY
Coronary flow reserve (CFR), the ratio of MBF during near-maximal coronary vasodilation to basal MBF, is an integrated measure of flow through both the large epicardial coronary arteries and the microcirculation and has been proposed as an indirect parameter to evaluate the function of the coronary circulation. An abnormal CFR can be due to narrowing of the epicardial coronary arteries or, in the absence of angiographically demonstrable atherosclerotic disease, may reflect dysfunction of the coronary microcirculation. The latter can be caused by structural (e.g., vascular remodeling with reduced lumen-to-wall ratio) or functional (e.g., vasoconstriction) changes, which may involve neurohumoral factors or endothelial dysfunction. Furthermore, an abnormal CFR may also reflect changes in coronary or systemic hemodynamics as well as changes in extravascular coronary resistance (e.g., increased intramyocardial pressure).
Because of its ability to provide noninvasive regional absolute quantification of MBF, PET has been widely used to assess CFR in healthy volunteers (56,57), in asymptomatic subjects with cardiovascular risk factors (58–60), in patients with coronary artery disease (CAD) (61), and other cardiac diseases (62–64). Furthermore, measurement of CFR has been used as a “surrogate endpoint” to assess the efficacy of therapeutic interventions such as α- (65) and β-adrenoceptor blockade (66), lipid-lowering strategies (67), cardiovascular conditioning (68), and coronary angioplasty (69,70).
MBF and CFR in Healthy Humans
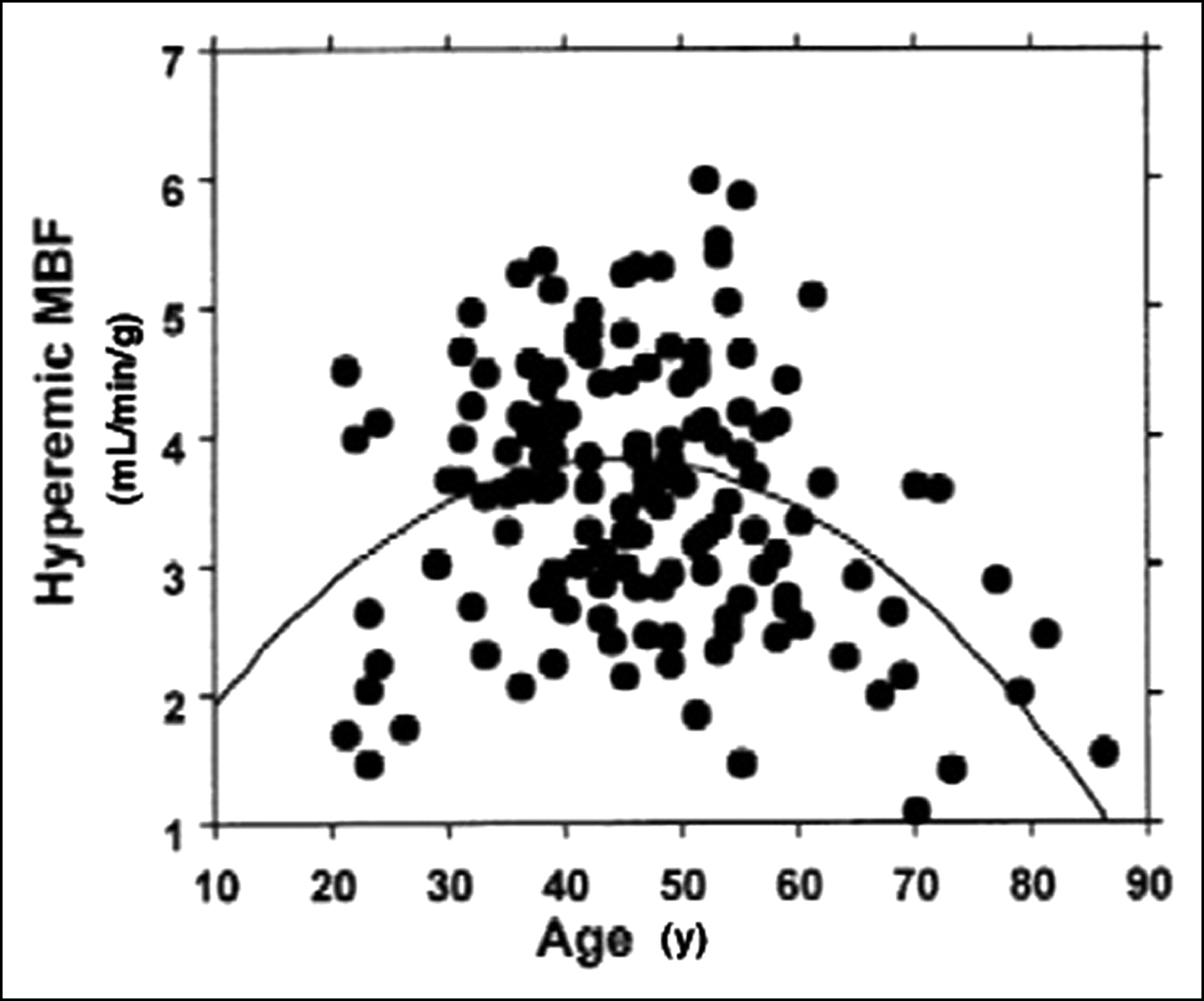
In a recent article, Chareonthaitawee et al. (71) have investigated the range of resting and hyperemic MBF in a large population (n = 160) of healthy men and women over a broad range of ages (21–86 y old). They found that baseline and hyperemic MBF are heterogeneous both within and between individuals. Baseline and hyperemic MBF exhibit a similar degree of spatial heterogeneity, which appears to be temporally stable. Resting myocardial perfusion ranged from 0.59 to 2.05 mL/min/g (average, 0.98 ± 0.23 mL/min/g) and hyperemic perfusion ranged from 1.85 to 5.99 mL/min/g (average, 3.77 ± 0.85 mL/min/g). Significant differences within subjects were found comparing different segments with each other, except for anterior versus lateral. MBF was significantly higher in women than in men. There was a significant linear association between age and baseline MBF, partly related to changes in the external cardiac workload with age. Hyperemic MBF declines in subjects who are >65 y old. These results are in agreement and extend previous animal and human studies that have provided evidence for significant heterogeneity of global and regional baseline and hyperemic MBF (56,72). Interestingly, in the study by Chareonthaitawee et al. (71), hyperemic MBF was found to reach higher values in subjects in whom the standard dose of adenosine (140 mg/kg/min) was used compared with those in whom dipyridamole (0.56 mg/kg infused over 4 min) was used as a hyperemic stressor. Both age and the type of vasodilator used determine the degree of hyperemia achieved and, thus, are sources of variability. As a result, the interindividual variability of hyperemic MBF is greater than that observed for baseline MBF. Several considerations suggest that this heterogeneity reflects a true biologic phenomenon rather than a methodologic artifact. First, there is spatial heterogeneity of MBF within each individual to support the overall variability of global MBF. Second, a similar degree of spatial heterogeneity was found in several studies in experimental animals (72–77) and in humans (12,56,78) despite the use of different techniques with different spatial resolution to estimate MBF. Third, the spatial heterogeneity exhibits remarkable temporal stability. Fourth, the temporal variability of PET is small as demonstrated by the high repeatability of PET perfusion measurements assessed 20 min apart in the same subjects (16,17). Small temporal fluctuations in baseline MBF may be attributed to “twinkling” of capillary flows (79) or time-dependent changes in regional metabolic requirements coupled to local autoregulatory changes in MBF (75,76,80). The spatial heterogeneity of MBF may be also attributed to the fractal nature of regional flow distribution (81,82) and different neural regulatory modulation in different regions and layers of the left ventricle (74,80). Differences in transmural MBF distribution across the left ventricle may also contribute to the spatial heterogeneity, although their magnitude is smaller than the overall heterogeneity (83–87).
In healthy humans, baseline MBF does not predict hyperemic MBF. This results in a wide range of regional CFR, which has important clinical implications. Measurements of regional CFR of <2.5 are often interpreted as impaired vasodilator capacity. However, many normal left ventricular segments have a CFR of <2.5. This may, at least in part, explain the patchy distribution of myocardial injury within a given region and suggests that there may be regions operating near maximal capacity in the baseline state, becoming more susceptible to injury when demand exceeds supply (88).
As stated earlier, baseline MBF increases with age, which is explained, in part, by the linear association between age and the rate·pressure product. As hyperemic MBF declines in subjects who are >65 y old (56,57,71,78), the combination leads to a significant decrease in CFR (Fig. 1). These changes are likely to result from the combination of several mechanical and neurohumoral factors associated with aging, including increased arterial impedance, thickening of left ventricular myocardium, reduced lusitropism, reduced catecholamine responsiveness, endothelial dysfunction, and deficient neuroendocrine regulation (71,89,90). Gender differences in baseline MBF have been repeatedly reported (57,71,91,92). Baseline MBF is higher in females even after correction for the rate·pressure product. This difference might be explained, at least in part, by the well-known effects of estrogens on vascular tone (93). In a recent study (71), a slightly higher hyperemic MBF was found in females than in males but due to the females’ higher baseline flow, overall their CFR was lower than in males. This should be considered when assessing CFR in females.
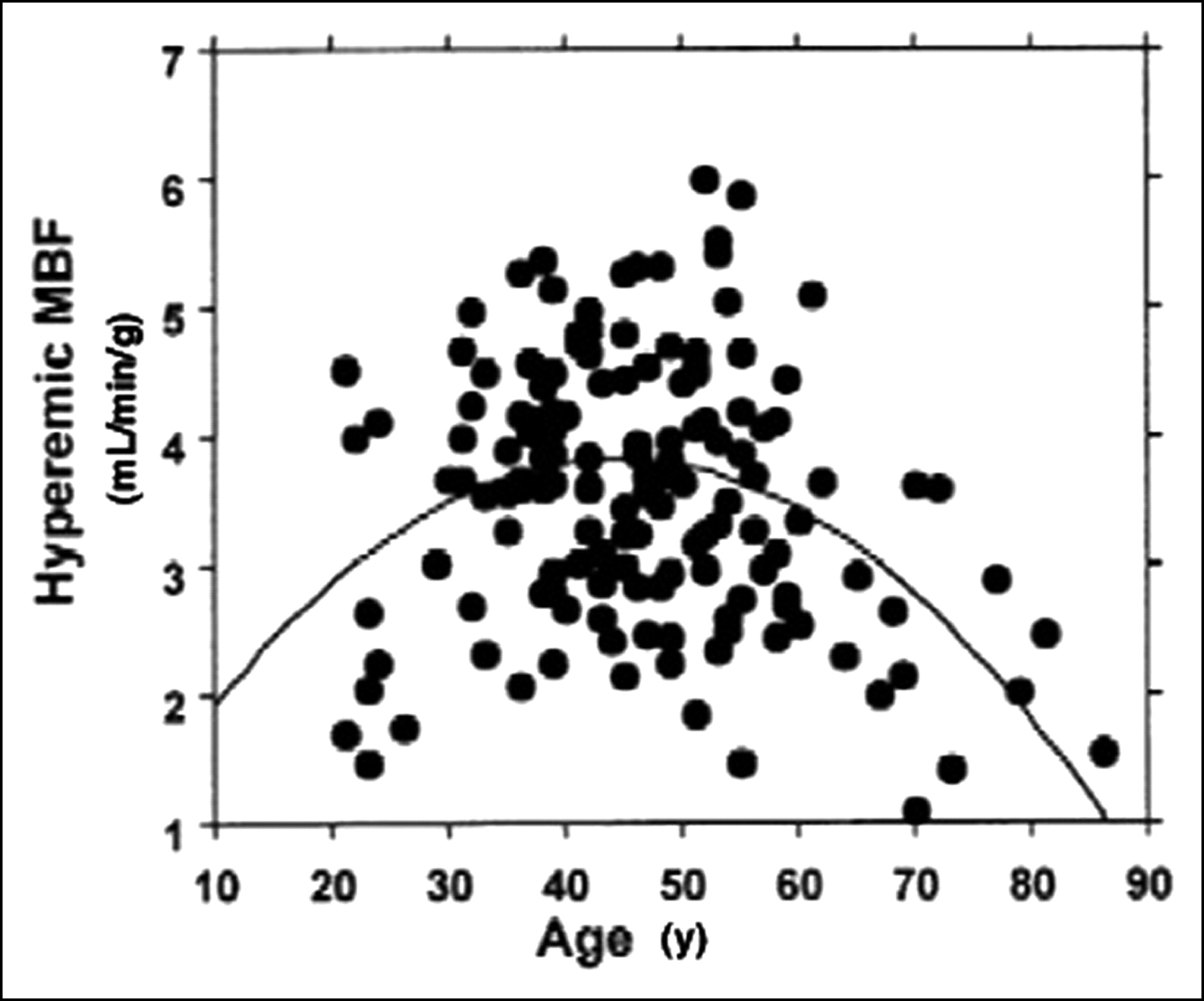
Scatterplot shows relationship between age and hyperemic MBF, illustrating lower hyperemic MBF in subjects >55 y old. Similarly, in the few youngest subjects, hyperemic MBF was also lower than the group mean. (Reprinted with permission of (71).)
In addition to pharmacologic stress, the cold pressor test (CPT) has been used to induce a hyperemic MBF response. The demonstration of a correlation between changes in MBF during intravascular acetylcholine infusion and CPT (94) supports the use of a CPT as a noninvasive test to study coronary endothelial function with PET. The flow response to the CPT is based on activation of the sympatho-adrenomedullary system induced by nociception. This, in turn, is supposed to increase myocardial oxygen demand, which, in the normal heart, induces an increase in MBF largely due to endothelium-dependent flow-mediated vasodilation. However, in patients with endothelial dysfunction, such as smokers or diabetics, the release of nitric oxide (NO), which follows the increase in shear stress and accounts for the vasodilation of the large epicardial arteries, is impaired and, hence, the response to the CPT is absent (95,96) or paradoxical (i.e., a reduction in MBF is observed).
MBF and CFR in Subjects with Risk Factors for CAD
Coronary endothelial and microcirculatory dysfunction have been demonstrated in patients with coronary risk factors such as hypercholesterolemia (60,97–99), essential hypertension (100,101), diabetes mellitus (102), and smoking (103–106). In other words, CFR provides a way to document how risk factors translate into measurable damage to the coronary circulation. As mentioned earlier, the most widely used hyperemic MBF stimulus for assessing CFR with PET is adenosine. Until recently, the vasodilator effect of adenosine was thought to be based solely on direct stimulation of A2-purinergic receptors on vascular smooth muscle cells, which mediate an increase in the second messenger cyclic adenylate (cyclic adenosine monophosphate) by stimulating adenylate cyclase. Therefore, this agent has been used frequently in animal as well as human studies to evaluate endothelium-independent vasodilation (107). However, adenosine can act also as an endothelial-dependent vasodilator (108), not only via flow-mediated dilation (109) but also by directly stimulating A1-purinergic receptors (110) and other purinergic receptors (111) on endothelial cells. Thus, adenosine-induced hyperemic MBF response (and CFR) is not entirely endothelium dependent or independent but, rather, a mixed response of both pathways.
Smoking.
Cigarette smoking is a well-established risk factor for cardiovascular disease (112), affecting both the coronary and the peripheral circulation (113). Endothelial dysfunction in brachial (103) and coronary (104) arteries has been demonstrated in long-term smokers and even in passive smokers (105,106).
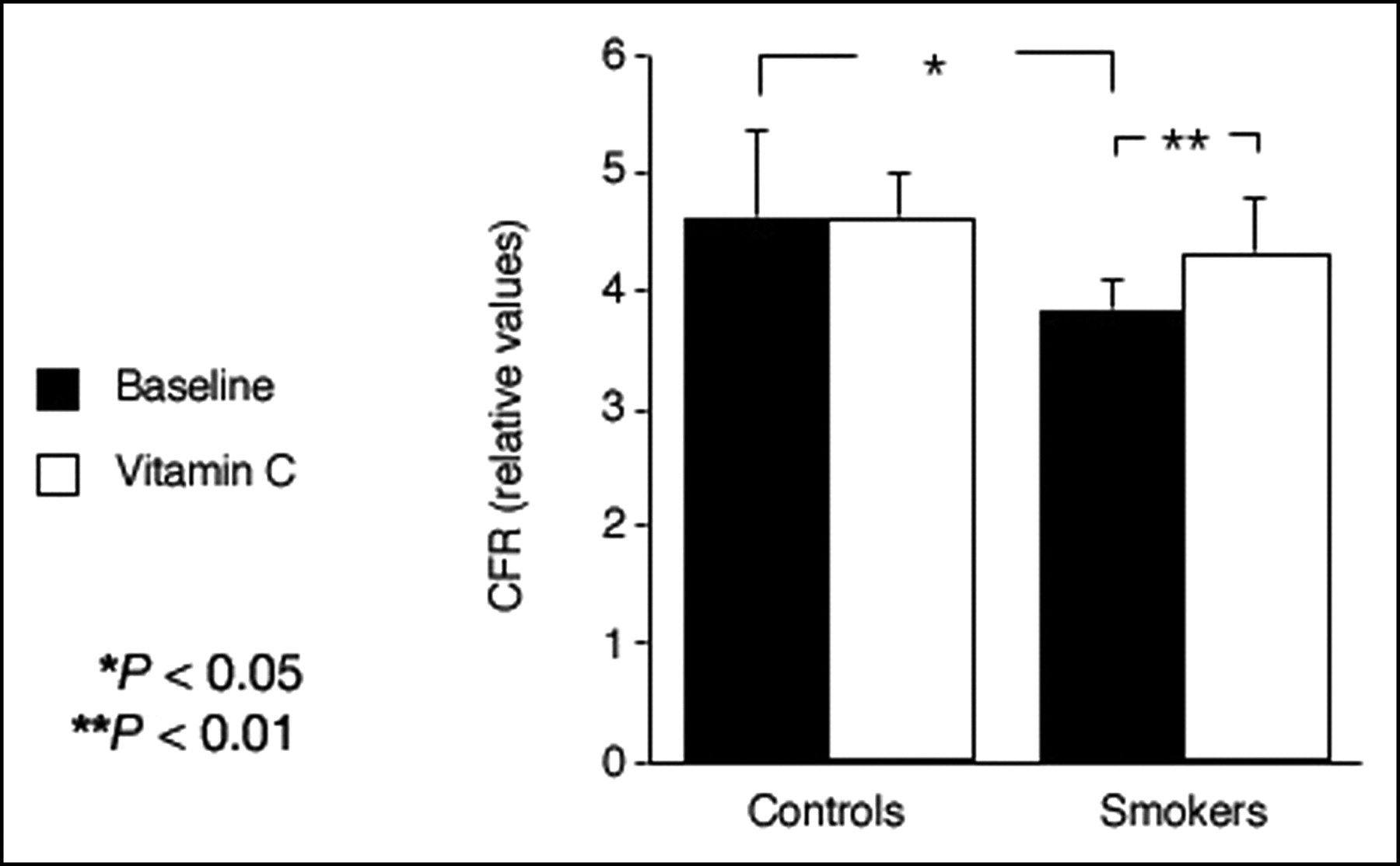
The findings of a recent PET study extend these observations and demonstrate that the noxious pro-oxidant effects of smoking extend beyond the epicardial arteries to the coronary microcirculation affecting the regulation of MBF (59). In smokers, adenosine-induced hyperemia was reduced by 17% and CFR was reduced by 21% compared with nonsmoking controls (Fig. 2). Although the mechanisms of smoking-associated vascular damage are not fully established, several factors may be involved. Nicotine has been shown to produce structural damage in aortic endothelial cells of animals (114). Smoking is associated with a direct toxic effect on human endothelial cells (115). The gas phase of cigarette smoke contains large amounts of free radicals and pro-oxidants lipophilic quinones (116), which can form the highly reactive hydroxyperoxide radical. These oxidants may increase the amount of oxidized low-density lipoprotein (LDL), which is markedly more effective than native LDL in impairing NO synthase (117). Short-term administration of the antioxidant vitamin C restored coronary microcirculatory responsiveness and normalized CFR in smokers (Fig. 2) without any significant effect in nonsmoking control subjects, lending support to the hypothesis (95) that the damaging effect of smoking is explained, at least in part, by an increased oxidative stress. This is in line with the results of a previous investigation in which another antioxidant, reduced glutathione (118), was shown to improve endothelial dysfunction in patients with cardiovascular risk factors, but had no effect in subjects without risk factors. Furthermore, vitamin C has been reported to attenuate abnormal coronary vasomotor reactivity in patients with vasospastic angina by scavenging oxygen free radicals (119).
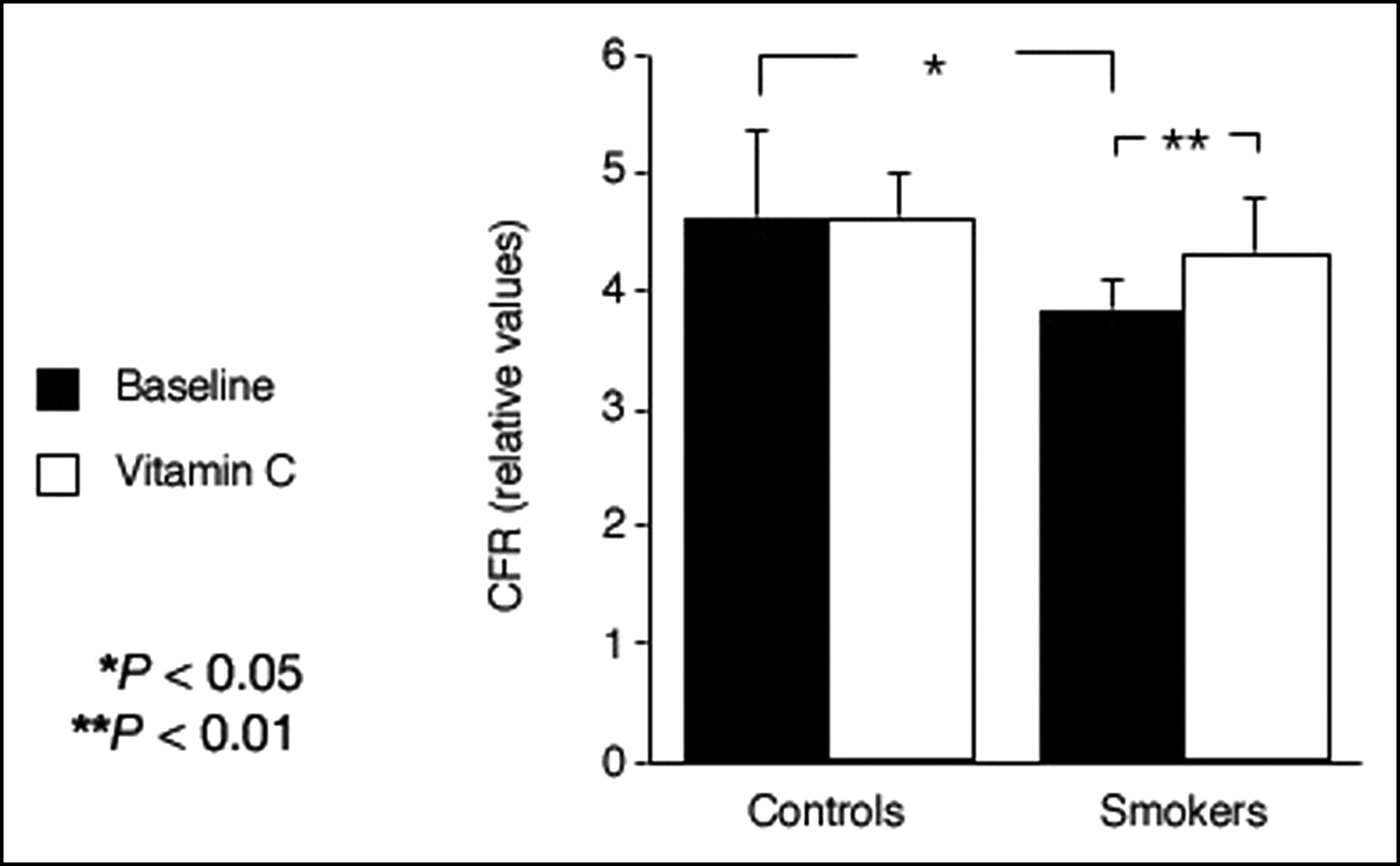
CFR is significantly lower in asymptomatic smokers than in healthy control subjects. This can be reversed by short-term infusion of high-dose vitamin C. (Reprinted with permission of (59).)
Hyperlipidemia.
In subjects with angiographically normal coronary arteries, hypercholesterolemia has been shown to impair endothelial-mediated coronary dilation (97,120). This is, at least in part, reversible by l-arginine infusion (121,122) and therapy with lipid-lowering drugs (123–126) or calcium channel blockers (98). A reduction in CFR in asymptomatic hypercholesterolemic subjects with angiographically normal coronary arteries, as well as its reversibility with cholesterol-lowering strategies (67,68,127), has been documented by means of PET (58,128). However, results from in vitro studies suggest that endothelial dysfunction is due to reduced NO release or increased production of superoxide anion by oxidized LDL cholesterol (117), or both, rather than by an increase in total cholesterol (TC). In fact, single LDL apheresis in humans has been shown to improve endothelium-dependent vasodilation in hypercholesterolemic patients (129). In a recent study in a population that included asymptomatic subjects with normal or elevated TC (60), no difference in either rest and hyperemic MBF or CRF was found based on TC. When all subjects (i.e., with normal and abnormal TC) were considered, there was a weak correlation between CFR and high-density lipoprotein cholesterol but not between CFR and LDL cholesterol. However, when only the subjects with high TC were considered, CFR was inversely related to the LDL subfraction (−0.61, P < 0.01). Similarly, previous studies had demonstrated a significant inverse correlation between CFR and lipid subfractions, including LDL cholesterol (58,128,130). The latter studies, however, also found a correlation between CFR and TC. Some of these discrepancies may be explained by different patient selection criteria, the numbers of the study cohort with a narrow, nonrepresentative range of cholesterol levels, and differences in concomitant medication. As an example, the study of Dayanikli et al. (58) included 16 hypercholesterolemic men, 12 of whom were being treated with lipid-lowering agents and 3 were smokers. Although Yokoyama et al. (130) found a significantly reduced CFR in patients with familial hypercholesterolemia, it seems that secondary and familial hypercholesterolemia do not necessarily have the same impact on endothelial function, as the latter could represent an epiphenomenon whereby the primary disease would directly affect the vascular bed. This is supported by the results of Pitkänen et al. (131), who found a correlation between TC and CFR in patients with familial combined hyperlipidemia and the phenotype IIB, but not in those with the phenotype IIA, despite increased TC in both groups. The same group provided evidence of linkage to a subchromosomal region (1q21-23) in familial combined hyperlipidemia (132) and therefore suggested that genetic factors behind familial combined hyperlipidemia may cause endothelial or smooth muscle dysfunction, or both, by mechanisms unrelated to lipid metabolism.
In the study of Kaufmann et al. (60), no relation between TC and MBF or CFR could be demonstrated, although the LDL cholesterol subfraction correlated inversely with CFR in those with high TC, supporting a direct pathogenic role of this subfraction in the development of coronary microcirculatory dysfunction. These in vivo results are in agreement with the previous observations identifying the LDL subfraction as a cause of endothelial dysfunction and extend these findings to the coronary microcirculation in humans. Furthermore, this provides pathophysiologic support for a clinical strategy (133) aimed at the treatment of the entire lipid profile rather than targeting TC reduction alone. In fact, risk assessment without taking the LDL subfraction into account seems to provide unreliable results (134). It is important to remember that the benefits of treating any risk factor depend not only on the absolute risk of future disease but also on the degree to which the risk factor in question contributes to this risk (135).
Diabetes.
It is a well-known finding of the Framingham study that patients with diabetes mellitus have an increased risk for development of micro- and macro-angiopathy and cardiac disease (136). In addition, angina or silent ischemia appears to be frequent in diabetic patients who have been shown to have angiographically normal coronary arteries. Although much of the excess CAD risk can be accounted for by the presence of diabetes-associated coronary risk factors such as obesity, dyslipidemia, and hypertension, a significant proportion of it remains unexplained (137). A direct deleterious effect of diabetes on vascular and, in particular, on endothelial function has been suggested, thereby increasing the potential for vasoconstriction and thrombosis. There is consistent evidence that in patients with diabetes coronary vascular function is impaired and that this may be an early marker of atherosclerosis as it precedes clinically overt CAD (102,138–140). Forearm blood flow studies have shown that acute local hyperglycemia as well as oral glucose load significantly attenuated flow-mediated dilation and forearm blood flow response to methacholine, independent of the systemic insulin concentration (141,142) in healthy volunteers and in subjects with impaired glucose tolerance. A recent PET study (96) has found markedly impaired coronary microvascular function in response to adenosine (reflecting partly endothelium-independent vasodilation) and to CPT (reflecting primarily endothelium-dependent vasodilation) in young subjects with uncomplicated diabetes. The findings were very similar in type 1 and type 2 diabetes, although patients with type 1 diabetes are insulin deficient (rather than insulin resistant, which is the hallmark of type 2 diabetes). This provides further support for a key role of hyperglycemia in the pathogenesis of vascular dysfunction in diabetes.
MBF AND CFR AS SURROGATE MARKERS
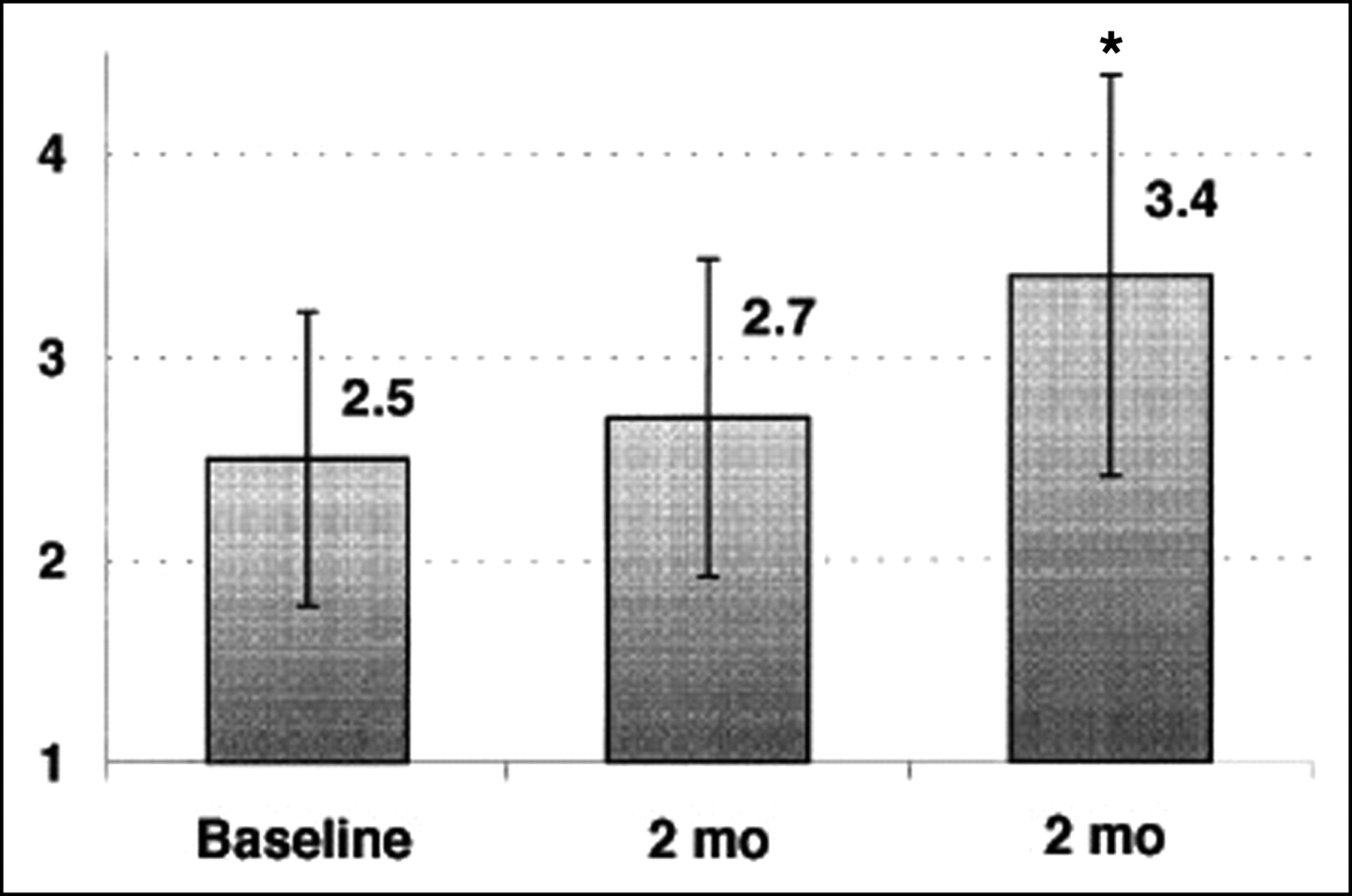
Because PET flow studies have been shown to be accurate, the method appears appropriate for the study of the effects of any intervention with each subject used as his or her own control. This was confirmed in recent reproducibility studies (16,17). Thus, PET is a unique noninvasive tool to follow changes in MBF and CFR during risk factor modification before structural alterations of the vascular bed may be found. Improvement in myocardial perfusion abnormalities by PET has been documented after short-term and long-term intense risk factor modification (67,68,143). It has been shown that lowering lipid levels is associated with improvement of CFR. This improvement, however, has been shown to be delayed as compared with the decrease in plasma lipid levels (127) (Fig. 3). Similarly, acute reductions in cholesterol, such as those achieved with lipid apheresis, were not associated with improvement of CFR, indicating that vascular reactivity is not only related to plasma cholesterol levels but also may represent a complex adaptation of vascular structures to high concentrations of circulating lipids. The measurement of CFR with PET in asymptomatic hypercholesterolemic subjects may not only help to identify those at highest risk but also provide a “target” to assess the functional effectiveness of lipid-lowering treatment (60).
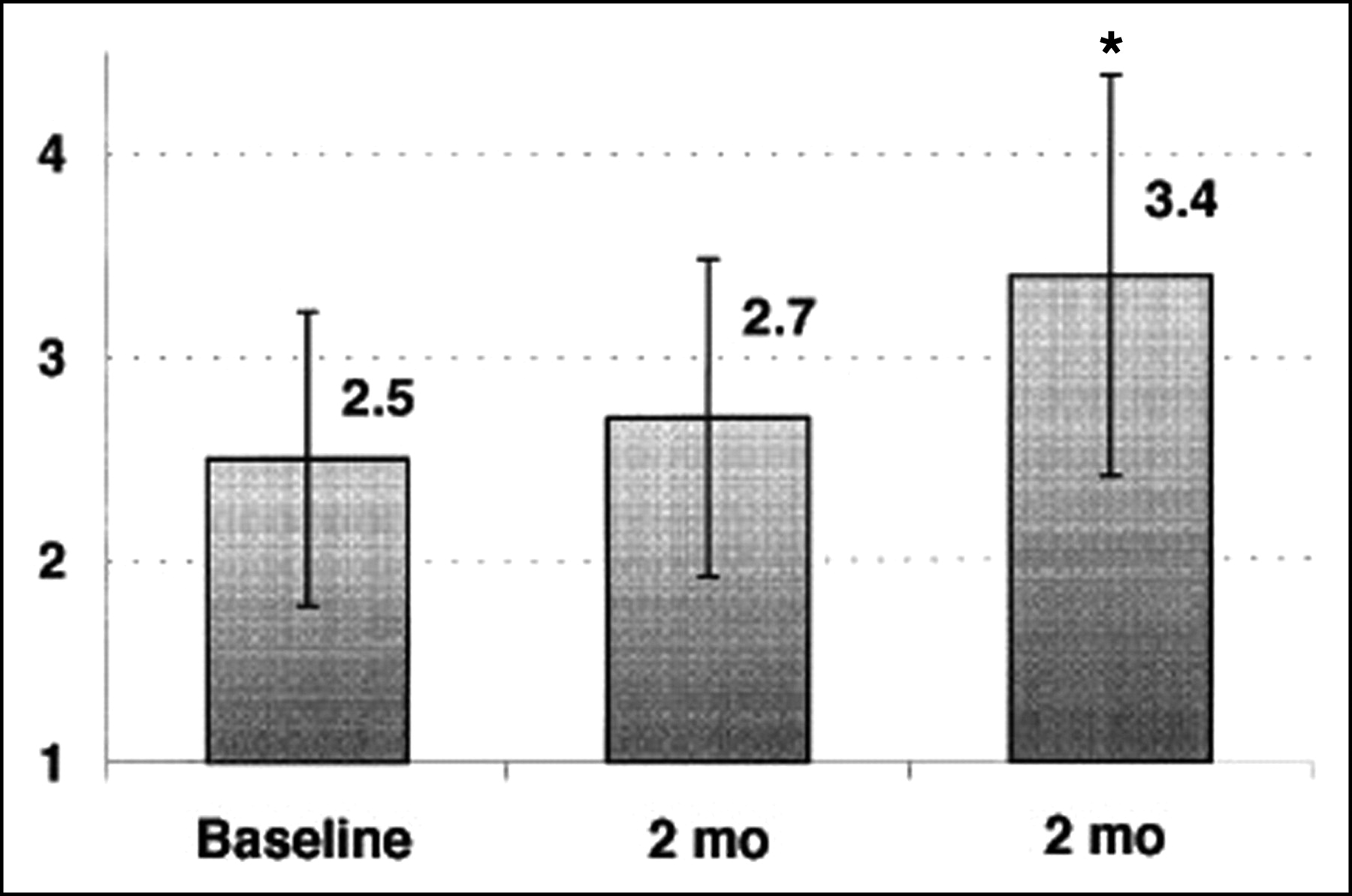
CFR (arbitrary units) during therapy with fluvastatin. *P < 0.05 vs. baseline. (Reprinted with permission of (127).)
EMERGING CONCEPT OF MICROVASCULAR DISEASE
Until quite recently, many of the most important forms of cardiovascular disease were considered to involve primarily large vessels, particularly the conduit coronary arteries. However, recent advances have highlighted the crucial involvement of the microcirculation in many cardiovascular conditions. A new concept has emerged in which microvascular disease is a well-defined condition that often precedes the development of full-blown diseases and may bear independent prognostic value.
Under normal circumstances, the small coronary arterioles <450 μm in diameter are the principal determinants of coronary vascular resistance (144–146). According to Chilian et al. (147), a 50% drop in perfusion pressure, relative to aortic, may be observed in vessels between 70 and 440 μm in diameter, which is consistent with 40%–50% of total coronary vascular resistance being located in prearterioles >100 μm. These vessels receive autonomic innervation and their diameter may be altered by stimulation of these nerves (148). Nearly all of the remaining resistance lies in vessels <100 μm in diameter that are also those responsible for autoregulation of MBF (145). In addition to intravascular resistances, myocardial perfusion is also influenced by extravascular forces, particularly due to the intramyocardial pressure, which is generated throughout the contractile cycle (149). Intramyocardial pressure is maximal in systole and in the subendocardial layers where it exceeds aortic pressure (150).
Although direct visualization of the coronary microcirculation has been achieved in experimental animal preparations using intravital microscopy and stroboscopic epi-illumination of the heart (151,152), no technique enables the direct visualization of the coronary microcirculation in humans in vivo. The resistive vessels in the coronary circulation are not generally visible on angiography and are too small to be amenable to selective catheterization. Therefore, study of the human coronary microcirculation is indirect and relies on assessing parameters that reflect its functional status, such as MBF and CFR. These are principally regulated by the coronary microcirculation and, thus, in the absence of coronary stenoses, their measurement provides an index of microvascular function (153).
Microvascular Disease in Primary Cardiomyopathies
Hypertrophic Cardiomyopathy (HCM).
HCM is a genetically determined disease with a wide range of clinical manifestations and pathophysiologic substrates (154–166), which has been estimated to occur in 0.02%–0.2% of the population. Symptoms and signs of myocardial ischemia are often found in patients with HCM despite angiographically normal coronary arteries. Myocardial ischemia can contribute to some of the lethal complications of HCM, including ventricular arrhythmias, sudden death, progressive left ventricular remodeling, and systolic dysfunction (167–172). In the past decade, several PET studies (173–176) have demonstrated that in HCM patients the vasodilator response to dipyridamole and, hence, CFR are markedly impaired not only in the hypertrophied septum but also in the least hypertrophied left ventricular free wall. In the absence of coronary stenoses, this finding is indicative of a diffuse microvascular dysfunction, which is in line with the autoptic evidence of widespread remodeling of the intramural coronary arterioles (172,177). This inadequate hyperemic MBF response to demand in patients with HCM is clinically relevant in that it predisposes them to myocardial ischemia, which, in turn, has been implicated in the pathogenesis of syncope, abnormal blood-pressure response to exercise, left ventricular systolic dysfunction, and sudden death (164,178–180).
A recent PET study investigated the impact of myectomy on regional MBF in HCM (64). After surgical myectomy, septal CFR was significantly higher in surgically treated than in medically treated patients, although CFR in both groups remained significantly reduced compared with control subjects. Myectomy seems to exert its beneficial effect on CFR by reducing extravascular compressive forces as a result of improved diastolic relaxation, reduced force of contraction, and, thus, reduced compression of the coronary arteries.
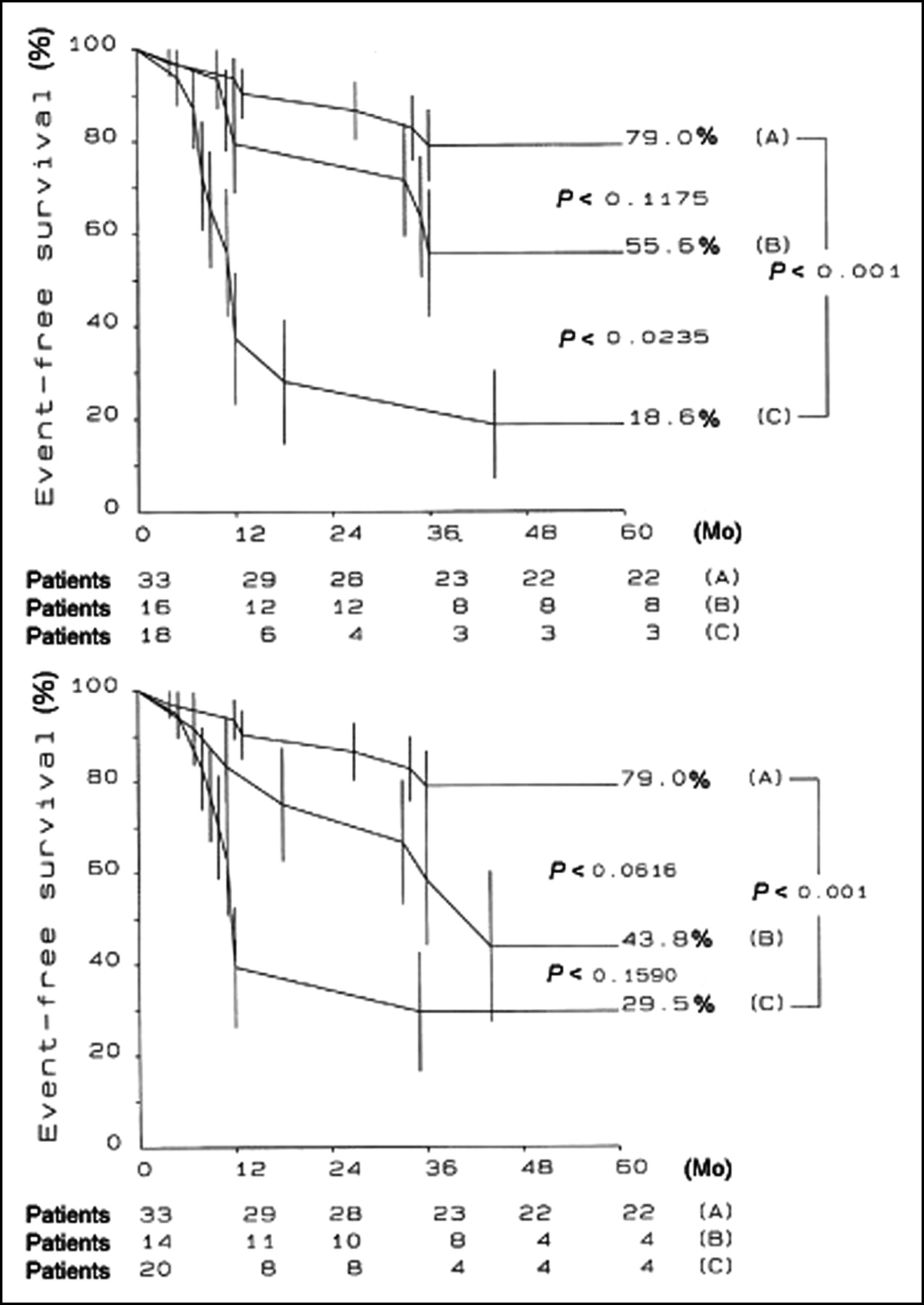
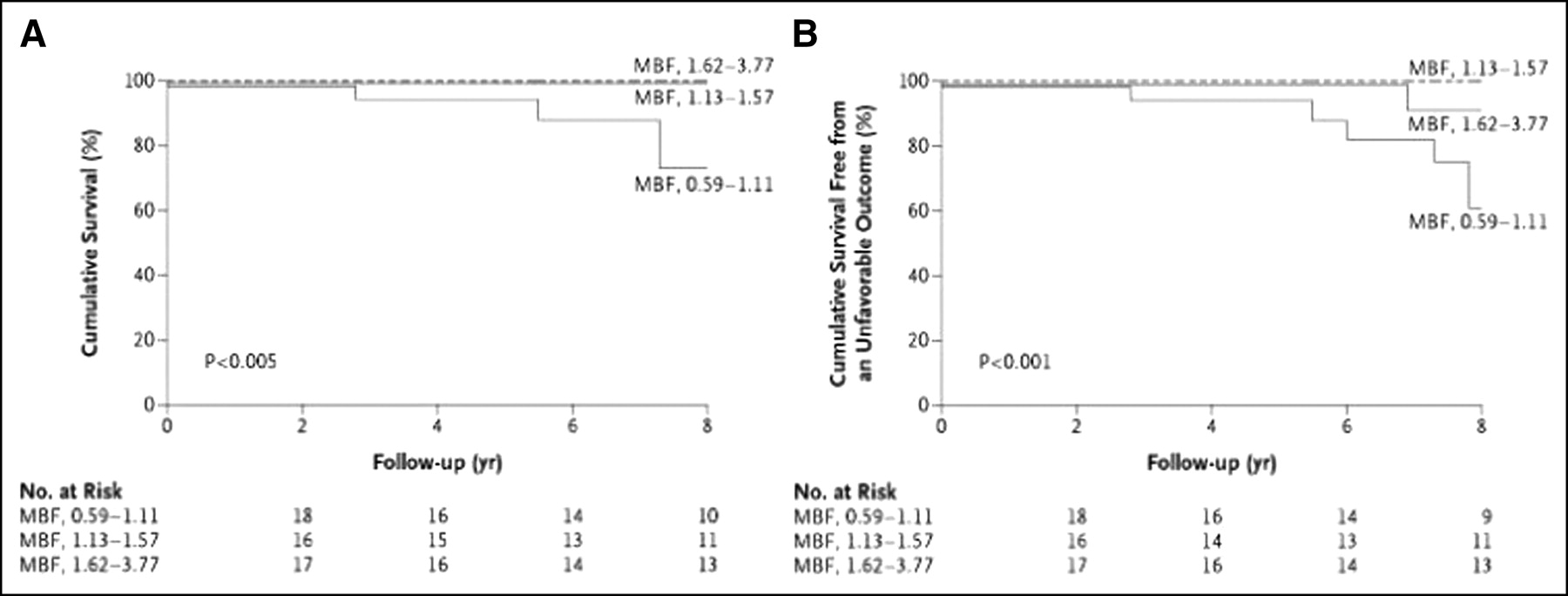
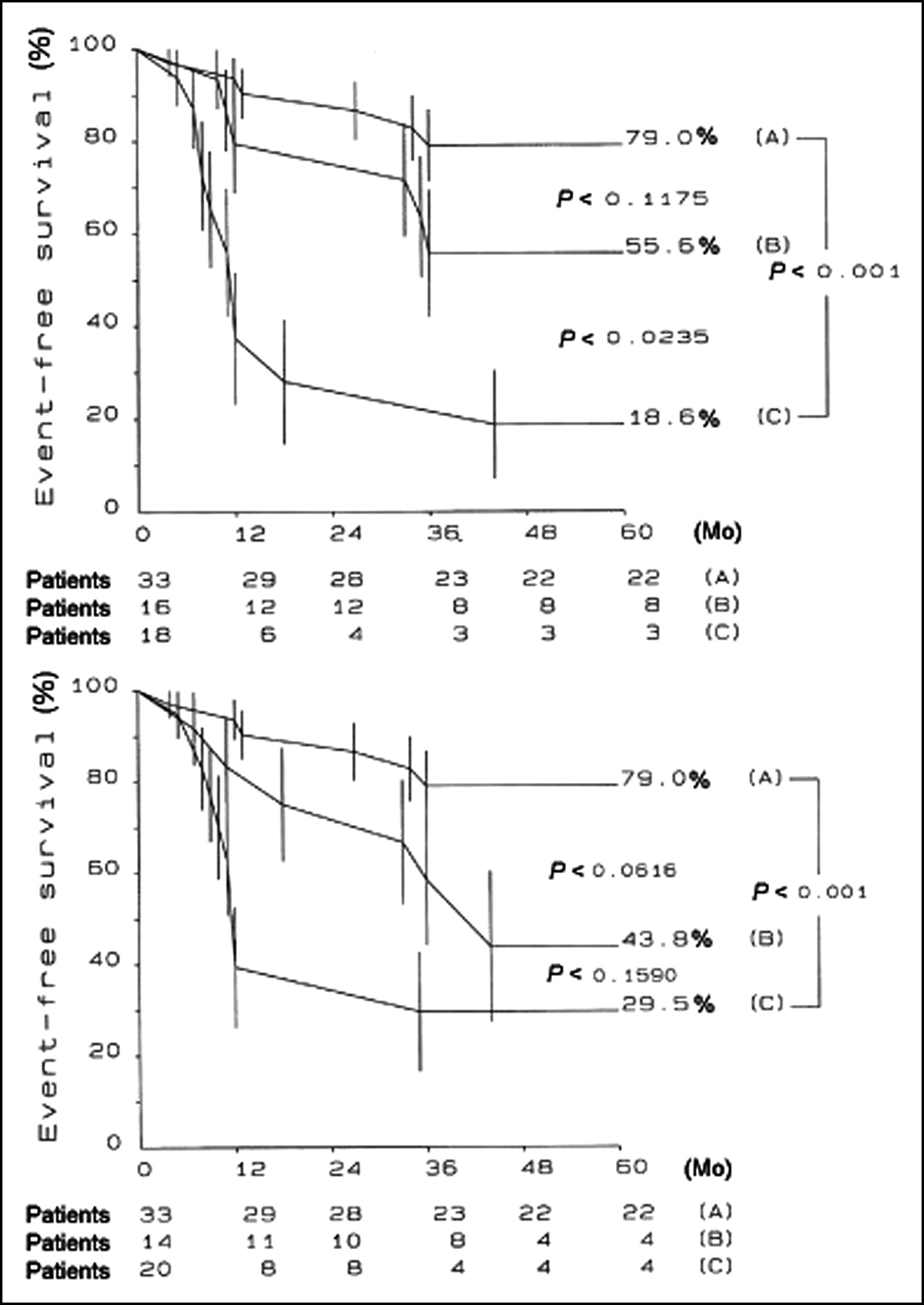
Several factors have been associated with an unfavorable outcome, but the identification of patients at risk for sudden death or progression to heart failure remains a formidable challenge (165,181,182). A recent PET study has documented that the severity of coronary microvascular dysfunction, assessed by PET, is an independent predictor of long-term clinical deterioration and death from cardiovascular causes in patients with HCM (62) (Fig. 4).

MBF values after dipyridamole infusion and long-term prognosis in HCM. Patients were divided into 3 equal groups according to MBF after dipyridamole infusion. (A) Overall cumulative suvival. (B) Cumulative survival free from unfavorable outcome. (Reprinted with permission of (62).)
Dilated Cardiomyopathy (DCM).
The incidence of DCM is reported to be about 5–8 cases per 100,000 population per year. In dilated DCM, impaired MBF at rest (183,184) and during hyperemia as well as impaired CFR (184–186) have been demonstrated in several studies (187). In a recent PET study, coronary microcirculatory dysfunction has been documented in patients with DCM as compared with healthy control subjects (188). Interestingly, in contrast to healthy volunteers, there was no correlation between PET findings of impaired coronary microcirculation and flow-mediated dilation of the brachial artery. This suggests that the assessment of endothelial function made in the peripheral circulation cannot necessarily be extrapolated to the coronary circulation and is in line with a previous report, which showed that peripheral flow response to transient arterial forearm occlusion does not reflect myocardial perfusion reserve either in healthy subjects or in patients with syndrome X or CAD (189). Impaired vasodilator capacity has been shown to be an independent predictor of subsequent cardiac events and is associated with an increased relative risk of death (Fig. 5) and further progression of heart failure (190).
(Top) Kaplan-Meier plots obtained in DCM patients with dipyridamole MBF of >1.36 mL·min−1·g−1 irrespective of New York Heart Association (NYHA) class (A) and in patients with dipyridamole MBF of ≤1.36 mL·min−1·g−1 subdivided according to absence (NYHA class I) (B) or presence (NYHA classes II and III) (C) of heart failure symptoms at enrollment. (Bottom) Kaplan-Meier plots obtained in patients with dipyridamole MBF of >1.36 mL·min−1·g−1 irrespective of left ventricular end-diastolic dimension (A) and in patients with dipyridamole MBF of ≤1.36 mL·min−1·g−1 subdivided according to left ventricular end-diastolic dimension of ≤60 mm (B) or >60 mm (C) at enrollment. (Reprinted with permission of (190).)
Isolated Ventricular Noncompaction (IVNC).
IVNC is thought to be a morphogenetic abnormality involving an arrest of compaction of the loose myocardial meshwork during fetal ontogenesis (191,192)—thus, present at birth in all patients (193,194). Heart failure, arrhythmia, embolism, and sudden cardiac death are common clinical manifestations of IVNC (195). Echocardiographic characteristics of IVNC have been validated at autopsy (196) and include, in the absence of any coexisting lesion, segmental thickening of the left ventricular myocardial wall consisting of 2 layers: a thin, compacted epicardial layer and an extremely thick endocardial layer with prominent trabeculations and deep recesses. IVNC has an estimated prevalence of 0.014% in patients referred to echocardiography. Abnormalities of MBF may play a crucial role in the pathophysiology IVNC, with ischemia possibly resulting from underlying abnormalities of the coronary microcirculation, as suggested by postmortem analysis of IVNC hearts with ischemic subendocardial lesions (195,196). A recent PET study (63) found a decreased hyperemic flow response to adenosine in IVNC. The microcirculatory dysfunction was not confined to the trabeculated segments but was also found in many nontrabeculated segments, indicating that the hypertrophy, per se, is unlikely to account for the decreased CFR, as the latter was also found in nonhypertrophic segments. In most of these segments, a wall motion abnormality was documented. By contrast, however, in most of the segments without a wall motion abnormality, CFR was preserved. This provides evidence that the microcirculatory dysfunction may be associated with wall motion abnormalities and, thus, with heart failure in patients with IVNC. The causal relationship between microcirculatory and contractility dysfunction in IVNC could not be identified with certainty from that study. Nevertheless, it appears reasonable to assume that the microvascular dysfunction might be responsible for the contractile impairment, particularly during high-demand situations, explaining the subendocardial scar, as found in histologic preparations of patients with IVNC (196), and reflected by fixed ammonia defects, despite no evidence of a previous myocardial infarction.
Microvascular Disease in Secondary Cardiomyopathies
Hypertensive Heart Disease.
Abnormal CFR, despite angiographically normal coronary arteries, has been demonstrated in several studies in patients with essential hypertension (197–199). This observation has often been attributed to the effects of left ventricular hypertrophy secondary to hypertension. These include increased extravascular compressive forces with elevated systolic or diastolic wall stress and impaired relaxation and structural alterations such as myocyte hypertrophy, interstitial fibrosis, and rarefaction of coronary microvasculature leading to reduced MBF (198). However, impairment of CFR in hypertensive patients is not necessarily related to the presence or degree of left ventricular hypertrophy (200). Impairment of CFR was found to be mainly caused by a reduction in the vasodilating capacity of the coronary resistance vessels rather than by effects linked to hypertrophy alone. This may be a consequence of vascular remodeling, such as media thickening, perivascular fibrosis, or functional alterations linked to endothelial dysfunction (201–204). The duration and severity of hypertension may have an important effect on CFR (205). A PET study has provided new insights into the complex interactions among hypertension, left ventricular hypertrophy, and impaired CFR (206). The authors found a significantly impaired CFR in hypertensive patients compared with normotensive control subjects. The global impairment, however, was not directly linked to the presence or degree of left ventricular hypertrophy as assessed by echocardiography. In addition, the abnormality of hyperemic MBF was found to be regionally heterogeneous. In patients with heterogeneous regional MBF, the flow impairment affected only a few areas, whereas others seemed unaffected. By contrast, in patients with homogenous MBF, the whole myocardium displayed a reduced CFR, possibly indicating a more advanced stage of the disease. The presence of left ventricular hypertrophy represented an indicator for those hypertensive patients who showed a stress-induced heterogeneous MBF. Local functional alterations such as hemodynamic pressure overload and local vasoactive substances may play an important role in the genesis of regional perfusion abnormalities. The PET study by Gimelli et al. (206) may provide some explanations for the well-known evidence of increased incidence of sudden cardiac death and arrhythmias in hypertensive patients with left ventricular hypertrophy. It is possible that regionally impaired vasodilating response may predispose to abnormal patterns of myocardial electrical depolarization and repolarization or regional myocardial ischemia, or both, during high flow demand conditions. Such a substrate could represent a focus for inducing clinically relevant arrhythmias. Thus, spatial flow heterogeneity during pharmacologic coronary vasodilation as assessed by PET, most often observed in hypertrophic hearts, may be the pathophysiologic mechanism of malignant arrhythmias.
Aortic Stenosis.
Development of left ventricular hypertrophy in patients with aortic valve stenosis (AS) is an adaptive response, which attempts to reduce wall stress in the left ventricle (207). Development of left ventricular hypertrophy also affects the coronary circulation, and patients with AS have a reduced CFR despite angiographically normal coronary arteries (208). This impairment of CFR is mainly due to a curtailment in maximal MBF (209). Hyperemia is hindered by a series of unfavorable hemodynamic changes, including high left ventricular cavity pressure, low coronary perfusion pressure, and increased extra-vascular compressive forces that lead to increased minimal coronary resistance (210). In addition, characteristic pathologic changes that contribute to impaired microvascular function have been described in the hypertrophied ventricle of patients with AS. These consist of perimyocytic fibrosis (211) and reduction in the number of resistance vessels per unit weight.
A recent PET study, using a state-of-the-art camera, has demonstrated that CFR was more severely impaired in the left ventricular subendocardium in patients with cardiac hypertrophy due to severe AS. Severity of impairment was related to the aortic valve area, hemodynamic load imposed, and diastolic perfusion time (DPT) rather than to the left ventricular mass (212). Furthermore, in a subsequent study, the same authors demonstrated that changes in coronary microcirculatory function in patients with AS after aortic valve replacement are not directly dependent on regression of the left ventricular mass. Reduced extravascular compression and increased DPT are proposed as the main mechanisms for improvement in MBF and CFR after aortic valve replacement (213).
CORONARY ARTERY DISEASE (CAD)
Functional Significance of Coronary Stenoses
The ability to make quantitative measurements of MBF with PET allows determination of the functional significance of epicardial coronary lesions. In patients with single-vessel CAD, chronic stable angina, and no previous history of myocardial infarction, CFR in response to a standard dose of dipyridamole was found to be markedly reduced in the myocardial regions supplied by the stenosed coronary artery compared with those regions supplied by angiographically normal vessels (214).
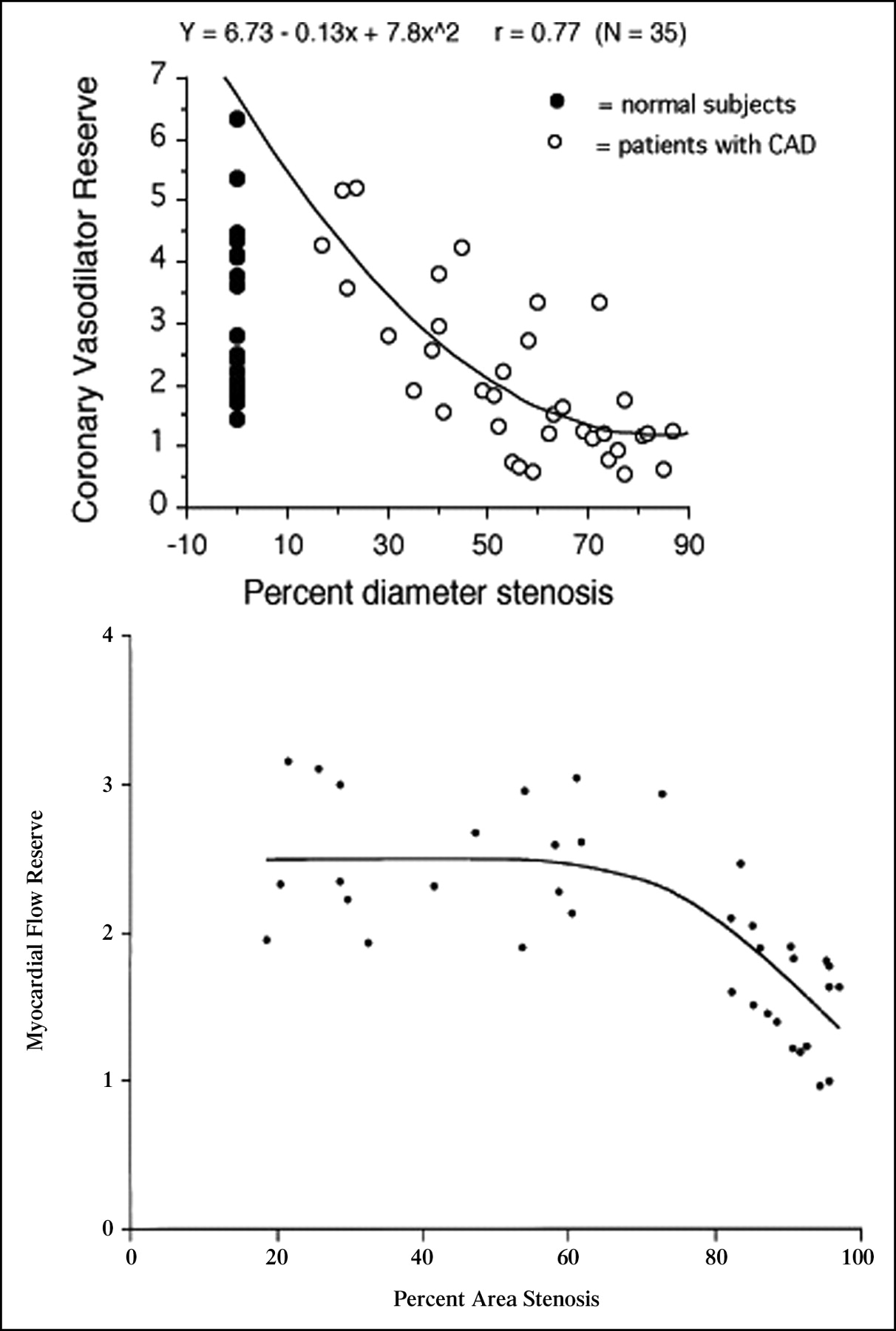
Other studies using H215O (61) or 13NH3 (215) with PET have evaluated the relationship between stenosis severity, measured by quantitative coronary angiography, and regional MBF and CFR. Different from the canine model (216,217), one study (61) showed that in humans resting MBF was preserved up to 95% diameter stenosis. Similar to the studies in dogs, the hyperemic response to dipyridamole and adenosine became attenuated at >40% diameter stenosis and was abolished at >80% stenosis (61,215) (Fig. 6). Although the inverse relation between stenosis severity and CFR was highly significant, a certain degree of variability was observed mainly at stenoses of intermediate severity. Variability was significantly less when minimal coronary resistance was plotted against stenosis severity, indicating the importance of accounting for interindividual differences in perfusion pressure (61).

(Top) No significant association was found between MBF at rest and degree of stenosis. Hyperemic MBF decreased significantly as stenosis severity increased. (Reproduced with permission of (61).). (Bottom) Scatterplot of relation between myocardial flow reserve and quantitative coronary angiographic measurements of percent area stenosis (r = 0.78, root mean square error = 0.61, P < 0.00001). (Reprinted with permission of (215).)
The relationship between regional MBF, measured with 13NH3 and PET, and regional wall motion, assessed by echocardiography, has been investigated in patients after dipyridamole infusion (218). Dipyridamole MBF in regions subtended by stenotic vessels was lower in the presence of inducible wall motion abnormalities compared with regions without dysfunction.
“Normal” Myocardium Remote from Ischemia
Measurement of MBF and CFR in remote, normally contracting myocardium supplied by angiographically normal coronary arteries in patients with a significant CAD elsewhere has produced mixed results (214,219–221). In one study dipyridamole CFR measured with H215O and PET was 2.7 ± 0.9 in remote myocardium compared with 4.1 ± 1.0 in healthy subjects (219). Similarly, in another study dipyridamole CFR measured with 13NH3 was 2.3 ± 0.8 in remote myocardium compared with 3.6 ± 0.9 (P < 0.01) in healthy subjects (214). Recently, reduced CFR in anatomically normal coronary arteries due to elevated baseline MBF in men with old myocardial infarction has been reported (221). However, Fujiwara et al. found preserved CFR in regions supplied by angiographically normal coronary arteries with 1-vessel CAD without coronary risk factors (220).
It is possible that angiographically undetectable atherosclerosis results in endothelial dysfunction, which could be responsible for a blunted CFR long before plaque starts to encroach on the luminal area of the vessel (222). This hypothesis is supported by a recent study evaluating CFR in asymptomatic subjects with hyperlipidemia and family history of CAD (58). Similar data were reported in asymptomatic males with hyperlipidemia in whom cardiovascular conditioning, including dietary changes and lowering of plasma cholesterol levels, did improve coronary vasodilator capacity (68).
MBF in Chronically Dysfunctional Myocardium in Patients with CAD
The debate on whether resting MBF to hibernating myocardium is reduced has attracted much interest and has contributed to stimulate new research on heart failure in patients with CAD. PET with H215O or 13NH3 has been used for the absolute quantification of regional MBF in human hibernating myocardium. When hibernating myocardium is properly identified—that is, a dysfunctional segment subtended by a stenotic coronary artery that improves function on coronary revascularization—the following conclusions can be reached based on the available literature (54): (a) in the majority of these studies resting MBF in hibernating myocardium is not different from either flow in remote tissue in the same patient or MBF in healthy volunteers; (b) a reduction in MBF of ∼20% compared with MBF in remote myocardium or age-matched healthy subjects has been demonstrated in a minority of truly hibernating segments; (c) hibernating myocardium is characterized by a severely impaired CFR that improves after revascularization in parallel with contractile function. Thus, the pathophysiology of hibernation in humans is more complex than initially postulated. The recent evidence that repetitive ischemia in patients with CAD can be cumulative and lead to more severe and prolonged stunning lends further support to the hypothesis that, at least initially, stunning and hibernation are 2 facets of the same coin.
CONCLUSION
Cardiac PET with 13NH3 or H215O is a mature, robust, and reproducible technique to obtain noninvasive quantitative measurements of MBF and CFR in humans in vivo. Like radioactive microspheres in animals, PET has provided a wealth of new information in the field of cardiac physiology and pathophysiology and remains the gold standard against which new techniques should be tested.
Acknowledgments
This work was supported by grant PP00A-68835 from the Swiss National Science Foundation.
Footnotes
Received Apr. 15, 2004; revision accepted Sep. 13, 2004.
For correspondence or reprints contact: Paolo G. Camici, MD, Medical Research Council Clinical Sciences Centre, Imperial College, Hammersmith Hospital, Du Cane Rd., London W12 0NN, United Kingdom.
E-mail: paolo.camici{at}csc.mrc.ac.uk
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Determining Hemodynamically Significant Coronary Artery Disease: Patient-Specific Cutoffs in Quantitative Myocardial Blood Flow Using [15O]H2O PET Imaging
- Effect of Sex Difference of Coronary Microvascular Dysfunction on Long-Term Outcomes in Deferred Lesions
- Sex Differences in Coronary Microvascular Dysfunction and Its Relationship With Outcome
- Sex Differences in Coronary Microvascular Function in Individuals With Type 2 Diabetes
- Invasive minimal Microvascular Resistance Is a New Index to Assess Microcirculatory Function Independent of Obstructive Coronary Artery Disease
- Zooming in on the Microvasculature in Heart Failure With Preserved Ejection Fraction
- Myocardial Perfusion Reserve and Strain-Encoded CMR for Evaluation of Cardiac Allograft Microvasculopathy
- Effect of Sex Differences on Invasive Measures of Coronary Microvascular Dysfunction in Patients With Angina in the Absence of Obstructive Coronary Artery Disease
- [18F]Fluorodeoxyglucose Uptake in Atherosclerotic Plaques Is Associated With Reduced Coronary Flow Reserve in Mice
- Quantitative Assessment of Myocardial Perfusion in the Detection of Significant Coronary Artery Disease: Cutoff Values and Diagnostic Accuracy of Quantitative [15O]H2O PET Imaging
- Angina Pectoris and Myocardial Ischemia in the Absence of Obstructive Coronary Artery Disease: Practical Considerations for Diagnostic Tests
- Invasive measurement of coronary microvascular resistance in patients with acute myocardial infarction treated by primary PCI
- Post-Traumatic Stress Disorder and Incidence of Coronary Heart Disease: A Twin Study
- Diagnostic Value of 13N-Ammonia Myocardial Perfusion PET: Added Value of Myocardial Flow Reserve
- Spin-Labeling Magnetic Resonance Imaging Detects Increased Myocardial Blood Flow After Endothelial Cell Transplantation in the Infarcted Heart
- Habitual dietary sodium intake is inversely associated with coronary flow reserve in middle-aged male twins
- Clinical Value of Absolute Quantification of Myocardial Perfusion With 15O-Water in Coronary Artery Disease
- Very high coronary calcium score unmasks obstructive coronary artery disease in patients with normal SPECT MPI
- Simplified Quantification of Myocardial Flow Reserve with flurpiridaz F 18: Validation with Microspheres in a Pig Model
- Inflammation is Related to Coronary Flow Reserve Detected by Positron Emission Tomography in Asymptomatic Male Twins
- Intrapericardial, But Not Extrapericardial, Fat Is an Independent Predictor of Impaired Hyperemic Coronary Perfusion in Coronary Artery Disease
- PET Assessment of Myocardial Perfusion Reserve Inversely Correlates with Intravascular Ultrasound Findings in Angiographically Normal Cardiac Transplant Recipients
- Relationship Between Regional Myocardial Oxygenation and Perfusion in Patients With Coronary Artery Disease: Insights From Cardiovascular Magnetic Resonance and Positron Emission Tomography
- Long-Term Prognostic Value of 13N-Ammonia Myocardial Perfusion Positron Emission Tomography: Added Value of Coronary Flow Reserve
- The Clinical Value of Myocardial Blood Flow Measurement
- Evaluation of the Novel Myocardial Perfusion Positron-Emission Tomography Tracer 18F-BMS-747158-02: Comparison to 13N-Ammonia and Validation With Microspheres in a Pig Model
- A Novel 18F-Labeled Tracer for the Quantification of Myocardial Blood Flow and Infarct Size With Positron-Emission Tomography: Another Way to Avoid the Need of an On-Site Cyclotron
- Impaired Tissue Perfusion: A Pathology Common to Hypertension, Obesity, and Diabetes Mellitus
- Spatial Relationship Between Coronary Microvascular Dysfunction and Delayed Contrast Enhancement in Patients with Hypertrophic Cardiomyopathy
- Initial Characterization of an 18F-Labeled Myocardial Perfusion Tracer
- Improvement in coronary vascular dysfunction produced with euglycaemic control in patients with type 2 diabetes
- Typical Chest Pain and Normal Coronary Angiogram: Cardiac Risk Factor Analysis Versus PET for Detection of Microvascular Disease
- Systolic and Diastolic Dyssynchrony in Patients With Diastolic Heart Failure and the Effect of Medical Therapy
- Smoking Cessation Normalizes Coronary Endothelial Vasomotor Response Assessed with 15O-Water and PET in Healthy Young Smokers
- Reproducibility of Measurements of Regional Myocardial Blood Flow in a Model of Coronary Artery Disease: Comparison of H215O and 13NH3 PET Techniques
- Novel Doppler Assessment of Intracoronary Volumetric Flow Reserve: Validation Against PET in Patients With or Without Flow-Dependent Vasodilation