


Abstract
The epidermal growth factor receptor (EGFR) is an attractive target for the design of radiotherapeutic agents for breast cancer because it is present on almost all estrogen receptor-negative, hormone-resistant tumors with a poor prognosis. In this study, we describe the antitumor effects and normal tissue toxicity of the novel Auger electron-emitting radiopharmaceutical 111In-labeled diethylenetriaminepentaacetic acid-human epidermal growth factor (111In-DTPA-hEGF) administered to athymic mice bearing EGFR-positive human breast cancer xenografts. Methods: Mice bearing subcutaneous MDA-MB-468 or MCF-7 human breast cancer xenografts were treated with 5 weekly doses of 111In-DTPA-hEGF (total, 27.7–92.5 MBq or 5–17 μg). Treatment was commenced 6 wk after tumor cell implantation (established tumors) or 1 wk after implantation (nonestablished tumors). Antitumor effects were assessed by use of the slope of the tumor growth curve. Normal tissue toxicity was assessed by use of plasma alanine transaminase and creatinine levels, hematologic indices (leukocytes, platelets, erythrocytes, and hemoglobin), histopathologic examination of the liver and kidneys, and changes in body weight. The uptake of 111In-DTPA-hEGF in tumors of different sizes (<5–200 mm3) was investigated, and microdosimetry estimates were calculated. Results: 111In-DTPA-hEGF exhibited strong antitumor effects against established MDA-MB-468 xenografts, decreasing their growth rate 3-fold compared with that in normal saline-treated mice (slopes, 0.0225 and 0.0737 d−1, respectively; P = 0.002). The antitumor effects of 111In-DTPA-hEGF were much more profound in mice with small, nonestablished MDA-MB-468 tumors, which regressed, than in saline-treated mice (slopes, −0.009 and 0.0297 d−1, respectively; P < 0.001). The growth of MCF-7 xenografts, with a 100-fold-lower level of EGFR expression, was modestly inhibited by 111In-DTPA-hEGF compared with that in saline-treated mice (slopes, 0.0250 and 0.0488 d−1, respectively; P = 0.051). There was a 1.4- to 2-fold decrease in leukocyte and platelet counts with 111In-DTPA-hEGF treatment, but these counts remained in the normal ranges. There was no change in other biochemical or hematologic parameters or body weight. There was no evidence of morphologic damage to the liver or kidneys. A strong inverse relationship was observed between radiopharmaceutical uptake and tumor size, with small tumors (<5 mm3) accumulating >30% of the injected dose (%ID) per gram, compared with 5 %ID/g for tumors measuring 6–30 mm3. Exceptionally high uptake (>80 %ID/g) was achieved in tumors measuring 1–2 mm3. Microdosimetry estimates indicated that the nucleus of an MDA-MB-468 cell would receive 90–1,400 cGy, depending on the level of radiopharmaceutical uptake. Conclusion: 111In-DTPA-hEGF exhibited strong antitumor effects against MDA-MB-468 breast cancer xenografts overexpressing EGFR. The highest tumor localization, radiation-absorbed doses, and growth inhibition were achieved for small, nonestablished tumors, suggesting that the radiopharmaceutical may be most valuable for the treatment of small-volume metastatic breast cancer or occult micrometastases in an adjuvant setting.
Improvements in the early diagnosis of breast cancer through mammographic screening of populations at risk have improved the prognosis for patients with operable disease, but the outlook for patients with disseminated breast cancer remains poor. Patients with estrogen receptor (ER)-positive tumors are candidates for hormonal therapy with tamoxifen or aromatase inhibitors, but they represent less than half of patients with advanced disease (1). Patients with ER-negative tumors may be treated with chemotherapy, but its effectiveness is severely restricted by dose-limiting toxicity to normal tissues and the development of multidrug resistance (2). The overexpression of the epidermal growth factor (EGF) receptor (EGFR) is inversely correlated with ER expression in breast cancer. More than 90% of ER-negative breast cancers express EGFR, and these represent hormone-resistant tumors with a poor prognosis (3). The EGFR is a 170-kDa transmembrane tyrosine kinase that specifically binds the 53-amino-acid peptide ligand EGF and the 50-amino-acid autocrine growth factor transforming growth factor α (TGFα) (4). The binding of EGF or TGFα to the receptor activates an intracellular signaling pathway that promotes cell division (5).
EGFR overexpression represents an attractive target for the design of novel anticancer therapeutic agents because the receptor has been implicated in the pathogenesis of many epithelial cell-derived tumors (6,7). Approaches to targeting EGFR include anti-EGFR monoclonal antibodies (mAbs) (e.g., C225), which block the binding of EGF to the extracellular ligand-binding domain of the receptor (8); specific tyrosine kinase inhibitors (e.g., Iressa), which interfere with signal propagation (9); or EGF- or TGFα-conjugated cell toxins, which exploit receptor-mediated ligand internalization to import highly potent inhibitors of protein synthesis into the cytoplasm of cancer cells (10).
We have been exploring the novel approach of using EGF as a specific vehicle for inserting the short-range Auger electron-emitting radionuclide 111In into EGFR-overexpressing breast cancer cells for targeted radiotherapy of the disease. Reilly et al. previously reported that 111In-labeled diethylenetriaminepentaacetic acid (DTPA)-human EGF (hEGF) (111In-DTPA-hEGF) was highly and selectively radiotoxic in vitro to MDA-MB-468 human breast cancer cells overexpressing EGFR, reducing their surviving fraction by more than 95% at <111–148 mBq per cell (11). The radiopharmaceutical was rapidly internalized into the cytoplasm of the cells, and some of the internalized 111In-DTPA-hEGF molecules were translocated to the cell nucleus, where the subcellular-range Auger electron emissions were damaging to DNA. 111In-DTPA-hEGF was not radiotoxic to MCF-7 human breast cancer cells, which exhibited a 100-fold-lower level of EGFR expression. Furthermore, in non-tumor-bearing mice administered large amounts of the radiopharmaceutical (3.7–111 MBq, equivalent on the basis of megabecquerels per square meter to amounts in humans of 740 MBq-21.3 GBq), there was no evidence of liver or kidney toxicity (11). The liver and kidneys exhibit moderate levels of EGFR (approximately 105 receptors per cell) (12,13), whereas most normal epithelial tissues have very low EGFR levels (<104 receptors per cell). Importantly, <3% of the bone marrow stem cell population has been reported to express EGFR (14).
In this study, we report for the first time the antitumor effects and normal tissue toxicity of 111In-DTPA-hEGF administered to mice implanted subcutaneously with MDA-MB-468 or MCF-7 breast cancer xenografts. Our results clearly demonstrate that the radiopharmaceutical has potent and selective tumor growth-inhibitory effects against EGFR-overexpressing breast cancer xenografts in vivo and is associated with minimal normal tissue toxicity. These findings suggest a potential future role for targeted Auger electron radiotherapy with 111In-DTPA-hEGF in the management of ER-negative and hormone-resistant advanced breast cancer in humans.
MATERIALS AND METHODS
Breast Cancer Cells
MDA-MB-468 and MCF-7 breast cancer cells were obtained from the American Type Culture Collection and were cultured in Dulbecco minimum essential medium (Ontario Cancer Institute) containing penicillin at 100 U/mL, streptomycin at 100 μg/mL, and l-glutamine at 2 mmol/L and supplemented with 10% fetal calf serum (Sigma Chemical Co.). MDA-MB-468 cells express 1 × 106–2 × 106 EGFRs per cell (15), and MCF-7 cells express 1 × 104 EGFRs per cell (16).
Radiopharmaceutical
hEGF (Upstate Biotechnology Inc.) was derivatized with DTPA and labeled to a high specific activity (5.6–7.4 MBq/μg; 3.4 × 104–4.4 × 104 MBq/μmol) with 111In-acetate as previously described (17). The radiochemical purity of 111In-DTPA-hEGF was 95%–98%, as measured by instant thin-layer chromatography (ITLC-SG; Pall Corp.) with sodium citrate at 100 mmol/L (pH 5.0). 111In-DTPA-hEGF exhibited preserved receptor-binding properties against MDA-MB-468 cells in a direct radioligand-binding assay (Ka, 7.5 × 108 L/mol; Bmax, 1.3 × 106 EGFRs per cell) (16). 111In-DTPA-hEGF was sterilized by filtration through a Millex GV 0.22-μm-pore-size filter (Millipore Corp.).
Effects of Tumor Size on Radiopharmaceutical Uptake
Ten female athymic mice were injected subcutaneously at multiple sites with 5 × 105 to 1 × 107 MDA-MB-468 human breast cancer cells in 100 μL of culture medium. After 4 wk, tumors of different sizes (diameter, 2–7 mm; volume, 5–200 mm3) were established (4 or 5 tumors per animal). The mice were then injected subcutaneously (at a site remote from that of tumor implantation) with 1.85 MBq (0.3 μg) of 111In-DTPA-hEGF. At 24 h after injection, the mice were sacrificed by cervical dislocation. The tumors were excised and weighed, and counts for tumors along with those for a standard of the injected radiopharmaceutical were obtained with a γ-counter (Cobra Quantum; Packard). For very small tumor xenografts, multiple tumors were combined and weighed, and the average weight and radioactivity were determined. The tumor uptake of 111In-DTPA-hEGF was expressed as a percentage of the injected dose (%ID) per gram, and the relationship between radiopharmaceutical uptake and tumor size was examined. All animal studies were conducted under a protocol approved by the University Health Network Animal Care Committee (number 94-036) and in accordance with Canadian Council on Animal Care guidelines.
Pharmacokinetics of 111In-DTPA-hEGF After Subcutaneous Injection
The pharmacokinetics of 111In-DTPA-hEGF (1.85 MBq; 0.3 μg) administered by subcutaneous injection were studied with 3 non-tumor-bearing athymic mice anesthetized by subcutaneous injection of ketamine:xylazine:acepromazine (2.5 mg:0.12 mg:0.025 mg per mouse). Blood samples (22 μL) were collected in heparinized microcapillary tubes (Fisher Scientific Co.) at 5, 10, 20, and 30 min and at 1, 3, 6, and 24 h after radiopharmaceutical injection by nicking the tail with a sterile scalpel blade. Counts for blood samples were obtained with a γ-counter, and the concentration of 111In (counts per minute per microliter) was determined. The elimination phase for the blood radioactivity-versus-time curve was fitted to a 2-compartment pharmacokinetic model by use of Prism software (GraphPad Software Inc.) (18) and standard pharmacokinetic parameters: distribution half-life, elimination half-life, volume of distribution for the central compartment (V1), volume of distribution at steady state (Vss), and systemic clearance (CLs) were calculated. For comparison, in a separate experiment, the blood and normal tissue concentrations of radioactivity at 24 h after intravenous (tail vein) or subcutaneous injection of 111In-DTPA-hEGF (1.85 MBq; 0.3 μg) into athymic mice were compared.
Treatment of Mice with MDA-MB-468 or MCF-7 Breast Cancer Xenografts
Female athymic mice were injected subcutaneously at multiple sites with 5 × 106 MDA-MB-468 breast cancer cells in 100 μL of culture medium. After 6 wk, established subcutaneous tumors were visible (diameter, 3 mm; volume, 14–15 mm3). Groups of 5 or 6 mice bearing a total of 15 tumors per group were treated with 5 weekly subcutaneous injections of 5.5 MBq (1.0 μg), 11.1 MBq (2.0 μg), or 18.5 MBq (3.4 μg) of 111In-DTPA-hEGF or normal saline (control). The total amounts of 111In-DTPA-hEGF administered were 27.7 (5.0 μg) to 92.5 (17 μg) MBq. The tumor diameter (d) was measured every 2–3 d for 7 wk by use of a precision caliper, and the tumor volume was calculated as (4/3)π(d/2)3. A tumor growth index was calculated by dividing the tumor volume at each time point by the initial tumor volume. The mean tumor growth indices were plotted against the time since the start of treatment to obtain the tumor growth curve.
In a separate experiment, female athymic mice bearing smaller but established subcutaneous MDA-MB-468 xenografts (volume, 4–5 mm3) were treated with a total of 92.5 MBq (3.4 μg) of 111In-DTPA-hEGF, unlabeled DTPA-hEGF (3.4 μg), or normal saline in 5 divided weekly doses. The tumor diameter was measured every 2–3 d, and the tumor volume and growth index were calculated. Normal tissue toxicity was evaluated by measuring body weight once per week and by obtaining peripheral blood cell counts and plasma alanine transaminase (ALT) and creatinine (CR) levels at the end of a 7-wk observation period. A body weight index was calculated by dividing the body weight at each time point by the initial body weight. Blood samples were collected from the tail vein into heparinized capillary tubes and pooled within each group to provide a sufficient volume for analysis. Plasma was obtained by centrifuging the blood samples at 1,000g (3,000 rpm) for 5 min. Peripheral blood cell counts and hemoglobin, plasma ALT, and CR levels were measured at the Clinical Biochemistry Laboratory at the Hospital for Sick Children (Toronto, Ontario, Canada). Finally, the mice were sacrificed, and the morphology of the liver and kidneys was examined by a clinical pathologist by light and electron microscopy.
The effects of early treatment with 111In-DTPA-hEGF on the growth of MDA-MB-468 breast cancer xenografts were determined with groups of 5 mice bearing a total of 15 or 16 tumors (volume, 10 mm3). Radiopharmaceutical treatment was started 1 wk after tumor implantation (these tumors are referred to hereafter as nonestablished). The mice received 5 weekly subcutaneous injections of 111In-DTPA-hEGF (18.5 MBq or 3.4 μg each) or normal saline (control). The tumor diameter was measured every 2–3 d, and the tumor volume and growth index were determined. The mean tumor growth index was plotted against the time since the start of treatment to obtain the tumor growth curve.
MCF-7 breast cancer xenografts were established with groups of 5 or 6 athymic mice by subcutaneous injection of 107 MCF-7 cells at multiple sites. The mice were implanted with an intradermal 60-d sustained-release β-estradiol pellet (Innovative Research of America) required for growth of the MCF-7 tumors. After 6 wk, a total of 9 or 10 tumors (volume, 11–17 mm3) was established in each group of animals. The mice were then treated with 5 weekly doses of 18.5 MBq (3.4 μg) of 111In-DTPA-hEGF (total amount, 92.5 MBq or 17 μg) or normal saline (control). Tumor diameter was measured every 2–3 d for 7 wk, and the tumor volume was determined. The tumor growth index was calculated, and the mean tumor growth indices were plotted against the time since the start of treatment to obtain the tumor growth curve.
Tumor Microdosimetry Estimates for 111In-DTPA-hEGF
The radiation-absorbed dose for 111In-DTPA-hEGF in the nucleus in MDA-MB-468 cells was estimated on the basis of a tumor uptake of 5, 30, or 80 %ID/g, representing the range observed for tumors with a volume of 1–2, 5, or 6–30 mm3, respectively (see below). A total administered dose of 92.5 MBq of 111In-DTPA-hEGF was assumed, and the cumulative radioactivity in the tumor (becquerels·seconds) was calculated by dividing the uptake in 1 g of tumor (becquerels) by the decay constant for 111In (3 × 10−6 s−1) and by assuming rapid accumulation and no biologic elimination of radioactivity from the tumor. Earlier biodistribution studies (16) had shown that 111In-DTPA-hEGF was rapidly accumulated by MDA-MB-468 xenografts (maximum uptake within 4 h after injection) and was stably retained in the tumors for at least 72 h. The cumulative radioactivity in each MDA-MB-468 cell (becquerels·seconds) was calculated by assuming that 1 g of tumor with a spheric geometry contains 2 × 109 cells, on the basis of a tissue density of 1 g/cm3 and a cellular volume of 5 × 10−10 cm3 (diameter, 10−3 cm). The reported subcellular distribution for 111In-DTPA-hEGF in MDA-MB-468 cells in vitro (11) was used to determine the cumulative radioactivity in each source compartment (cell surface, cytoplasm, and nucleus) (ÃS; becquerels·seconds). The mean radiation-absorbed dose in the nucleus (grays) was estimated by use of the cellular microdosimetry model of Goddu et al. (19) as ÃS times the mean radiation-absorbed dose in the nucleus per unit of cumulative radioactivity in a subcellular source compartment. The latter values for the cell surface, the cytoplasm, and the nucleus were 1.78 × 10−4, 3.18 × 10−4, and 60.30 × 10−4 Gy/Bq·s, respectively (19).
Statistical Comparisons
Data were expressed as mean ± SEM. Statistical comparisons of means were made by use of the Student t test (P ≤ 0.05). The rate of tumor growth in animals treated with 111In-DTPA-hEGF or control animals was determined from the slope of the tumor growth index-versus-time curve fitted by linear regression. Statistical comparisons of slopes were made by use of the F test (P < 0.05).
RESULTS
Effects of Tumor Size on Radiopharmaceutical Uptake
There was a strong inverse correlation between the tumor uptake of 111In-DTPA-hEGF and the tumor mass for small tumors (<5 mg) (Fig. 1). The mean tumor uptake of 111In-DTPA-hEGF in tumors of <5 mg (31.6 ± 7.5 %ID/g) was much higher than that in tumors of 6–30 mg (5.5 ± 0.5 %ID/g) (P < 0.001), and very small tumors (1–2 mg) showed an exceptionally high accumulation of the radiopharmaceutical (>80 %ID/g). There was also a small but significant decrease (data not shown) in 111In-DTPA-hEGF uptake in tumors of 31–200 mg (3.4 ± 0.5 %ID/g) (P = 0.028) compared with that in tumors of 6–30 mg. On the basis of the assumption of a spheric geometry and a tissue density of 1 mg/mm3, the range of tumor weights examined (<5 mg, 6–30 mg, and 31–200 mg) corresponded to tumors with diameters of <2, 2–4, and 4–7 mm and with volumes of <5, 6–30, and 31–200 mm3, respectively.
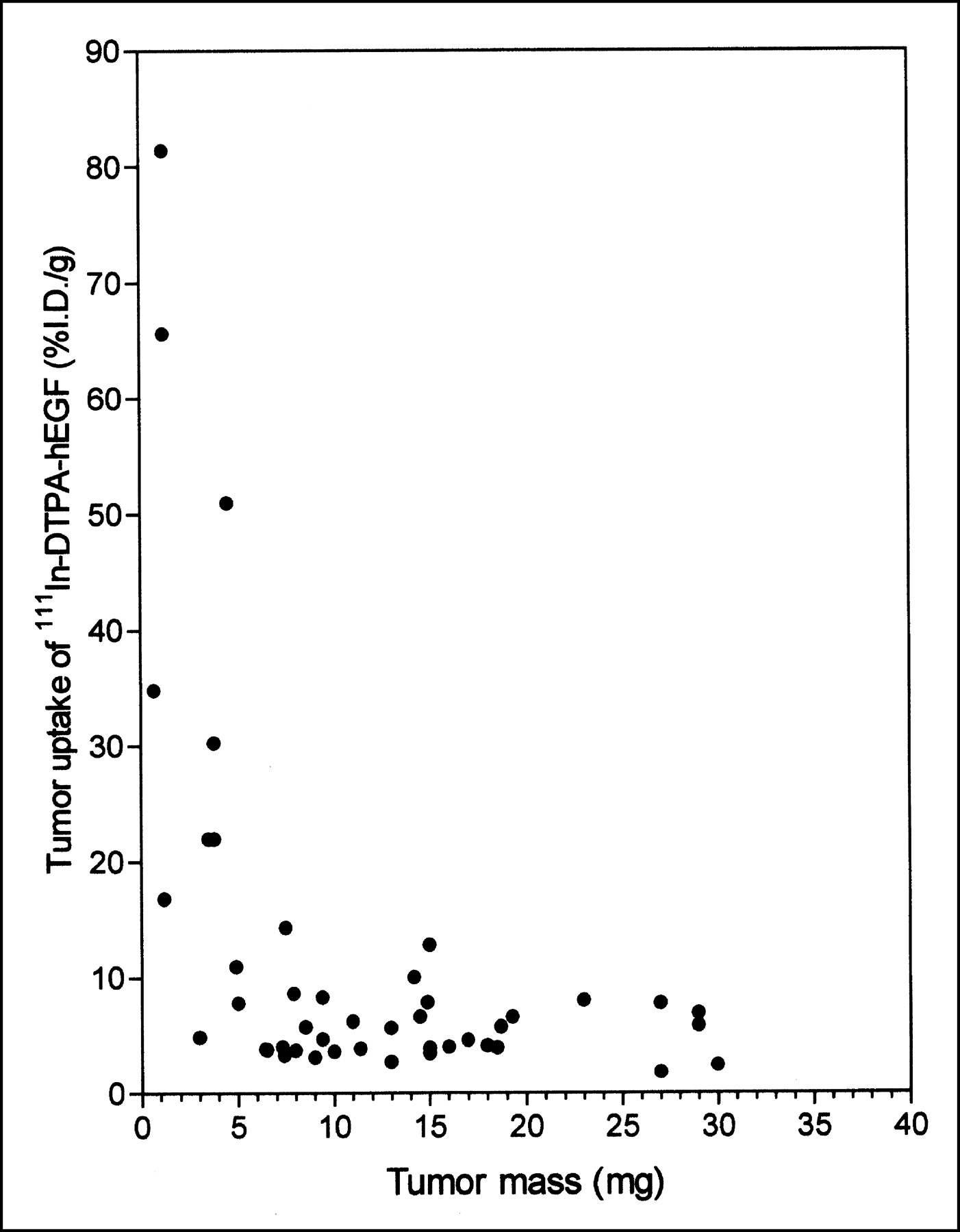
Relationship between tumor accumulation (%ID/g) and tumor mass for 111In-DTPA-hEGF at 24 h after subcutaneous administration to athymic mice bearing subcutaneous MDA-MB-468 human breast cancer xenografts. Site of radiopharmaceutical injection was remote from that of tumor implantation.
Pharmacokinetics of 111In-DTPA-hEGF After Subcutaneous Injection
111In-DTPA-hEGF was rapidly absorbed within 20 min after subcutaneous administration and quickly eliminated with biphasic kinetics (Fig. 2). The elimination phase was well described by a 2-compartment pharmacokinetic model. The α-phase (distribution) half-life was 1.5 h, and the β-phase (elimination) half-life was 146.7 h (Table 1). The V1 was 13.2 mL (528 mL/kg for a 25-g mouse), and the Vss was 34.2 mL (1,368 mL/kg). The V1 and Vss values were about 7 and 18 times higher, respectively, than the expected blood volume for a mouse (70–80 mL/kg) (20). V1 represents the volume in which the radiopharmaceutical is initially distributed (volume of the central compartment), whereas Vss represents the maximum volume of distribution. The CLs of 111In-DTPA-hEGF was 0.16 mL/h (6.4 mL/kg·h). There were no significant differences in the concentrations of radioactivity in the blood at 24 h after subcutaneous (0.17 ± 0.04 %ID/g) or intravenous (0.27 ± 0.05 %ID/g) administration of 111In-DTPA-hEGF (P = 0.141) (Table 2). Similarly, there were no significant differences between the 2 routes of administration in the liver or kidney uptake of 111In-DTPA-hEGF (Table 2).
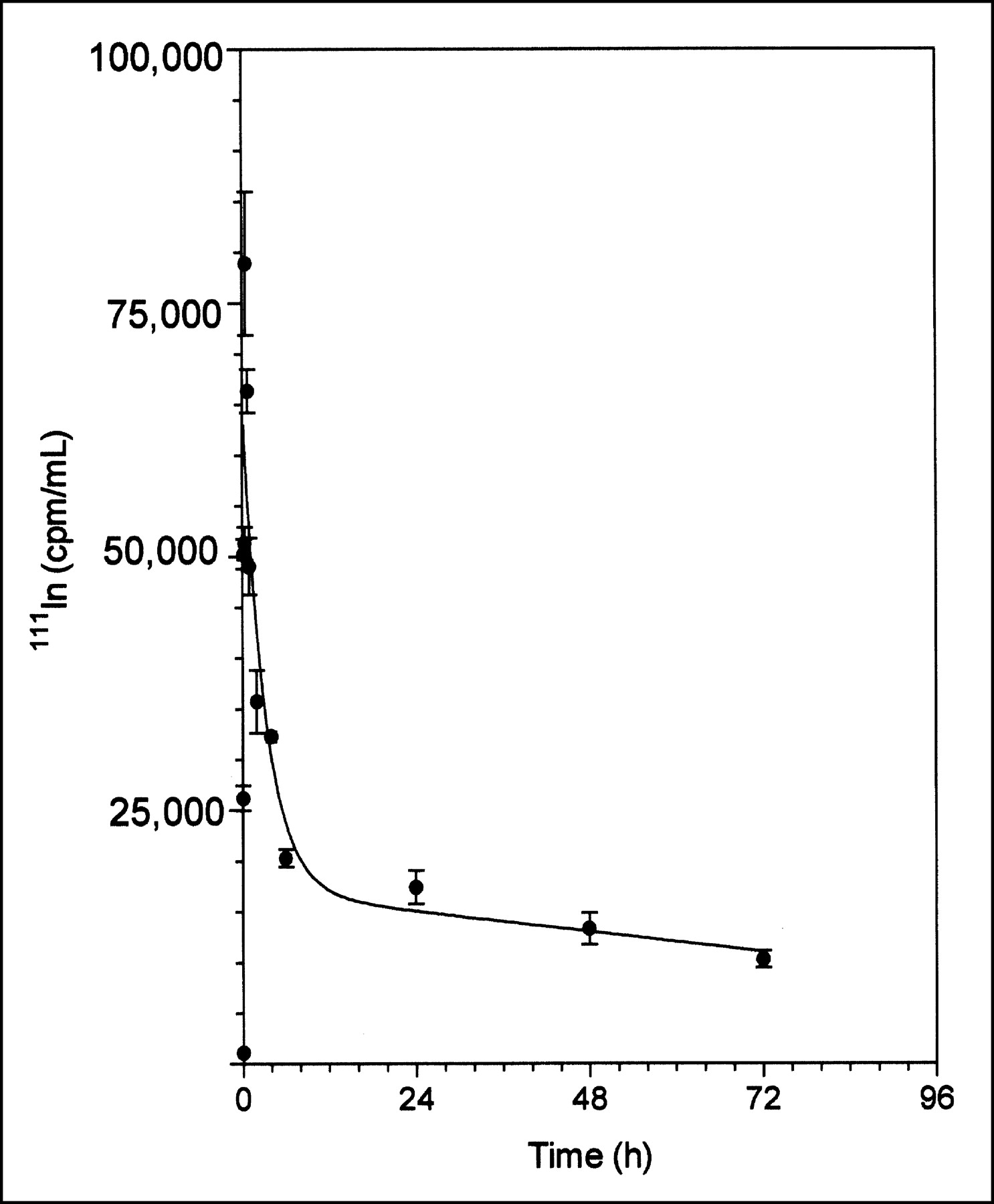
Absorption and elimination of 111In-DTPA-hEGF from blood after subcutaneous administration to non-tumor-bearing athymic mice. Error bars indicate SEMs.
Pharmacokinetic Parameters for 111In-DTPA-hEGF Administered Subcutaneously to Non-Tumor-Bearing Athymic Mice
Normal Tissue Accumulation of 111In-DTPA-hEGF 24 Hours After Subcutaneous or Intravenous Injection in Non-Tumor-Bearing Athymic Mice
Treatment of Mice with MDA-MB-468 or MCF-7 Breast Cancer Xenografts
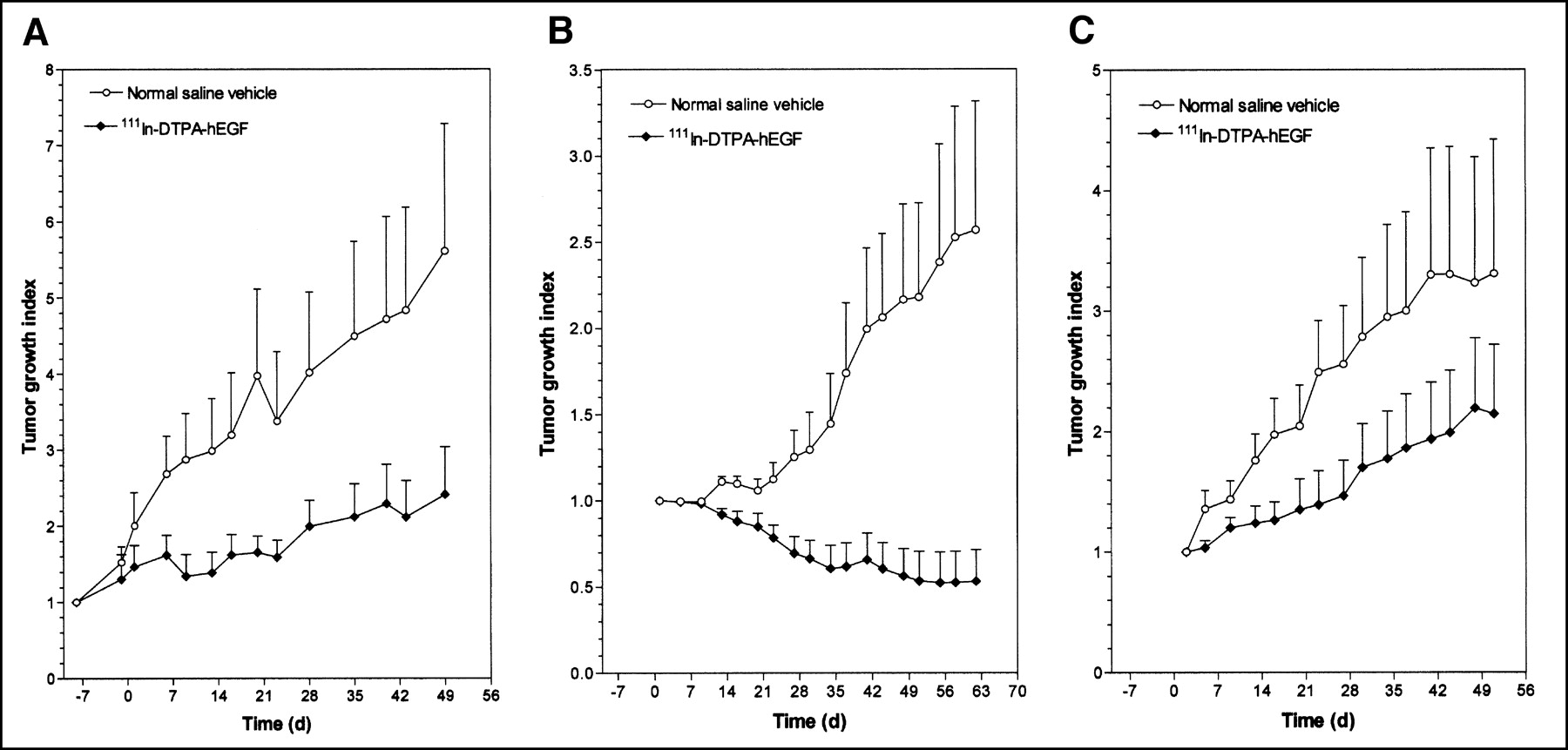
The effects of treatment of athymic mice with 5 weekly injections of 18.5 MBq (3.4 μg) of 111In-DTPA-hEGF (total, 92.5 MBq or 17 μg) on the growth of established MDA-MB-468 breast cancer xenografts (initial tumor volume, 14–15 mm3) are shown in Figure 3A. Linear regression analysis of the tumor growth curves (data not shown) revealed that the rate of tumor growth in mice treated with 111In-DTPA-hEGF was 3-fold less than that in control mice treated with normal saline (slopes of tumor growth curves, 0.0225 and 0.0737 d−1, respectively; F test; P = 0.002). Unlike established MDA-MB-468 tumors, the growth of which was inhibited by 111In-DTPA-hEGF, nonestablished tumors (initial volume, 10 mm3) treated with 111In-DTPA-hEGF showed regression (Fig. 3B) compared with tumors in normal saline-treated control mice, which exhibited rapid growth (slopes, −0.009 and 0.0297 d−1, respectively; F test; P < 0.001). The tumor growth indices at 62 d were 0.53 ± 0.18 for 111In-DTPA-hEGF treatment and 2.57 ± 0.75 for normal saline treatment. 111In-DTPA-hEGF appeared to inhibit the growth of MCF-7 breast cancer xenografts (Fig. 3C), but the difference in the slopes of the tumor growth curves for 111In-DTPA-hEGF-treated mice and normal saline-treated mice did not quite reach statistical significance (slopes, 0.0250 and 0.0488 d−1, respectively; F test; P = 0.051). The tumor growth indices at 51 d were 2.15 ± 0.57 for 111In-DTPA-hEGF treatment and 3.31 ± 1.11 for normal saline treatment.

Tumor growth index versus time (days) for athymic mice implanted with established 14- to 15-mm3 subcutaneous MDA-MB-468 human breast cancer xenografts (A), nonestablished 10-mm3 subcutaneous MDA-MB-468 breast cancer xenografts (B), or established 11- to 17-mm3 subcutaneous MCF-7 human breast cancer xenografts (C). Data were obtained after treatment with 5 weekly subcutaneous doses of 111In-DTPA-hEGF (cumulative dose, 92.5 MBq or 17 μg) or normal saline vehicle. Treatments were started on day 0. Site of radiopharmaceutical injection was remote from that of tumor implantation. Error bars indicate SEMs.
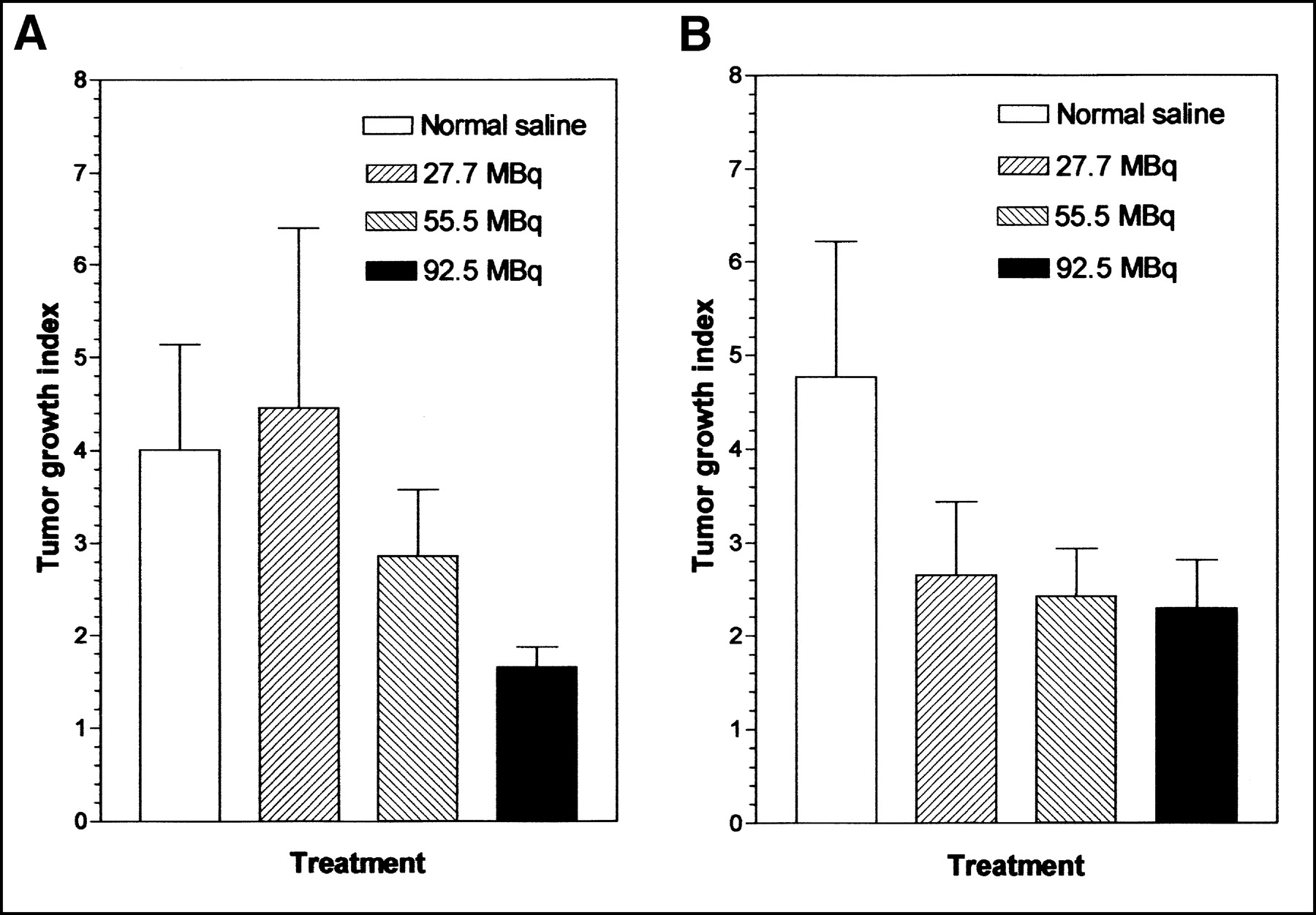
Tumor growth inhibition by 111In-DTPA-hEGF was dose related. The tumor growth indices at 20 d after the start of treatment for mice administered totals of 27.7, 55.5, and 92.5 MBq of 111In-DTPA-hEGF were 4.45 ± 1.94, 2.88 ± 0.69, and 1.65 ± 0.21, respectively, compared with 3.98 ± 1.14 for control mice treated with normal saline (Fig. 4A). The tumor growth indices at 49 d for mice treated with totals of 27.7, 55.5, and 92.5 MBq of 111In-DTPA-hEGF were 3.37 ± 0.98, 2.58 ± 0.64, and 2.42 ± 0.62, respectively, compared with 5.61 ± 1.67 for control mice treated with normal saline (Fig. 4B). In a separate experiment with mice bearing smaller but established MDA-MB-468 tumors (initial tumor volume, 4–5 mm3), 111In-DTPA-hEGF (92.5 MBq; 17 μg) strongly inhibited tumor growth compared with that in normal saline-treated control mice (slopes, 0.0112 and 0.0668 d−1, respectively; F test; P < 0.001) (Fig. 5A). DTPA-hEGF (17 μg) stimulated the growth of these small tumors (slope, 0.123 d−1). The tumor growth indices 50 d after the start of treatment with 111In-DTPA-hEGF, DTPA-hEGF, and normal saline were 1.54 ± 0.79, 6.34 ± 2.95, and 4.12 ± 1.43, respectively. There were no significant decreases in whole-body weight in mice treated with 111In-DTPA-hEGF, DTPA-hEGF, and normal saline over 50 d (Fig. 5B), suggesting no generalized normal tissue toxicity from the radiopharmaceutical. The body weight indices at 50 d were 1.05 ± 0.02, 1.06 ± 0.03, and 1.04 ± 0.02 for 111In-DTPA-hEGF, DTPA-hEGF, and normal saline treatments, respectively. There were also no significant differences in plasma ALT and CR levels in mice treated with 111In-DTPA-hEGF and normal saline (Table 3), suggesting no significant liver or renal toxicity from the radiopharmaceutical. These results were confirmed by histopathologic examination of the liver and kidneys by a clinical pathologist by light and electron microscopy, which revealed no evidence of morphologic changes (data not shown). There was a decrease in leukocyte (WBC) and platelet counts in mice treated with 111In-DTPA-hEGF compared with normal saline-treated mice (Table 3), but these counts remained in the normal ranges (20).

Tumor growth index for athymic mice with established MDA-MB-468 human breast cancer xenografts implanted subcutaneously at increasing doses of 111In-DTPA-hEGF. Data were obtained at 20 d (A) or 49 d (B) after subcutaneous administration of 5 weekly doses of 111In-DTPA-hEGF (total cumulative doses, 27.7–92.5 MBq or 5–17 μg) or normal saline vehicle. Error bars indicate SEMs.

Tumor growth index versus time (days) (A) or body weight index versus time (days) (B) for athymic mice implanted with small 4- to 5-mm3 established subcutaneous MDA-MB-468 human breast cancer xenografts. Data were obtained after treatment with 5 weekly subcutaneous doses of 111In-DTPA-hEGF (92.5 MBq; 17 μg). Treatments were started on day 0. Error bars indicate SEMs.
Clinical Biochemistry and Hematology Test Results in Athymic Mice Implanted with MDA-MB-468 Breast Cancer Xenografts and Treated with 111In-DTPA-hEGF
Tumor Microdosimetry Estimates for 111In-DTPA-hEGF
Estimates of the radiation-absorbed doses of 111In-DTPA-hEGF to the cell nucleus in MDA-MB-468 cells, assuming a tumor uptake of 5, 30, or 80 %ID/g, are shown in Table 4. Estimates of the radiation-absorbed doses in the cell nucleus were 88 cGy at 5 %ID/g, 529 cGy at 30 %ID/g, and 1,402 cGy at 80 %ID/g.
Tumor Microdosimetry Projections for 111In-DTPA-hEGF Administered Subcutaneously to Athymic Mice Bearing MDA-MB-468 Human Breast Cancer Xenografts
DISCUSSION
There is currently considerable interest in Auger electron-emitting radiopharmaceuticals as potential targeted radiotherapeutic agents for cancer because of the extremely short, subcellular range of the electrons, which theoretically restricts their radiotoxicity to cells that specifically bind to and internalize the radiopharmaceuticals (21). In this study, we demonstrated that hEGF labeled with the Auger electron emitter 111In has potent, dose-related, and selective tumor growth-inhibitory effects on human breast cancer xenografts overexpressing EGFR. Treatment of mice with 5 weekly doses of 111In-DTPA-hEGF (27.7–92.5 MBq; 5–17 μg) significantly decreased (F test; P = 0.002) the rate of growth of established MDA-MB-468 tumors (1 × 106–2 × 106 EGFRs per cell)—up to 3-fold—compared with that in normal saline-treated control mice. Furthermore, tumor regression was achieved for small, nonestablished tumors when treatment was started within 1 wk of tumor cell implantation. 111In-DTPA-hEGF (92.5 MBq; 17 μg) also appeared to have weaker antiproliferative effects (1.5-fold) on MCF-7 breast cancer xenografts, which exhibited a 100-fold-lower level of EGFR expression, but the magnitude of tumor growth rate inhibition compared with that in normal saline-treated mice did not quite reach statistical significance (F test; P = 0.051). The total doses of 111In-DTPA-hEGF administered to mice (27.7–92.5 MBq; 5–17 μg) corresponded to 5.3–17.8 GBq (0.9–3.3 mg) in humans, on the basis of body surface areas of 0.009 and 1.73 m2, respectively. These doses of radioactivity are lower than those (4.7–160 GBq) recently evaluated in a phase I trial of 111In-pentetreotide for targeted Auger electron radiotherapy of somatostatin receptor-positive tumors in humans (22).
The stronger antiproliferative effects of 111In-DTPA-hEGF on nonestablished MDA-MB-468 tumors were likely attributable to the higher accumulation of the radiopharmaceutical in these small tumors. Tumor uptake of 111In-DTPA-hEGF was approximately 5 %ID/g for tumors of 6–30 mm3; that for tumors of <5 mm3 was >30 %ID/g. Exceptionally high tumor uptake (up to 80 %ID/g) was achieved in very small tumors, of 1–2 mm3. Biodistribution studies of radiolabeled monoclonal antibodies (mAbs) in mice have similarly shown an inverse correlation between tumor accumulation and tumor size (23). Tumor accumulation is one of the major predictors of tumor response to radioimmunotherapy in mouse tumor xenograft models (24), and it appears from the present study that this may also be the case for small radiolabeled peptides, such as 111In-DTPA-hEGF.
Microdosimetry estimates for MDA-MB-468 tumors were calculated on the basis of the assumption that the intracellular distribution of the radiopharmaceutical in MDA-MB-468 cells in vivo was the same as that determined in vitro (i.e., 20% on the cell membrane, 65% in the cytoplasm, and 15% in the nucleus) (11). Microdosimetry estimates for a total administered dose of 111In-DTPA-hEGF of 92.5 MBq revealed that approximately 90 cGy would be delivered to the nucleus in an MDA-MB-468 cell on the basis of the assumption of a tumor uptake of 5 %ID/g, whereas a tumor uptake of 30 or 80 %ID/g would deliver >500 or >1,400 cGy, respectively (Table 4). Chen et al. recently determined that the 50% effective dose for the treatment of MDA-MB-468 cells in vitro with γ-radiation was approximately 400 cGy and that the 90% effective dose was 600 cGy (25). The Auger electron emissions from 111In may be more damaging than γ-radiation because of their high linear energy transfer characteristics and greater propensity to generate double-strand breaks in DNA (26). On the basis of the antiproliferative response to γ-radiation, a radiation-absorbed dose of 90 cGy would be expected to have minimal antiproliferative effects on MDA-MB-468 tumors, whereas a dose of 500 cGy would decrease the growth rate 2-fold and a dose of 1,400 cGy would decrease the growth rate >10-fold. The observed 3-fold decrease in the tumor growth rate in established MDA-MB-468 tumors after treatment with the largest amount (92.5 MBq) of 111In-DTPA-hEGF and the tumor regression observed for small, nonestablished tumors would be consistent with the projected microdosimetry estimates.
In a separate study, using the MIRD schema (27), we recently estimated the radiation-absorbed dose in normal tissues in humans on the basis of studies of the biodistribution of 111In-DTPA-hEGF in mice (unpublished data, November 2002). It was possible only to calculate macrodosimetry estimates for normal tissues because the subcellular distribution of 111In-DTPA-hEGF in normal cells is not known. The highest radiation-absorbed doses would be delivered to the spleen and kidneys (approximately 1 mGy/MBq), whereas the liver would receive 0.3 mGy/MBq and the whole-body radiation-absorbed dose would be approximately 0.07 mGy/MBq.
High concentrations of hEGF are inhibitory to the growth of MDA-MB-468 cells, and this effect is mediated by p21WAF-1/CIP-1-induced cell cycle arrest and apoptosis (15,28). The paradoxical antiproliferative effects of EGF on EGFR-overexpressing cancer cells have also been demonstrated for A431 epidermoid carcinoma cells (29) and for MX-1 and UM-1 breast cancer xenografts in athymic mice (30). 111In-labeled hEGF is much more cytotoxic, however, than unlabeled hEGF against MDA-MB-468 cells. In an earlier study, Chen et al. determined that the 50% inhibitory concentration (IC50) for DTPA-hEGF against MDA-MB-468 cells in vitro was 500 pmol/L and that the 90% inhibitory concentration (IC90) was 2 nmol/L (compared with IC50 and IC90 values of 70 and 200 pmol/L, respectively, for 111In-DTPA-hEGF) (25). On the basis of a V1 of 13.2 mL for 111In-DTPA-hEGF (Table 1), the initial concentration in blood after the administration of a single dose of the radiopharmaceutical (5.5–18.5 MBq; 1.0–3.4 μg) would be 13–43 nmol/L, approximately 26- to 86-fold higher than the IC50 and 6- to 21-fold higher than the IC90 for DTPA-hEGF. On the assumption of an α-phase half-life of 1.5 h (Table 1), the initial concentration of 111In-DTPA-hEGF in blood (nanomoles per liter) would exceed the IC50 of unlabeled DTPA-hEGF for 7–10 h and the IC90 for 4–8 h. These transiently high concentrations of circulating 111In-DTPA-hEGF also likely contributed to the antiproliferative effects of the radiopharmaceutical on MDA-MB-468 tumors.
Although theoretically targeted Auger electron-emitting radiopharmaceuticals require receptor-mediated internalization and nuclear translocation to produce a radiotherapeutic effect (because of the subcellular range of the electrons), it has been shown that radiation can induce a “bystander effect,” in which radiobiologically damaged cells convey messages to nonirradiated viable cells to induce cell death in these “bystanders.” This phenomenon is thought to be mediated through the release of cytokines and free radicals from the radiated cells. The bystander effect for γ-irradiated tumor cells can occur at radiation-absorbed doses as low as 1 cGy (31). Xue et al. (32) recently demonstrated a strong bystander effect in vivo for the Auger electron-emitting radiopharmaceutical 125I-iododeoxyuridine at radiation-absorbed doses as low as 2.3–6.9 cGy. It is possible that 111In-DTPA-hEGF targeted to EGFR-positive breast cancer cells similarly induces a bystander effect in neighboring nontargeted cells, thereby achieving a radiotherapeutic effect stronger than that expected from the microdosimetry estimates predicted by the Auger electron emissions. Such an effect may explain the tumor growth-inhibitory properties of the radiopharmaceutical at relatively modest radiation-absorbed doses (90–1,400 cGy). The bystander effect associated with Auger electron-emitting radiopharmaceuticals is an intriguing area that warrants further investigation.
There were no significant changes in body weight over 7 wk in mice administered a total of 92.5 MBq (17 μg) of 111In-DTPA-hEGF. There were no significant increases in the plasma ALT and CR levels, indicating no major toxicity to the liver or kidneys, the normal tissues of which have moderate levels of EGFR expression. Histopathologic examination confirmed that there were no morphologic changes characteristic of hepatotoxicity (steatosis, proliferation of smooth endoplasmic reticulum, or alterations in mitochondria) or renal toxicity (changes in renal tubular structure). These results are consistent with the previous finding (11) that high doses of 111In-DTPA-hEGF (up to 111 MBq; equivalent to 21.3 GBq in humans on the basis of megabecquerels per square meter) can be safely administered to mice. There were no changes in hemoglobin and erythrocyte counts in mice administered 111In-DTPA-hEGF, but there were moderate (1.4- to 2-fold) decreases in WBC and platelet counts compared with those in normal saline-treated control mice (Table 3). Nevertheless, the WBC and platelet counts remained in the normal ranges (20). Because <3% of the bone marrow stem cell population expresses EGFR (14), hematopoietic toxicity was not anticipated in this study. We hypothesize that the unexpected decreases in WBC and platelet counts may have been attributable to nonspecific radiotoxicity to bone marrow stem cells caused by the low linear energy transfer but penetrating γ-emissions of 111In (energy of γ-emissions, 172 and 247 keV). Behr et al. (33) similarly found that mAb CO17-1A, which does not specifically bind to bone marrow stem cells, was 23-fold less radiotoxic to bone marrow in mice when labeled with 111In than when labeled with 90Y because of the absence of a “cross-fire” effect for the Auger electron emissions; however, hematopoietic toxicity was nevertheless dose limiting for 111In-labeled CO17-1A, possibly because of the γ-radiation.
The explanation for the antiproliferative effects of 111In-DTPA-hEGF on MCF-7 xenografts is not clear, because MCF-7 cells are resistant to 111In-DTPA-hEGF in vitro. Again, the penetrating γ-emissions of 111In-DTPA-hEGF may have produced nonspecific radiotoxicity to MCF-7 cells. Govindan et al. (34) demonstrated that 111In-labeled specific mAb LL1 was 24-fold more cytotoxic to CD74-positive Raji B-cell lymphoma cells in vitro than a 111In-labeled nonspecific mAb, but the nonspecific mAb was nevertheless able to achieve killing of Raji cells at higher concentrations (99% cell killing at 0.5 and 12.3 MBq/mL, respectively). Our observations and those of Govindan et al. (34) and Behr et al. (33) suggest that the radiotoxicity of targeted radiotherapeutic agents labeled with 111In to antigen- or receptor-positive cells depends on the amount of radioactivity administered, with a decrease in specificity at higher doses, presumably because of the effects of the γ-emissions from 111In. These findings have important implications for predicting normal organ toxicity in studies of targeted Auger electron radiotherapy of malignancies in humans for which large amounts of 111In-labeled radiopharmaceuticals (>20 GBq) are required (22).
CONCLUSION
The novel targeted Auger electron-emitting radiopharmaceutical 111In-DTPA-hEGF had potent, dose-related, and selective antiproliferative effects on EGFR-overexpressing human breast cancer xenografts implanted into athymic mice. The radiopharmaceutical was well tolerated, demonstrating minimal normal tissue toxicity and causing modest decrease in WBC and platelet counts. No hepatotoxicity or renal toxicity was associated with 111In-DTPA-hEGF. The highest tumor localization, radiation-absorbed doses, and growth inhibition were achieved for small, nonestablished tumors, suggesting that this radiopharmaceutical may have the most value as a treatment for small-volume metastatic breast cancer or even for occult micrometastases in an adjuvant setting.
Acknowledgments
This study was supported by grants from the U.S. Army Breast Cancer Research Program (grant DAMD17-98-1-8171) and the Susan G. Komen Breast Cancer Foundation (grant 9749). Parts of this study were presented at the 48th Annual Meeting of the Society of Nuclear Medicine, Toronto, Ontario, Canada, June 23–27, 2001.
Footnotes
Received Oct. 24, 2002; revision accepted Feb. 13, 2003.
For correspondence or reprints contact: Raymond M. Reilly, PhD, Leslie Dan Faculty of Pharmacy, University of Toronto, 19 Russell St., Toronto, Ontario, Canada M5S 2S2.
E-mail: raymond.reilly{at}utoronto.ca
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Interrogating Tumor Metabolism and Tumor Microenvironments Using Molecular Positron Emission Tomography Imaging. Theranostic Approaches to Improve Therapeutics
- ErbB-2 Blockade and Prenyltransferase Inhibition Alter Epidermal Growth Factor and Epidermal Growth Factor Receptor Trafficking and Enhance 111In-DTPA-hEGF Auger Electron Radiation Therapy
- Cellular Dosimetry of 111In Using Monte Carlo N-Particle Computer Code: Comparison with Analytic Methods and Correlation with In Vitro Cytotoxicity
- Cancer Nanotargeted Radiopharmaceuticals for Tumor Imaging and Therapy
- Therapeutic Targeting of Nuclear Protein Import in Pathological Cell Conditions
- Improvement of Biodistribution and Therapeutic Index via Increase of Polyethylene Glycol on Drug-carrying Liposomes in an HT-29/luc Xenografted Mouse Model
- Trastuzumab-Resistant Breast Cancer Cells Remain Sensitive to the Auger Electron-Emitting Radiotherapeutic Agent 111In-NLS-Trastuzumab and Are Radiosensitized by Methotrexate
- Relationship Between Induction of Phosphorylated H2AX and Survival in Breast Cancer Cells Exposed to 111In-DTPA-hEGF
- Epidermal Growth Factor Receptor Inhibition Modulates the Nuclear Localization and Cytotoxicity of the Auger Electron Emitting Radiopharmaceutical 111In-DTPA Human Epidermal Growth Factor
- Carbon Nanotubes: Potential Benefits and Risks of Nanotechnology in Nuclear Medicine
- Preclinical Pharmacokinetic, Biodistribution, Toxicology, and Dosimetry Studies of 111In-DTPA-Human Epidermal Growth Factor: An Auger Electron-Emitting Radiotherapeutic Agent for Epidermal Growth Factor Receptor-Positive Breast Cancer
- Nuclear Localizing Sequences Promote Nuclear Translocation and Enhance the Radiotoxicity of the Anti-CD33 Monoclonal Antibody HuM195 Labeled with 111In in Human Myeloid Leukemia Cells
- Auger Electrons: Lethal, Low Energy, and Coming Soon to a Tumor Cell Nucleus Near You
- A Kit Formulated Under Good Manufacturing Practices for Labeling Human Epidermal Growth Factor with 111In for Radiotherapeutic Applications
- Cancer Therapy with Auger Electrons: Are We Almost There?