


Abstract
The intracellular distribution of Auger electron–emitting radiopharmaceuticals is a determinant of cytotoxicity. However, the mechanisms by which these agents are routed through the cell are ill understood. The aim of this study was to investigate how trafficking of 111In-labeled human epidermal growth factor (111In-DTPA-hEGF) relates to that of the EGF receptor (EGFR) and whether coadministration of agents that modulate EGFR signaling alters the efficacy of 111In-DTPA-hEGF. Methods: The spatiotemporal interaction between AlexaFluor488-EGF (AF488-EGF) and Cy3-conjugated anti-EGFR antibody (Cy3-anti-EGFR) was studied in the breast cancer cell line MDA-MB-468 using fluorescence resonance energy transfer and 2-photon fluorescence lifetime imaging. 111In internalization and nuclear fractionation assays were performed to investigate the effect of the ErbB-2–blocking antibody trastuzumab and a prenyltransferase inhibitor, L-778,123, on the subcellular localization of 111In-DTPA-hEGF in MDA-MB-468 (1.3 × 106 EGFR per cell; ErbB-2 negative) and 231-H2N (0.2 × 106 EGFR per cell; 0.4 × 105 ErbB-2 per cell) cell lines. The cytotoxicity of 111In-DTPA-hEGF (0–64 nM) plus trastuzumab (0–50 μg/mL) or L-778,123 (0–22.5 μM) was measured using clonogenic assays in a panel of breast cancer cell lines that express different levels of EGFR and ErB-2. Clonogenic survival data were used to calculate combination indices. Tumor growth inhibition was measured in vivo in 231-H2N xenograft–bearing mice treated with 111In-DTPA-hEGF plus trastuzumab or L-788,123. Results: Using fluorescence resonance energy transfer, we showed that EGF interacts with EGFR in the cytoplasm and nucleus after internalization of the ligand–receptor complex in MDA-MB-468 cells. Nuclear localization of 111In-DTPA-hEGF is enhanced by trastuzumab and L-788,123. Trastuzumab and L-788,123 sensitized 231-H2N cells to 111In-DTPA-hEGF. Nuclear localization and cytotoxicity of 111In-DTPA-hEGF were significantly increased in 231-H2N xenografts by cotreatment with L-788,123 (P < 0.0001). Conclusion: The therapeutic efficacy of 111In-DTPA-hEGF is increased through the coadministration of selected molecularly targeted drugs that modulate EGFR signaling and trafficking.
The epidermal growth factor (EGF) and its receptor (EGFR) are involved in the regulation of cellular proliferation, apoptosis, cell motility, and many other cellular processes (1). Overexpression of EGFR and the resulting upregulation of EGFR-mediated signaling have been observed in several tumor types, including breast cancer, non–small cell lung cancer, and head and neck squamous cell carcinoma. Reilly's group has exploited the EGF–EGFR axis in a Trojan horse approach to deliver the Auger electron–emitting isotope 111In, as 111In-labeled human EGF (111In-DTPA-hEGF), to the nucleus of EGFR-overexpressing cancer cells for molecularly targeted radiation therapy (2–5). Good correlation between EGFR expression and 111In-DTPA-hEGF uptake, nuclear localization, DNA damage, and cell kill was demonstrated in a panel of breast cancer cell lines (2). In the in vivo setting, 111In-DTPA-hEGF inhibited growth of MDA-MB-468 xenografts in athymic mice. A kit for DTPA-hEGF was formulated under good manufacturing practice conditions (6).
Auger electron therapy is a promising form of molecular radiotherapy and has recently made the transition from the laboratory bench to the clinic (7–9). The main advantage of Auger electron–induced radiation damage over β-emitting radioisotopes such as 90Y or 131I, used in the approved drugs ibritumomab tiuxetan and tositumomab (10), or α-emitters such as 213Bi, is the limited track length of Auger electrons, which is in the nanometer to micrometer range (8,11). This reduced track length limits crossfire from targeted cells to nearby normal cells, thereby avoiding normal tissue toxicity, and also accounts for the importance of nuclear localization for the manifestation of the cytotoxic effects of Auger electron emitters. Therefore, an increase in the nuclear localization of existing Auger electron–emitting radiopharmaceuticals would result in enhancement of their cytotoxic effects. Various approaches to increase nuclear localization of Auger electron–emitting radiopharmaceuticals have been explored, including their modification through attachment of a peptide with a nuclear localization sequence (7) or coadministration with other pharmaceuticals that enhance their nuclear localization (12,13). In this paper, it was shown that EGF remains bound to EGFR as it translocates to the nucleus, providing an opportunity to alter 111In-DTPA-hEGF trafficking and the extent of its nuclear localization through coadministration of drugs that influence nuclear translocation of EGFR. Bailey et al. have shown previously that exposure of MDA-MB-468 cells to gefitinib (Iressa [AstraZeneca plc], an EGFR tyrosine kinase inhibitor) increases nuclear localization of 111In-DTPA-hEGF, resulting in increased DNA damage as measured by induction of γH2AX foci and reduced clonogenicity (12).
In this report, the effect of combining 111In-DTPA-hEGF with drugs that alter EGFR localization and signaling was investigated. The agents tested were trastuzumab (Herceptin [Genentech], a monoclonal antibody that binds to and blocks ErbB-2) and a prenyltransferase inhibitor (PTI; L-778,123, a peptidomimetic dual farnesyltransferase and geranylgeranyltransferase I inhibitor). The effect of combining 111In-DTPA-hEGF with trastuzumab or L-778,123 on clonogenic survival in vitro and on biodistribution and tumor growth inhibition in vivo was evaluated.
MATERIALS AND METHODS
Cell Culture
MDA-MB-468, MDA-MB-231, and MCF-7 breast cancer cells were obtained from the CRUK cell services laboratories. The MDA-MB-231 cell line stably transfected with the erbB-2 gene, 231-H2N, was kindly provided by Robert Kerbel (Sunnybrook Health Sciences Centre) (14). Cells were cultured in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich), supplemented with 10% fetal calf serum (Invitrogen) and antibiotics (penicillin–streptomycin, 100 units/mL, Invitrogen). EGFR expression levels were measured as described previously and are summarized in Table 1 (15).
Characteristics of Cell Lines and Combination Indices of 111In-DTPA-hEGF Plus Trastuzumab or L-788,123
Synthesis and Testing of Coadministered Agents
Cy3-anti-EGFR was synthesized using a Cy3-tetrafluorophenyl ester kit, according to the manufacturer's instructions (GE Healthcare). For confocal microscopy and fluorescence resonance energy transfer (FRET) experiments, cells were seeded on coverslips (2 × 105 cells per coverslip) and left overnight to adhere. Growth medium was removed, cells were washed with phosphate-buffered saline (PBS), and EGF-biotin-streptavidin-AlexaFluor488 (AF488-EGF, 4 nM, Invitrogen) was added in DMEM (without fetal calf serum). At selected time points up to 24 h, cells were washed, fixed using 4% paraformaldehyde (10 min, room temperature), permeabilized using 1% Triton X-100 in PBS (10 min, room temperature), blocked using 2% bovine serum albumin in PBS with 0.1% Triton X-100 (1 h at 37°C), and stained with Cy3-anti-EGFR (0.5 μg/mL in PBS containing 2% bovine serum albumin) for 1 h at 37°C. Confocal microscopy was performed using a TCS SP2 microscope (Leica Microsystems). FRET by sensitized emission (FRET-SE) was calibrated for donor and acceptor signals and measured using Leica built-in software, correcting for bleed-through and crossover, with AF488 as the donor and Cy3 as the acceptor. The occurrence of FRET was confirmed by fluorescence lifetime imaging of AF488 (the donor fluorophore) on a modified TE200 inverted microscope (Nikon) (16,17). AF488 fluorescence lifetime in AF488-EGF–treated cells (1 h) with and without Cy3-anti-EGFR staining was analyzed using least squares–based fitting with the TRI2 software program (18).
Saturation Binding, Internalization, and Nuclear Localization of 111In-DTPA-hEGF
111In-DTPA-hEGF was synthesized as previously described, using a kit developed by one of the authors (6). Radiolabeling efficiency was more than 95%; specific activity was 6 MBq/μg. A specific activity of 6 MBq/μg is equivalent to one 111In atom per 44 EGF molecules. EGFR expression and the affinity of 111In-DTPA-hEGF for the EGFR (measured as the dissociation constant Kd) were measured in MDA-MB-468 and 231-H2N cells exposed for 16 h to PBS, trastuzumab (10 μg/mL), or L-778,123 (3 μM), by saturation binding assay, as previously described (19). To evaluate 111In-DTPA-hEGF internalization, an acid wash step was used, as previously described (20). Cells were incubated with PBS, trastuzumab (10 μg/mL), or L-778,123 (3 μM) for 16 h, then exposed to 4 nM 111In-DTPA-hEGF in DMEM for varying lengths of time. After incubation, cell medium was removed, and cells were washed with ice-cold PBS and exposed to an acid wash solution (0.1 M glycine·HCl, pH 2.5, for 6 min at 4°C) to strip 111In-DTPA-hEGF from the cell membrane (membrane-bound fraction). After being washed with ice-cold PBS, whole cells were lysed using NaOH (0.1 M), releasing the internalized 111In-DTPA-hEGF. The amount of 111In in the cell medium, membrane, and internalized fractions was measured using an automated γ-counter (Wizard; PerkinElmer). Nuclear localization was determined by selective lysis of the cell membrane as described previously by exposure of cells to NP-40 (0.1%) in PBS for 6 min at 4°C (21). Nuclei were isolated by centrifugation and washed, and the amount of 111In present was measured.
Clonogenic Assays and Combination Index Analysis
MDA-MB-468, MDA-MB-231, 231-H2N, and MCF-7 cell suspensions (106 cells/mL) were treated with combinations of 111In-DTPA-hEGF (0–64 nM) plus trastuzumab (0–50 μg/mL) or L-778,123 (0–22.5 μM) or with each drug alone. After incubation at 37°C for 24 h, an aliquot of cells was plated in DMEM with 10% fetal calf serum (20% for MDA-MB-468 cells) plus antibiotics and incubated at 37°C in 5% CO2. Colonies were counted after 1–2 wk, and the clonogenic survival fraction was calculated. The combination index of 111In-DTPA-hEGF plus trastuzumab or L-778,123 was calculated as described (22). Briefly, the combination index equaled (D)1/(Dx)1 + (D)2/(Dx)2, where (D)1 or (D)2 is the dose that causes a specific effect—that is, clonogenic survival—in the combination, and (Dx)1 or (Dx)2 is the dose of the same drug that will produce the identical effect by itself. Combination indices of 1, less than 1, or more than 1 indicate additivity, superadditivity, and antagonism, respectively.
Biodistribution of 111In-DTPA-hEGF and Tumor Growth Inhibition
All animal procedures were performed in accordance with the U.K. Animals (Scientific Procedures) Act of 1986 and with local ethical committee approval. 231-H2N xenografts were established in female BALB/c nu/nu mice (Harlan) by subcutaneous injection of 1.5 × 106 cells in the right flank in DMEM:matrigel 1:1 (Matrigel; BD). When xenografts reached a volume of approximately 100 mm3, animals (7/group) were treated with drug: PBS (control animals), trastuzumab (4 mg/kg), or L-778,123 (40 mg/kg) administered by intraperitoneal injection on days 1, 2, 3, 7, 8, and 9. 111In-DTPA-hEGF (3.4 μg, 6 MBq/μg) or PBS was administered intravenously on days 3 and 9. Tumor size was measured by caliper twice weekly. Tumor volume (V) was calculated as a2 × b, with a and b being the short and long axes, respectively.
In another group of mice, animals (3/group) bearing xenografts of approximately 500 mm3 were treated with drug (PBS, trastuzumab [4 mg/kg], or L-778,123 [40 mg/kg]) administered by intraperitoneal injection on days 1, 2, and 3. 111In-DTPA-hEGF (3.4 μg, 6 MBq/μg) was administered intravenously on day 3. Three hours after 111In-DTPA-hEGF administration, animals were euthanized, and blood, selected tissues, and tumor were removed. Tissues were washed, blot-dried, weighed, and counted for radioactivity. The amount of 111In in blood and tissues was expressed as percentage injected dose per gram of blood or tissue. The tumors were further processed by disaggregation, and nuclear localization of 111In-DTPA-hEGF was measured using a nuclear localization kit according to the manufacturer's instructions (NUC-201; Sigma).
Statistical Analysis
All statistical analyses and nonlinear regression were performed using GraphPad Prism (GraphPad Software Inc.). One-way or 2-way ANOVA was used for multiple comparisons. The F test was used to compare parameters between curves. Kaplan–Meier curves were generated for survival analysis. Log-rank tests were performed to compare Kaplan–Meier survival curves.
RESULTS
Confocal Microscopy, FRET-SE, and Fluorescence Lifetime Imaging
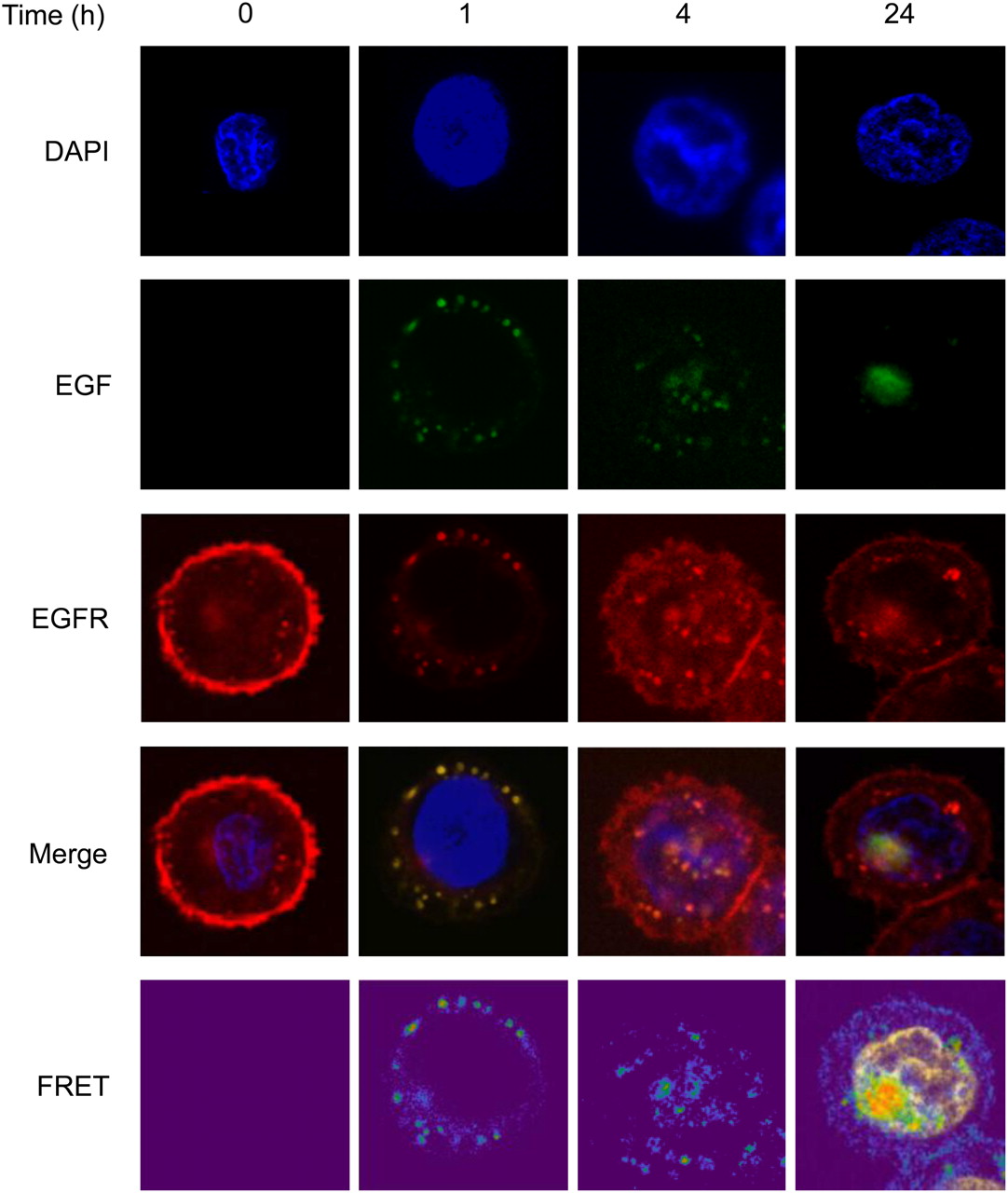
The uptake of AF488-EGF into MDA-MB-468 cells over time is shown in Figure 1. Confocal microscopy shows colocalization (yellow in merged images) of EGF (green) with EGFR (red). At baseline, EGFR is expressed predominantly at the cell surface. After the addition of AF488-EGF, at 1 h, EGFR and EGF are internalized into the cells and appear as distinct foci in the cytoplasm. Nuclear localization of EGF and EGFR is clearly seen by 4 h. At 24 h, EGFR is again observed at the cell membrane, with some residual staining in the nucleus also. In contrast, at 24 h AF488-EGF is seen exclusively in the nucleus. To investigate the interaction between EGF and EGFR, FRET-SE was used. FRET-SE analysis of MDA-MB-468 cells exposed to AF488-EGF and stained using Cy3-anti-EGFR showed FRET between EGF and EGFR in foci in the cytoplasm and nucleus at 1 and 4 h. FRET was still observed in a large focus in some nuclei 24 h after the start of exposure (bottom right image, Fig. 1).

MDA-MB-468 cells were exposed for various times to AF88-EGF (green) and stained for EGFR using Cy3-anti-EGFR antibody (red). Confocal images and FRET-SE images were acquired. Colocalization of EGF with EGFR results in yellow, seen in the nucleus at 4 and 24 h. DAPI = 4′,6-diamidino-2-phenylindole.
To validate FRET-SE findings, FRET was also measured using fluorescence lifetime imaging (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org). Fluorescence lifetime of the donor (AF488-EGF) in the absence of acceptor, Cy3, was calculated by global regression analysis to be 1.96 ± 0.28 ns. When cells were also stained with Cy3-anti-EGFR, AF488 lifetime was significantly reduced to 1.52 ± 0.05 ns (P = 0.01), showing that FRET between AF488 and Cy3 is occurring, indicative of interaction between EGF and EGFR. Global biexponential analysis using Levenberg–Marquardt optimization was used (16). Examination of the fitting residuals and χ2 value indicated that biexponential analysis was most appropriate, revealing a second, faster, lifetime component of 0.39 ± 0.17 ns, which was unaffected by the addition of acceptor Cy3 (lifetime with Cy3 added was 0.33 ± 0.06 ns; P = 0.52) and is attributed to autofluorescence of the cells.
111In-DTPA-hEGF Saturation Binding, Internalization, and Nuclear localization
To investigate whether exposure of cells to L-788,123 or trastuzumab alters EGFR expression level, 111In-DTPA-hEGF saturation binding assays were performed in the presence of these agents (Supplemental Figs. 2A and 2B). Exposure of 231-H2N cells to L-788,123 for up to 24 h resulted in a minor reduction in EGFR expression at the cell membrane, which did not reach statistical significance. There was, however, a 22% decrease in EGFR expression after treatment with trastuzumab (P < 0.001) (Supplemental Fig. 2B). There was no change in EGFR expression in response to trastuzumab or L-788,123 in MDA-MB-468 cells (Supplemental Fig. 2A).

Internalization and nuclear localization of 111In-DTPA-hEGF. MDA-MB-468 (A and C) or 231-H2N (B and D) cells were exposed to L-788,123 (3 μM) or trastuzumab (10 μg/mL) and to 111In-DTPA-hEGF. (A and B) Cells were exposed to 111In-DTPA-hEGF (4 nM), and at selected time points the amount of 111In internalized into cells was determined. (C and D) Cells were exposed to 4 nM 111In-DTPA-hEGF. At selected time points, cell nuclei were isolated, and amount of 111In determined. Results are shown as averages of 3 experiments ± SD.
Exposure of MDA-MB-468 and 231-H2N cells to L-788,123 but not trastuzumab resulted in a significant reduction in the internalization rate of 111In-DTPA-hEGF (Figs. 2A and 2B). The 111In internalization rates (k) in untreated and in trastuzumab- and L-788,123–treated MDA-MB-468 cells were 0.48% ± 0.07%, 0.45% ± 0.06%, and 0.29% ± 0.04%/h, respectively (control vs. trastuzumab, P = 0.30; control vs. L-788,123, P < 0.0001). Internalization rates in untreated and in trastuzumab- and L-788,123–treated 231-H2N cells were 2.16% ± 0.30%, 2.62% ± 0.38%, and 1.42% ± 0.18%/h, respectively (control vs. trastuzumab, P = 0.25; control vs. L-788,123, P = 0.03). However, nuclear localization (calculated as the percentage of the total amount of 111In added to the culture medium that accumulates in the nucleus) was significantly increased in MDA-MB-468 and 231-H2N cells after exposure to both trastuzumab and L-788,123 (Figs. 2C and 2D). Nuclear localization at 1 h after the addition of 111In-DTPA-hEGF to control and to trastuzumab- and L-788,123–treated MDA-MB-468 cells was 1.2% ± 0.0%, 2.5% ± 0.3%, and 3.0% ± 0.8%, respectively (control vs. trastuzumab, P = 0.0002; control vs. PTI, P = 0.03). Nuclear localization at 1 h after the addition of 111In-DTPA-hEGF to control and to trastuzumab- and L-788,123–treated 231-H2N cells was 1.6% ± 0.04%, 2.4% ± 0.01%, and 2.1% ± 0.03%, respectively (P < 0.0001 for both). This degree of localization resulted in an increase of the area under the time–activity curve, a measure of the 111In dose delivered to the nuclei of cells, of 2.3- and 2.0-fold for trastuzumab and L-788,123 in MDA-MB-468 cells (P = 0.0007 and 0.023, respectively). The area under the time–activity curve was increased by 2.4- and 2.3-fold for trastuzumab and L-788,123, respectively, in 231-H2N cells (P < 0.0001 for both).
Clonogenic Assays and Combination Index Analysis
Trastuzumab alone was cytotoxic to MDA-MB-231, 231-H2N, and MCF7 cells. PTI alone was cytotoxic to all cell lines used in this study. 111In-DTPA-hEGF alone was very toxic to MDA-MB-468 cells and moderately toxic to MDA-MB-231, 231-H2N, and MCF7 cells. The combination indices for 111In-DTPA-hEGF plus trastuzumab and for 111In-DTPA-hEGF plus L-788,123 in a panel of breast cancer cell lines with differing EGFR and ErbB-2 expression were calculated from the results of clonogenic assays (Supplemental Fig. 3) and are summarized in Table 1 (22). Combination indices less than 1 indicate superadditivity of both trastuzumab and L-788,123 with 111In-DTPA-hEGF in 231-H2N cells. There was no superadditivity when MDA-MB-468, MDA-MB-231, or MCF-7 cells were exposed to 111In-DTPA-hEGF plus trastuzumab or L-788,123.

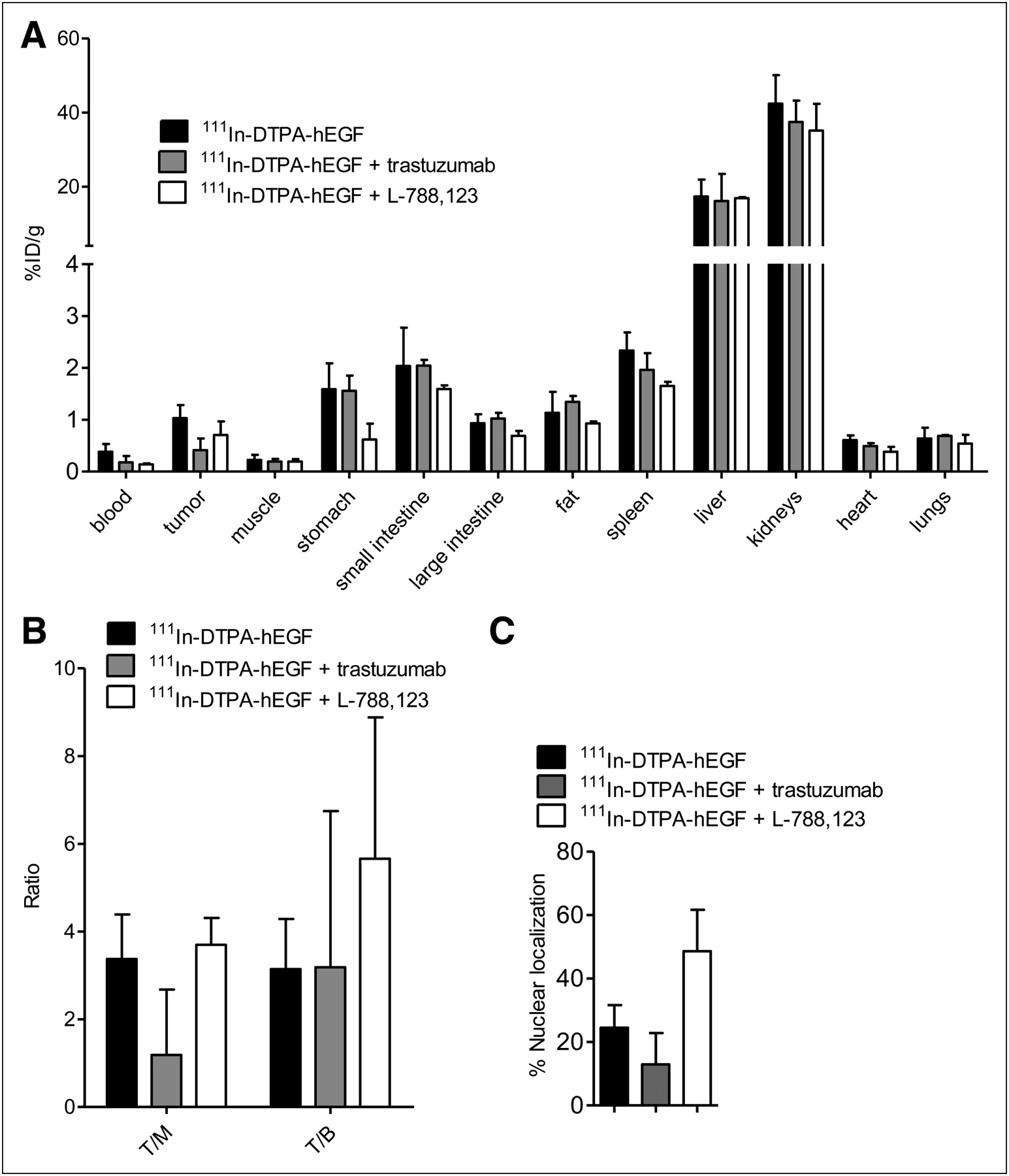
Mice bearing 231-H2N xenografts were treated daily for 3 d with PBS, trastuzumab, or L-788,123 and on day 3 with 111In-DTPA-hEGF (3.4 μg) intravenously. Tumors and other organs were removed 3 h later. (A) Distribution of 111In in tumor and normal organs is shown as percentage injected dose per gram of tissue. (B) Tumor-to-muscle (T/M) and tumor-to-blood (T/B) ratios of 111In-DTPA-hEGF were calculated. (C) Three animals were treated as in A, and tumors were removed and nuclei isolated. The ratio of 111In contained in cell nuclei vs. whole tumor was determined. Results are shown as mean ± SD.
Biodistribution of 111In-DTPA-hEGF and Tumor Growth Inhibition
Biodistribution results for 111In-DTPA-hEGF are shown in Figures 3A and 3B. Two-way ANOVA analysis with post hoc Bonferroni test showed that there is no significant change in the uptake of 111In-DTPA-hEGF (expressed as percentage injected dose per gram of blood or tissue) in various tissues after trastuzumab or L-788,123 administration (P > 0.05), except for the kidneys, which showed a small (5%) but statistically significant decrease in 111In uptake after L-788,123 treatment (P < 0.05). Also, there was no significant difference in the tumor-to-blood or tumor-to-muscle ratios of 111In-DTPA-hEGF after trastuzumab or L-788,123 administration (P > 0.05).
Nuclear localization of 111In-DTPA-hEGF in tumors was measured at 3 h after injection (Fig. 3C). Pretreatment of animals with L-788,123, compared with controls, resulted in a 2-fold increase in 111In localized in the nuclei of tumor cells (48% ± 13% and 24% ± 7%, respectively, P = 0.01). Pretreatment of animals with trastuzumab did not result in a significant change in the relative nuclear uptake of 111In (P > 0.05) (Fig. 3C).
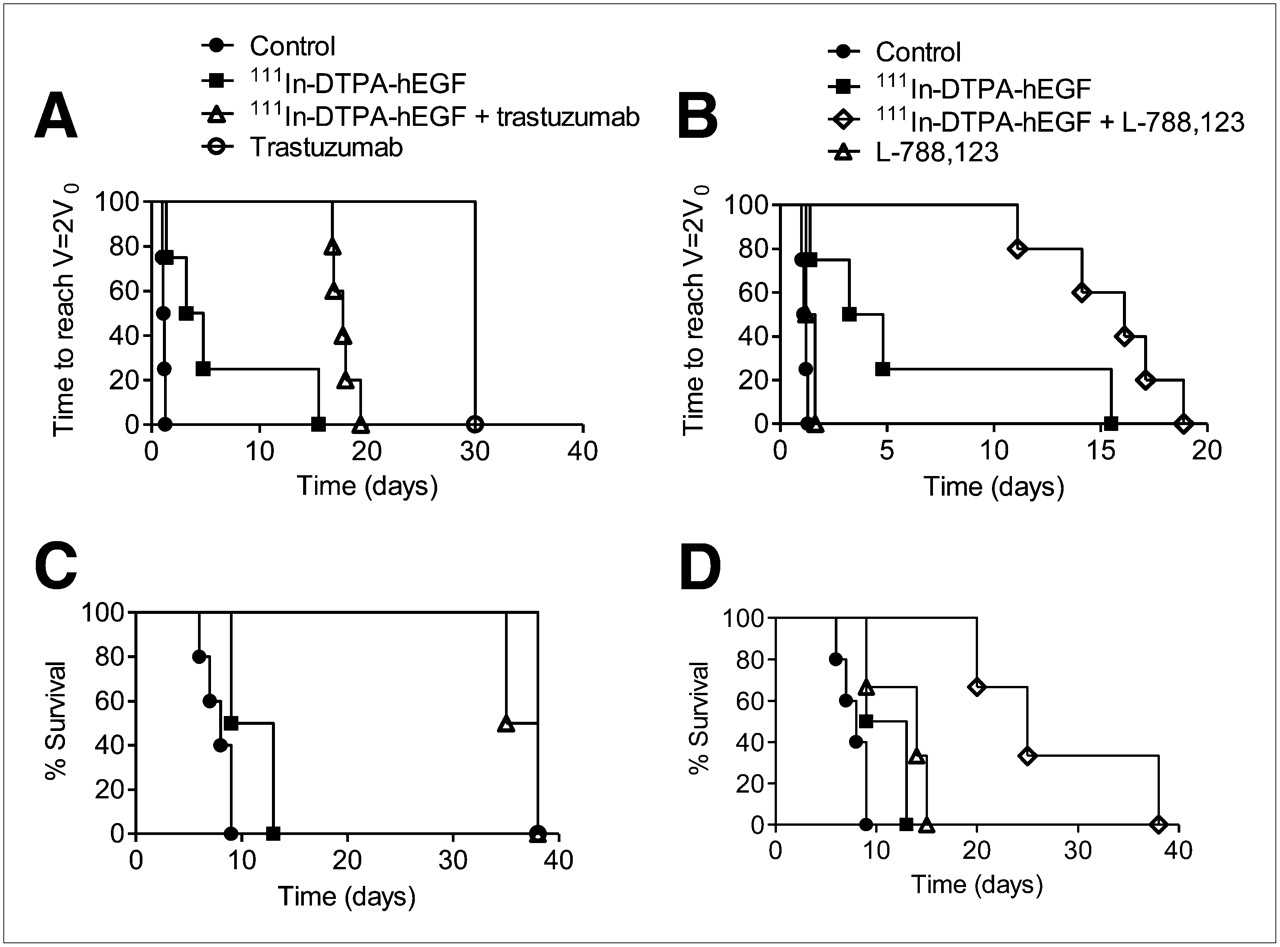
The results of 111In-DTPA-hEGF therapy alone or combined with L-788,123 or trastuzumab in mice bearing 231-H2N xenografts are shown in Figure 4. Log-rank analysis on doubling-time Kaplan–Meier curves (where an event was defined as V = 2 × V0) showed greater tumor growth inhibition with the combination of 111In-DTPA-hEGF plus L-788,123, but not 111In-DTPA-hEGF plus trastuzumab, than with any agent alone (Figs. 4A and 4B). Survival curves showed more prolonged survival of tumor-bearing mice with the combination of 111In-DTPA-hEGF plus L-788,123 than with either agent alone (Fig. 4D). Trastuzumab alone was effective at inhibiting tumor growth. The combination of trastuzumab plus 111In-DTPA-hEGF did not perform significantly better than trastuzumab alone (P > 0.05) (Figs. 4A and 4C).
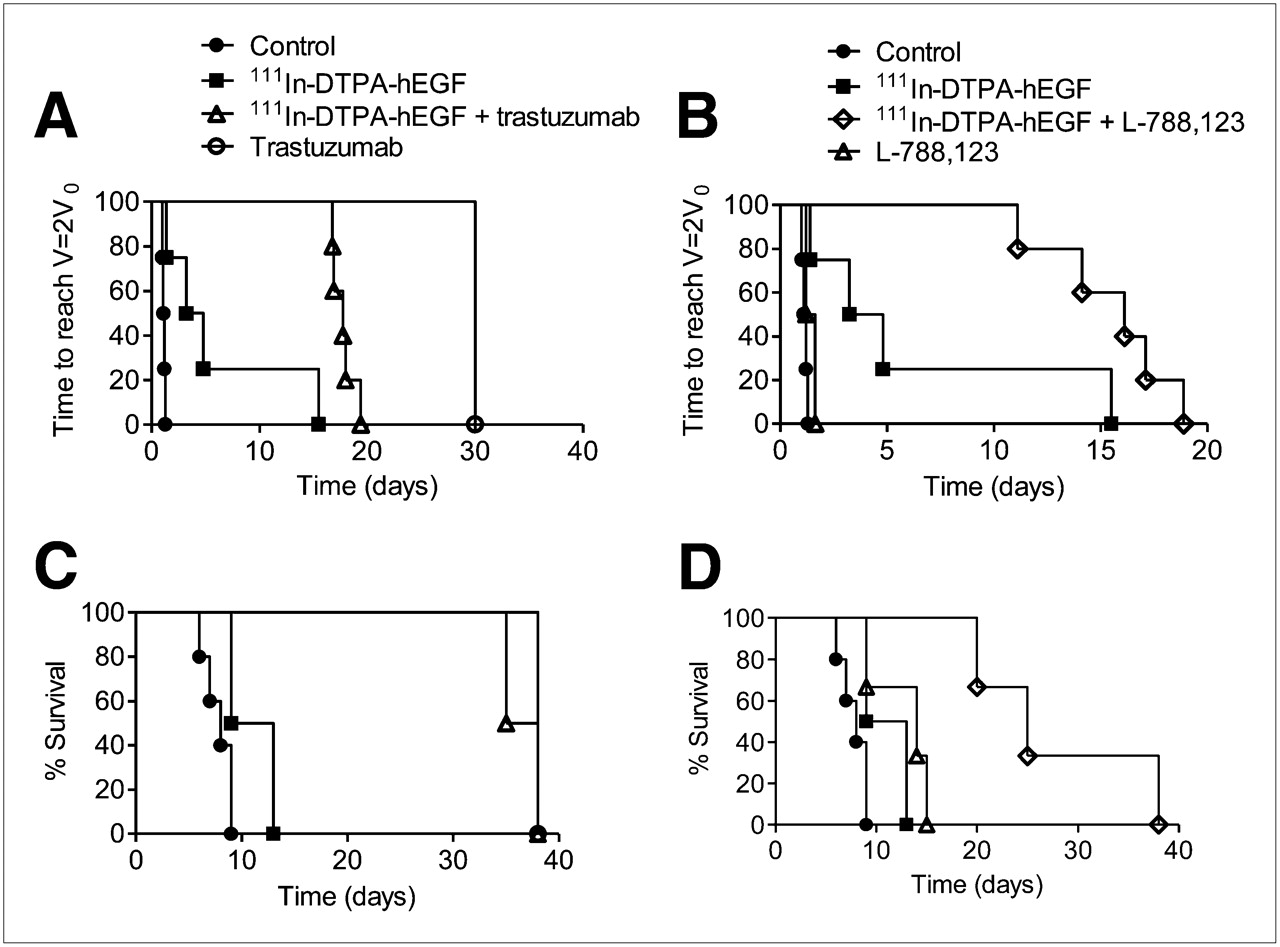
Mice, bearing 231-H2N xenografts, received PBS, trastuzumab, or L-788,123 with or without 111In-DTPA-hEGF, according to protocol described in “Materials and Methods” section. Tumor size was monitored twice weekly by caliper. Kaplan–Meier survival curves were generated using time taken for tumor volume to reach twice starting volume (A and B) or time for tumor to reach maximum volume permitted under terms of relevant animal welfare license (C and D).
DISCUSSION
111In-DTPA-hEGF, an Auger electron–emitting radiopharmaceutical, has been developed to target EGFR-overexpressing cancer cells. After ligand binding, EGFR internalizes into cells via clathrin-coated-pit endocytosis to reach the early and then late endosome (23). Once internalized, EGFR may recycle to the cell surface, may be ubiquitylated in preparation for lysosomal degradation, or is translocated to the nucleus. How EGFR escapes from the endosome to enter the nucleus is not fully understood, although nuclear import is thought to be mediated through the action of a nuclear localizing signal contained in the intracellular domain of the receptor and to be regulated by importin β (24). A recently proposed model suggests the possibility that EGFR remains membrane-associated as it transits through the golgi, endoplasmic reticulum, and nuclear pore complexes and that the Sec61b translocon plays a role in the release of EGFR from the lipid bilayer into the nucleus (25). In the nucleus, EGFR plays a role in transactivation of target genes (26). It is known that 111In-DTPA-hEGF localization in the nuclei of EGFR-overexpressing cells is necessary for its Auger electron–mediated DNA damage and cytotoxicity (2). The purpose of the current study was to clarify whether nuclear translocation of 111In-DTPA-hEGF results from continued interaction of the EGF moiety of the radiopharmaceutical with EGFR as it is routed through the cell. Evidence for such an interaction would provide a rationale for combining 111In-DTPA-hEGF with agents that promote nuclear accumulation of EGFR. The results of a FRET-SE study show that EGF and EGFR interact in the nucleus of MDA-MB-468 cells (Fig. 1), indicating that modulation of EGFR trafficking could affect nuclear uptake of 111In-DTPA-hEGF, absorbed radiation dose, DNA damage, and cell survival. The mechanism or relevance of the observed focal pattern of nuclear localization of EGF and EGFR at 24 h is not understood but would be expected to result in heterogeneity of the absorbed radiation dose from 111In-DTPA-hEGF in the nucleus if the localization of 111In-DTPA-hEGF mirrors that of unlabeled EGF (Fig. 1).
Bailey et al. reported that nuclear localization of 111In-DTPA-hEGF is enhanced and its cytotoxicity increased when it is combined with the EGFR tyrosine kinase inhibitor gefitinib (12). In the current report, pretreatment of breast cancer cells with drugs that target the EGFR cell-signaling pathway, trastuzumab and L-788,123, resulted in changes in internalization and nuclear uptake of 111In-labeled EGF. In 231-H2N cells but not MDA-MB-468 cells, EGFR-mediated internalization of radiolabeled EGF was modestly reduced after treatment with L-788,123 or trastuzumab (Supplemental Figs. 2A and 2B). This finding is consistent with a previous report that another farnesyltransferase inhibitor, R115777, was associated with reduced internalization of 123I-hEGF (19). L-788,123 is both a farnesyltransferase inhibitor and a geranylgeranyltransferase I inhibitor and has multiple guanosine triphosphatase substrates other than Ras, including RhoB (27). Perturbation of guanosine triphosphatase signaling is known to affect the function of the endocytic machinery and, therefore, trafficking of G-protein–coupled receptors such as the EGFR (28). For example, farnesyltransferase inhibition causes an increase in endosomal RhoB that leads, in turn, to inhibition of lysosomal degradation of EGFR (29). We speculated that a reduction in degradation of EGFR may result in a greater proportion of internalized receptor reaching the nucleus. Further, the inhibition of Ras signaling is known to sensitize cells in general to the effects of ionizing radiation (30). Thus, the 2- to 3-fold increase in nuclear accumulation of 111In that follows concurrent treatment with L-788,123 plus 111In-DTPA-hEGF, both in vitro (Fig. 2C and 2D) and in vivo (Fig. 3C), takes place in cells that have been sensitized to the adverse effects of ionizing radiation. This coincidence of events could account for the marked superadditivity of L-788,123 and 111In-DTPA-hEGF. Qayum et al. have recently reported that L-778,123 induces vascular normalization and improved tumor oxygenation leading to enhanced responsiveness to radiation therapy (31). It is possible that this phenomenon contributes to the marked tumor growth delay observed after the combination of L-788,123 and 111In-DTPA-hEGF in vivo (Table 1; Figs. 4B and 4D). PTIs are themselves capable of causing DNA damage, and this is thought to occur through the generation of reactive oxygen species. For example, inhibition of farnesyltransferase by manumycin induced γH2AX foci formation and DNA protein kinase–mediated phosphorylation of NBS1 and BRCA1 (33). Thus PTI-induced DNA damage may add to the clustered DNA damage typically associated with Auger electron radiation to produce a synergistic effect.
Superadditivity of L-788,123 and 111In-DTPA-hEGF was seen only in 231-H2N and not in the parental cell line, MDA-MB-231. Interestingly, resistance to farnestyltransferase inhibition by MDA-MB-231 in vitro and in vivo has been noted by others (34). This might be an indication that ErbB-2 expression plays a role in the response to farnesyltransferase inhibition through an as yet unknown mechanism. Superadditivity was not observed when 111In-DTPA-hEGF was combined with L-788,123 in MDA-MB-468 cells, even though this cell line has the highest EGFR density among the lines tested. MDA-MB-468 cells contain wild-type K-Ras, and this may explain the absence of additivity because farnesyltransferase inhibitors are known to be less effective in cells of this genotype (35). MCF-7 cells have low EGFR expression and are unresponsive to 111In-DTPA-hEGF; thus, they would not be expected to exhibit synergy when exposed to other agents (2).
ErbB-2 is an important heterodimerization partner of EGFR, which, when overexpressed, reduces the EGFR internalization rate constant and the rate of lysosomal targeting and promotes rapid recycling of activated EGFR (36,37). Trastuzumab is a humanized IgG1 antibody that binds to the ectodomain of the ErbB-2 receptor. One aim of the current study was to investigate the effect of combining trastuzumab with 111In-DTPA-hEGF on the delivery of 111In to the nuclei of EGFR-positive cells. The combination of trastuzumab plus 111In-DTPA-hEGF resulted in enhanced nuclear uptake of 111In in the EGFR- and ErbB-2–positive 231-H2N cell line in vitro, likely accounting for the low combination index (denoting superadditivity) for this combination (Fig. 2D; Table 1). Surprisingly, trastuzumab also resulted in an increase in the amount of 111In that was delivered to the nuclei of MDA-MB-468 cells, which are ErbB-2–negative (Fig. 2C). Although MDA-MB-468 cells do not express easily detectable levels of ErbB-2 protein, they are not null for the erbB-2 gene, and it is possible that binding of even small amounts of ErbB-2 by trastuzumab could result in perturbation of EGFR trafficking in this highly EGFR-dependent cell line. The enhancement of nuclear uptake of 111In that was seen in vitro in 231-H2N cells did not occur in 231-H2N xenografts in vivo (Fig. 3C). The reason for this difference is not clear but may account for the observed lack of synergism between trastuzumab and 111In-DTPA-hEGF in vivo (Figs. 4A and 4C). Also, trastuzumab was so effective at inhibiting 231-H2N xenograft growth when given as a single agent that the addition of 111In-DTPA-hEGF had little impact (Figs. 4A and 4C).
CONCLUSION
L-788,123 increases the cytotoxicity of 111In-DTPA-hEGF in 231-H2N tumors in vivo. The effect of combining 111In-DTPA-hEGF with molecularly targeted agents that alter EGFR trafficking is complex and dependent on cell context.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Deborah Scollard and Conrad Chan for DTPA-EGF kits and Thomas Brunner and Nadia Falzone for helpful discussion. This research was supported by Cancer Research UK (C14521/A6245), the Medical Research Council, the CRUK EPSRC Oxford Cancer Imaging Centre, and the National Institute for Health Research Oxford Biomedical Research Centre.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication November 3, 2010.
- Accepted for publication January 7, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}