


Abstract
The tyrosine kinase inhibitor erlotinib poorly penetrates the blood–brain barrier (BBB) because of efflux transport by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2), thereby limiting its utility in the treatment of non–small cell lung cancer metastases in the brain. Pharmacologic strategies to inhibit ABCB1/ABCG2-mediated efflux transport at the BBB have been successfully developed in rodents, but it remains unclear whether these can be translated to humans given the pronounced species differences in ABCG2/ABCB1 expression ratios at the BBB. We assessed the efficacy of two different ABCB1/ABCG2 inhibitors to enhance brain distribution of 11C-erlotinib in nonhuman primates as a model of the human BBB. Methods: Papio anubis baboons underwent PET scans of the brain after intravenous injection of 11C-erlotinib under baseline conditions (n = 4) and during intravenous infusion of high-dose erlotinib (10 mg/kg/h, n = 4) or elacridar (12 mg/kg/h, n = 3). Results: Under baseline conditions, 11C-erlotinib distribution to the brain (total volume of distribution [VT], 0.22 ± 0.015 mL/cm3) was markedly lower than its distribution to muscle tissue surrounding the skull (VT, 0.86 ± 0.10 mL/cm3). Elacridar infusion resulted in a 3.5 ± 0.9-fold increase in 11C-erlotinib distribution to the brain (VT, 0.81 ± 0.21 mL/cm3, P < 0.01), reaching levels comparable to those in muscle tissue, without changing 11C-erlotinib plasma pharmacokinetics. During high-dose erlotinib infusion, 11C-erlotinib brain distribution was also significantly (1.7 ± 0.2-fold) increased (VT, 0.38 ± 0.033 mL/cm3, P < 0.05), with a concomitant increase in 11C-erlotinib plasma exposure. Conclusion: We successfully implemented ABCB1/ABCG2 inhibition protocols in nonhuman primates resulting in pronounced increases in brain distribution of 11C-erlotinib. For patients with brain tumors, such inhibition protocols may ultimately be applied to create more effective treatments using drugs that undergo efflux transport at the BBB.
Non–small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers and is the leading cause of cancer-related death around the world (1,2). Gene mutations that activate epidermal growth factor receptor (EGFR) (e.g., the exon 19 deletion delE746-A750 or the exon 21-point mutation L858R) occur in 10%–20% of patients with NSCLC in North American and European populations and in up to 60% among Asian populations. In patients with activating mutations, EGFR-targeting tyrosine kinase inhibitors (TKIs), such as gefitinib, erlotinib, and afatinib, have led to remarkable tumor shrinkage and improvement in progression-free survival and quality of life compared with standard chemotherapy. Unfortunately, the central nervous system is a common site of progression, and 25%–40% of NSCLC patients develop brain metastases during or after systemic benefit with TKIs (1,2). The outcome is poor for NSCLC patients with brain metastases, for whom there are only few effective treatment options, such as radiation therapy with or without stereotactic radiosurgery (1,2). Insufficient drug delivery to the central nervous system, due to the poor permeability of the blood–brain barrier (BBB) to TKIs, is hypothesized to account for differences in clinical response to TKI treatment between the central nervous system and peripheral organs.
The BBB is essentially formed by the endothelial cells of brain capillaries that express high levels of adenosine triphosphate–binding cassette (ABC) efflux transporters, such as P-glycoprotein (rodents: Abcb1a; humans: ABCB1) and breast cancer resistance protein (rodents: Abcg2, humans: ABCG2), which can restrict the brain distribution of many drugs. It has been shown that most EGFR-targeting TKIs, including erlotinib and gefitinib, are substrates of both ABCB1 and ABCG2, which leads to very low brain concentrations of these drugs (3,4). Whereas there is a large body of evidence that the BBB is disrupted in necrotic areas of primary brain tumors as compared with healthy brain tissue (5), it remains controversial to what extent a BBB disruption in metastatic lesions affects chemotherapeutic drug exposure and efficiency (6).
A potentially powerful strategy to improve delivery of TKIs to the brain, including tumor tissue that is protected by an intact BBB, is the concomitant administration of ABC transporter inhibitors (5,7). Studies on transporter knockout mice showed that Abcb1a and Abcg2 work together in limiting the brain distribution of most TKIs (3,4). When Abcb1a alone or Abcg2 alone is knocked out, the other transporter restricts the access of dual substrates to the brain, so that dual substrates can penetrate the brain only when both Abcb1a and Abcg2 are absent (8). On the basis of this functional interplay between ABCB1 and ABCG2, delivery of dual ABCB1/ABCG2 substrates such as TKIs to the brain can most likely be significantly enhanced only when both transporters are simultaneously inhibited. The most potent currently available dual ABCB1/ABCG2 inhibitor is elacridar (GF120918). Numerous studies have shown that elacridar can effectively inhibit Abcb1a and Abcg2 at the mouse BBB (3). However, it is not currently clear to what extent ABCB1/ABCG2 inhibition at the BBB by elacridar can be translated to species other than rodents, including humans. Importantly, there are considerable species differences in relative expression levels of ABCB1 and ABCG2 at the rodent and human BBB, with ABCG2/ABCB1 expression ratios of approximately 1.3 in humans and 0.3 in mice (9). These species differences in transporter expression may lead to different sensitivities to transporter inhibition in rodents and humans.
PET can be used to noninvasively study the effect of transporters on the brain distribution of radiolabeled drugs. PET studies already have demonstrated that ABCB1 can be effectively inhibited at the human BBB with the third-generation ABCB1 inhibitor tariquidar, resulting in up to 5-fold increases in the brain distribution of the radiolabeled ABCB1 substrates (R)-11C-verapamil and 11C-N-desmethyl-loperamide (10,11). In contrast to these ABCB1-selective substrates, the brain distribution of dual ABCB1/ABCG2 substrate radiotracers was enhanced to a much smaller extent by tariquidar administration (12), supporting the assumption that both ABCB1 and ABCG2 need to be inhibited to increase the brain distribution of dual ABCB1/ABCG2 substrates.
The aim of the present study was to assess for the first time the effect of ABCB1/ABCG2 inhibition with elacridar on brain distribution of 11C-erlotinib in nonhuman primates that more closely resemble humans than rodents in terms of transporter expression levels at the BBB (13). In addition, on the basis of previous evidence that erlotinib at high doses can inhibit ABCB1 and ABCG2 (14), we investigated the effect of high-dose erlotinib administration on the brain delivery of 11C-erlotinib.
MATERIALS AND METHODS
Animals
All animal use procedures were in accordance with the recommendations of the European Community (86/809/CEE) and the French National Committees (law 87/848) for the care and use of laboratory animals. The experimental protocol was evaluated by a local ethics committee for animal use (CETA/APAFIS 892). Animal experiments were performed on 3 adult male Papio anubis baboons (24–27 kg in weight during the study) obtained from a primatology station (Celphedia Station of Primatology).
Chemicals
Erlotinib hydrochloride was obtained from Apollo Scientific Ltd. Elacridar hydrochloride and 6-O-desmethyl erlotinib were purchased from Syncom BV.
Radiochemistry
11C-erlotinib was synthesized by 11C-methylation of 6-O-desmethyl erlotinib following a previously described procedure (15). 11C-erlotinib was formulated in 0.9% aqueous saline with 10% ethanol (v/v) at an approximate concentration of 50 MBq/mL for intravenous injection into the animals. The radiochemical purity of 11C-erlotinib was greater than 98%, and the specific activity at the end of synthesis was 140–300 GBq/μmol (n = 11).
Experimental Conditions
The PET experiments were performed under 3 conditions. Baseline 11C-erlotinib kinetics (n = 4) were compared with those obtained using infusion of high-dose erlotinib at a dose of 10 mg/kg/h (n = 4) and using infusion of elacridar at a dose of 12 mg/kg/h (n = 3). The administered dose of erlotinib was based on previous experiments in rodents (16). The administered dose of elacridar was based on the maximal possible solubility of the compound. Each baboon underwent one PET scan under each condition. One animal additionally received a second baseline and high-dose erlotinib scan. In each baboon, only one PET scan was performed per study day, with an interval of at least 15 d between scans on the same baboon. Erlotinib for intravenous infusion was formulated at a concentration of 4 g/L. Briefly, erlotinib hydrochloride (400 mg) was solubilized in sterile water for injection (50 mL) containing 6% (w/v) Captisol (Ligand Pharmaceuticals, Inc.). This solution was then mixed with 5% sterile aqueous glucose solution (w/v, 50 mL). Elacridar was formulated for intravenous administration in sterile aqueous glucose solution (2.5%, w/v) at a concentration of 5 g/L using an in-house–developed cosolvent strategy resulting in a final tetrahydrofuran concentration of less than 5% (v/v). The infusion rate of the solutions was 58–67 mL/h, depending on body weight, starting 30 min before 11C-erlotinib injection and continuing until the end of PET scanning (total infusion time, 90 min).
PET Experiments
Each monkey underwent a T1-weighted brain MR scan using an Achieva 1.5-T scanner (Philips) while under ketamine anesthesia (10 mg/kg, intramuscularly; Virbac). The PET experiments were subsequently performed on an HR+ Tomograph (Siemens Healthcare), with the animal anaesthetized as described previously (17). In brief, the animal first received ketamine (10 mg/kg, intramuscularly) to induce anesthesia while the animal was being prepared the PET experiment. After the animal had been intubated, a catheter was inserted into a sural vein for injection of the radiotracer and tested compounds. Another sural vein catheter was dedicated to propofol infusion.
After the animal had been positioned under the camera, anesthesia was maintained using an intravenous bolus of propofol (2 mL) followed by a 16- to 22-mL/h intravenous infusion under oxygen ventilation. The tidal volume was adjusted to achieve a stable end-tidal CO2 tension (partial pressure of CO2) of between 38 and 42 mm Hg. Heart rate and rectal temperature were monitored throughout the experiment.
The head of the animal was positioned in the tomograph using a custom-designed stereotaxic head holder. Head transmission scans were acquired for 10 min using 3 retractable 68Ge rod sources. Then, the animal was intravenously injected with 11C-erlotinib (255 ± 64 MBq, corresponding to an injected erlotinib mass of 5.2 ± 3.9 μg) and scanned for 60 min. Dynamic PET data were acquired over the brain in 3-dimensional mode with the animal supine. The animal was extubated approximately 15 min after the end of PET scanning and then was continuously monitored for at least 60 min. No influence of erlotinib or elacridar infusion on partial pressure of CO2, heart rate, rectal temperature, or respiratory frequency could be detected.
Arterial Input Function and Metabolism
A femoral artery catheter was used to draw blood samples for arterial input function assessment. Arterial plasma samples (6 mL) were drawn immediately before the start of the PET scan to measure erlotinib or elacridar concentrations using newly developed high-performance liquid chromatography methods that apply ultraviolet detection. During the PET acquisition, arterial plasma samples (0.5 mL) were withdrawn at 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 3, 5, 7.5, 10, 15, 20, 30, 40, 50, and 60 min after radiotracer injection, centrifuged, and counted. Additional plasma samples drawn at 0, 5, 10, 15, 30, and 60 min were used to estimate the percentage of unmetabolized parent 11C-erlotinib by radio–high-performance liquid chromatography analysis. For each animal, a 2-exponential decay function was fitted to the percentage of unmetabolized 11C-erlotinib versus time and then applied to the corresponding total radioactivity time–activity curve in plasma. Time–activity curves of unmetabolized parent 11C-erlotinib in plasma were expressed as SUV ([radioactivity per mL of plasma/injected radioactivity] × body weight). Parent 11C-erlotinib plasma exposure was estimated under all tested conditions by calculating the area under the plasma time–activity curve (AUC) from 0 to 60 min. AUC, expressed as SUV × min, correlated inversely with 11C-erlotinib plasma clearance (dose/AUC).
Data Reconstruction and Analysis
Measured attenuation and scatter corrections were applied to the emission data. The data were reconstructed using a 3-dimensional reprojection algorithm with an axial and transaxial Hanning filter for which the cutoff was set to the Nyquist frequency.
In each baboon, the following volumes of interest were manually drawn on T1-weighted MR images: whole brain hemispheres (58.3 ± 3.3 cm3), cerebellum (10.3 ± 0.5 cm3), striatum (1.8 ± 0.2 cm3), frontal lobe (9.0 ± 0.1 cm3), occipital lobe (6.6 ± 0.2 cm3), temporal lobe (10.4 ± 0.2 cm3), parietal lobe (11.5 ± 3.4 cm3), and temporal muscles surrounding the brain (21.5 ± 2.4 cm3). The 11C-erlotinib PET images acquired during elacridar infusion (representing the highest brain PET signal) were coregistered to the corresponding MR images. The baseline PET images and the PET images obtained during high-dose erlotinib infusion were then coregistered to the respective PET images acquired during elacridar infusion. The volumes of interest were then applied to all coregistered dynamic PET data to generate corresponding time–activity curves.
Kinetic modeling was performed using Logan plot analysis with the corresponding arterial plasma input function to estimate the total volume of distribution (VT; mL/cm3) for each region under all tested conditions (18). VT can be considered an estimate of the tissue-to-plasma concentration ratio of erlotinib at steady state. Image analysis was performed using PMOD software (version 3.6; PMOD Technologies Ltd.).
Statistical Analysis
Values are given as mean ± SD. Differences between groups were assessed on log-transformed data. A 2-way ANOVA followed by Tukey post hoc testing was performed using “treatment” (baseline, erlotinib, and elacridar) and “region” (brain and muscle) as factors. The level of statistical significance was set at a P value of 0.05.
RESULTS
Plasma Pharmacokinetics
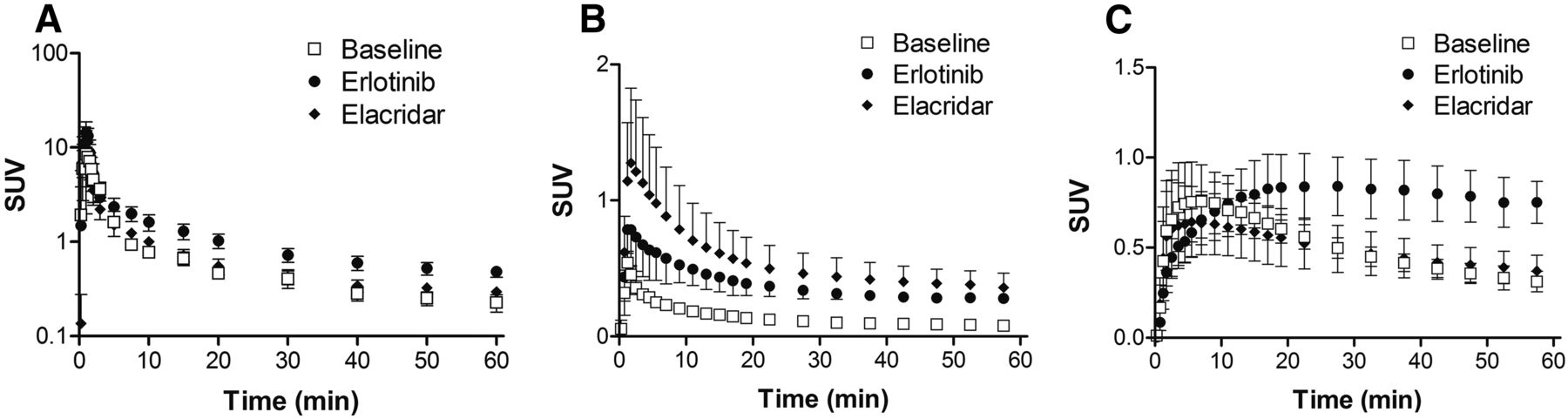
Infusion of high-dose erlotinib (10 mg/kg/h) resulted in an erlotinib concentration of 6.9 ± 0.3 μg/mL (approximately 16.0 μmol/L) in arterial plasma at the time of the PET experiment. In elacridar-treated animals, the elacridar concentration in plasma at the start of the PET experiment was 10.0 ± 1.5 μg/mL (∼16.7 μmol/L). 11C-erlotinib plasma pharmacokinetics were not influenced by elacridar coinfusion, with plasma AUC values of 43.0 ± 6.6 and 47.7 ± 6.6 SUV × min in baseline and elacridar-treated animals, respectively (Fig. 1A). Conversely, high-dose erlotinib significantly decreased 11C-erlotinib plasma clearance, giving a plasma AUC of 70.7 ± 6.6 SUV × min (P < 0.01; Fig. 1A). Time–activity curves of total radioactivity in plasma (i.e., the sum of parent 11C-erlotinib and radiolabeled metabolites) and the percentage of parent 11C-erlotinib with respect to total radioactivity in plasma under each condition are shown in Supplemental Figure 1 (supplemental materials are available at http://jnm.snmjournals.org). The percentage of parent 11C-erlotinib in plasma was lower in elacridar-treated animals than in baseline or high-dose erlotinib-treated animals, suggesting that elacridar may decrease the plasma clearance of radiolabeled metabolites of 11C-erlotinib (Fig. 1A and Supplemental Fig. 1).

Time–activity curves (SUVmean ± SD) in arterial plasma (A), brain (B), and temporal muscle surrounding skull (C) measured after injection of 11C-erlotinib in baseline (n = 4), high-dose erlotinib-treated (n = 4), and elacridar-treated (n = 3) animals. In A, time–activity curves are corrected for radiolabeled metabolites of 11C-erlotinib and display only unmetabolized parent 11C-erlotinib in plasma.
PET Experiments
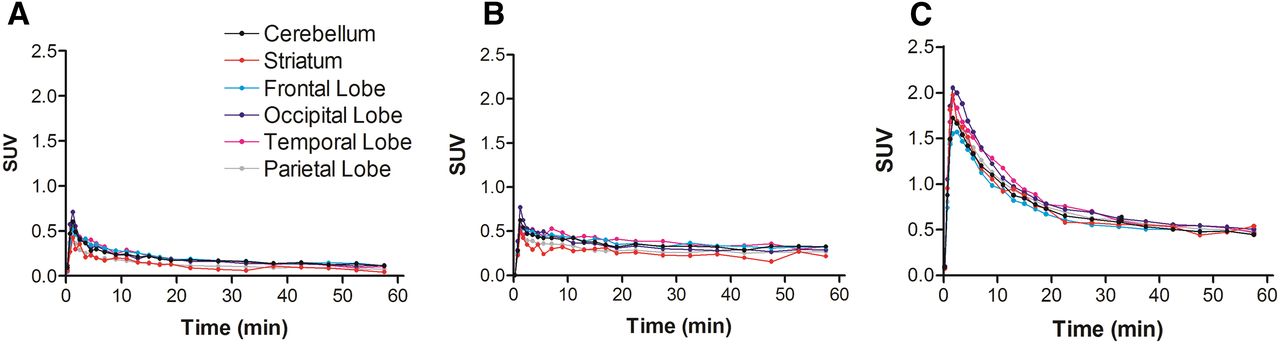
Baseline PET scans confirmed a very low brain distribution of 11C-erlotinib, with no regional retention of radioactivity (Figs. 2A and 3A). Brain radioactivity peaked rapidly (SUVmax, 0.54 ± 0.08; time of maximum uptake [Tmax], 1.25 min) followed by a rather fast decrease in brain radioactivity, with an uptake of 0.080 ± 0.01 at 60 min after injection (SUV60 min) (Figs. 1B and 2A). In high-dose erlotinib-treated animals, brain radioactivity peaked at a higher value (SUVmax, 0.78 ± 0.17; Tmax, 1.75 min), with a higher SUV60 min of 0.28 ± 0.02 (P < 0.05) (Figs. 1B and 2B). ABCB1/ABCG2 inhibition using elacridar resulted in a pronounced increase in SUVmax (1.28 ± 0.55; Tmax, 1.75 min), with a higher SUV60 min (0.36 ± 0.11) than the baseline SUV (P < 0.01) (Figs. 1B and 2C). Regional analysis showed that 11C-erlotinib kinetics were similar in different brain regions under all 3 tested conditions (Fig. 3), suggesting a nonspecific retention mechanism of 11C-erlotinib in monkey brain and lack of blood flow dependence on radiotracer uptake. Time–activity curves measured in temporal muscles surrounding the skull were similar in baseline (SUVmax, 0.76 ± 0.20; Tmax, 5.5 min; SUV60 min = 0.31 ± 0.06) and elacridar-treated animals (SUVmax, 0.64 ± 0.20; Tmax, 7 min; SUV60 min, 0.37 ± 0.09) (Fig. 1C). Radioactivity measured in the temporal muscles of high-dose erlotinib-treated animals peaked at higher concentrations (SUVmax, 0.84 ± 0.16), with a delayed Tmax of 27.5 min and a higher SUV60 min of 0.75 ± 0.12 (P < 0.001), than in baseline or elacridar-treated animals. This finding may mainly reflect changes in 11C-erlotinib plasma pharmacokinetics in this highly vascularized tissue (Fig. 1C).
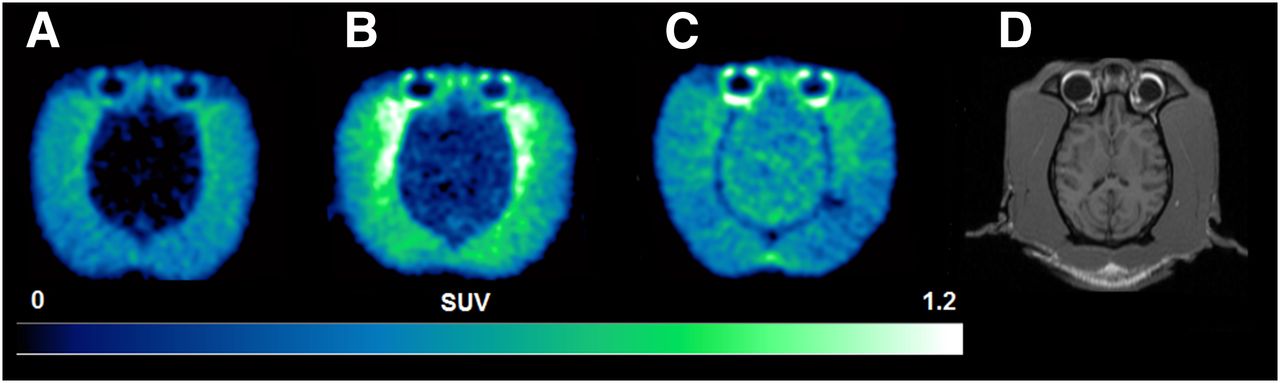
(A–C) Representative horizontal 11C-erlotinib PET summation images (10–60 min) obtained in brain of baseline (A), high-dose erlotinib-treated (B), and elacridar-treated (C) animals. Radioactivity concentration is normalized to injected dose per body weight, expressed as SUV, and set from 0 to 1.2. (D) Coregistered T1-weighted MR image that was used to delineate different brain regions.

Representative time–activity curves of 11C-erlotinib in different brain regions for baseline (A), high-dose erlotinib-treated (B), and elacridar-treated (C) animals.
The PET data were modeled to estimate the tissue distribution of 11C-erlotinib from arterial plasma under each tested condition (Supplemental Table 1). Baseline 11C-erlotinib distribution to the brain (VT, 0.22 ± 0.015 mL/cm3) was significantly enhanced by high-dose erlotinib (VT, 0.38 ± 0.033 mL/cm3; 1.7 ± 0.2-fold increase, P < 0.05) and by elacridar treatment (VT, 0.81 ± 0.21 mL/cm3; 3.5 ± 0.9-fold increase, P < 0.01) (Fig. 4). There were no significant differences in VT values in the temporal muscle between the baseline (VT = 0.86 ± 0.10 mL/cm3), high-dose erlotinib (VT, 1.04 ± 0.06 mL/cm3), and elacridar (VT, 0.92 ± 0.19 mL/cm3) conditions, indicating BBB-specific treatment effects (Fig. 4). VT values were significantly higher in the temporal muscle (P < 0.001) than in the brain for baseline and erlotinib-treated animals but not for elacridar-treated animals (Fig. 4).
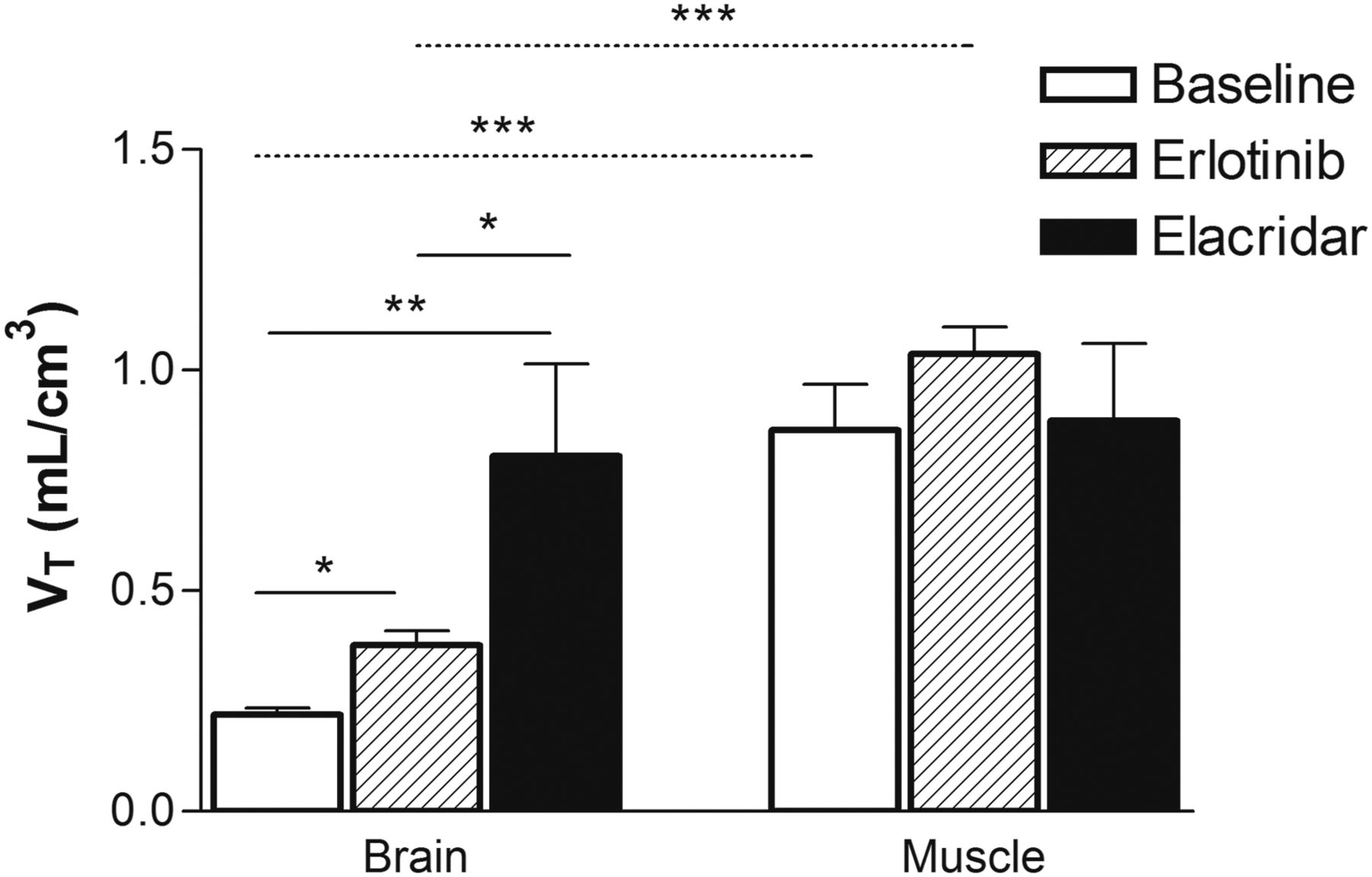
VT of 11C-erlotinib estimated with Logan graphical analysis in brain and temporal muscle surrounding skull for baseline, high-dose erlotinib-treated, and elacridar-treated animals. *P < 0.05, 2-way ANOVA with Tukey post hoc testing. **P < 0.01, 2-way ANOVA with Tukey post hoc testing. ***P < 0.001, 2-way ANOVA with Tukey post hoc testing.
DISCUSSION
Several previous PET studies on humans have examined the effect of ABCB1 inhibitors such as cyclosporine A or tariquidar on the brain distribution of radiotracers that are transported only by ABCB1 at the BBB, such as racemic 11C-verapamil, (R)-11C-verapamil, and 11C-N-desmethyl-loperamide (10,11,19). In contrast to the inhibition of ABCB1, the combined inhibition of both ABCB1 and ABCG2 has so far not been exploited at the human BBB because of the lack of suitable dual ABCB1/ABCG2 inhibitors for clinical use. Elacridar, a known ABCB1/ABCG2 inhibitor, was initially developed as a multidrug resistance reversal agent (20), but its further clinical development was stopped, most likely because of failure of the compound to improve the outcome in clinical oncology trials. Elacridar was available only as an oral formulation for administration to humans and suffered from poor bioavailability, which considerably limits plasma exposure (7,21). Numerous rodent studies have shown that intravenous administration of elacridar can substantially inhibit Abcb1a and Abcg2 at the rodent BBB, resulting in pronounced increases in the brain distribution of various dual ABCB1/ABCG2 substrate drugs (3,4).
This study is, to our knowledge, the first in which the effect of dual ABCB1/ABCG2 inhibition has been assessed in vivo at the nonhuman primate BBB. We studied the brain distribution of erlotinib, which was previously shown to be a substrate of both rodent and human ABCB1 and ABCG2 (8,22,23). Erlotinib can be straightforwardly labeled with 11C (15). Our previous study showed that the brain distribution of 11C-erlotinib in mice is restricted by Abcb1a and Abcg2 and that radiolabeled metabolites of 11C-erlotinib are not taken up by the brain (16). As compared with wild-type mice, distributional clearance of 11C-erlotinib to the brain was 4.6-fold higher in Abcb1a/b(−/−)Abcg2(−/−) mice, 5.3-fold higher in elacridar-pretreated wild-type mice, and 4.5-fold higher in high-dose erlotinib-pretreated wild-type mice, suggesting that both elacridar and high-dose erlotinib can inhibit Abcb1a- and Abcg2-mediated transport of 11C-erlotinib at the mouse BBB (16).
The most important finding of our study is that ABCB1/ABCG2 inhibition with elacridar can be achieved at the nonhuman primate BBB, resulting in an increase in brain distribution of 11C-erlotinib (3.5-fold increase in brain VT) remarkably similar to that in rodents (3.4-fold increase in brain-to-plasma concentration ratio at 25 min after radiotracer injection) (16). On the basis of earlier findings that the transporter inhibitory effect of elacridar at the BBB is rapidly reversible (Supplemental Fig. 2), we administered elacridar as a continuous intravenous infusion that was maintained for the duration of the PET scan, similar to PET studies with the ABCB1 inhibitor tariquidar (10,11). Importantly, elacridar administration had no influence on the plasma pharmacokinetics of 11C-erlotinib (Fig. 1A), indicating that increases in exposure of the brain to 11C-erlotinib were due only to transporter inhibition at the BBB. The limited impact of elacridar on 11C-erlotinib plasma pharmacokinetics is consistent with another study showing that total plasma clearance of erlotinib was comparable in wild-type and Abcb1a/b(−/−)Abcg2(−/−) mice (22). Elacridar also had no effect on 11C-erlotinib pharmacokinetics in muscle tissue surrounding the skull (Fig. 1C), further supporting the possibility that the effect of elacridar on 11C-erlotinib distribution specifically depends on the presence of ABCB1 and ABCG2 at the blood–tissue interface. During elacridar infusion, 11C-erlotinib brain distribution was on a level similar to 11C-erlotinib muscle distribution (Fig. 4). Moreover, during elacridar infusion, 11C-erlotinib VT in brain (0.81 ± 0.21 mL/cm3) was comparable to that in primary lung tumors of NSCLC patients without activating EGFR mutations (0.67–1.22 mL/cm3) (15). This indicates that the elacridar concentrations achieved in plasma in our study can effectively overcome the “sanctuary” property of the brain compared with peripheral tissues.
The second important finding of our study was that high-dose erlotinib coinfusion during the PET scan led to a significant 1.7-fold increase in 11C-erlotinib brain VT as compared with baseline experiments, in which only a microdose (<10 μg) of 11C-erlotinib was administered (Fig. 4). This finding suggests that erlotinib may partially saturate its own transport by ABCB1 and ABCG2 at the BBB, leading to improved brain penetration at higher doses. This possibility is supported by some clinical evidence showing that pulsatile high-dose erlotinib administration (up to oral doses of 2,000 mg) resulted in a partial but favorable treatment response in NSCLC patients with brain metastases (24,25). The increase in 11C-erlotinib brain VT during erlotinib infusion was markedly smaller than the VT increase observed in elacridar-treated animals, suggesting that erlotinib is a less potent transporter inhibitor than elacridar and, at the used dose, led to only to incomplete ABCB1 and ABCG2 inhibition at the BBB. In contrast to elacridar treatment, high-dose erlotinib infusion caused a significant 1.6-fold increase in plasma exposure of 11C-erlotinib (Fig. 1A), which might have been due to transporter inhibition in clearance organs such as the liver. Similar increases in 11C-erlotinib plasma exposure after high-dose erlotinib administration that were observed in a mouse study were attributed to inhibition of uptake transport by the liver (16). It is noteworthy that erlotinib plasma concentrations at the time of the PET scan (∼16.0 μmol/L) were in the same range as peak plasma concentrations in NSCLC patients treated with pulsatile high-dose erlotinib (14.5–22.7 μmol/L for an oral dose of 1,600 mg (24)). For comparison, peak erlotinib plasma concentrations after standard clinical dosing (150 mg oral) were reported as 3.7 ± 1.4 μmol/L (26), which is most likely too low to inhibit ABCB1/ABCG2 at the BBB. Our findings suggest that the improved response rates observed with high-dose erlotinib in NSCLC patients with brain metastases may at least partly be due to increases in erlotinib distribution to brain metastases by saturation of ABCB1- and ABCG2-mediated efflux at the BBB. High-dose erlotinib may thus achieve a significant degree of ABCB1/ABCG2 inhibition at the human BBB at clinically feasible doses. Given that erlotinib is a marketed drug that is available as an oral formulation, high-dose erlotinib, which proved to be well tolerated in cancer patients at doses of up to 2,000 mg (24), may thus offer an interesting alternative to elacridar for inhibition of ABCB1 and ABCG2 at the human BBB to increase brain distribution of drugs that are dual ABCB1/ABCG2 substrates (e.g., other TKIs). In combination with other anticancer drugs, high-dose erlotinib may additionally exhibit a synergistic antitumor effect. Further work is therefore needed to find out whether this new inhibition protocol will be equally effective for ABCB1/ABCG2 substrates other than erlotinib.
Because nonhuman primates closely resemble humans in transporter expression levels at the BBB (13), our nonhuman primate data strongly suggest that strategies to inhibit ABCB1 and ABCG2 at the BBB may be translatable to humans. A limitation of this study is the use of healthy animals without brain tumors. It is not known whether NSCLC patients with activating EGFR mutations whose systemic disease is controlled by TKI treatment and who have developed central nervous system metastases during the course of the disease may benefit from ABCB1/ABCG2 inhibition at the BBB. Overexpression of EGFR is also frequently found in glioblastoma and is often associated with the most malignant phenotype of the disease and a poor clinical outcome (27). The clinical efficacy of EGFR-targeted therapy such as erlotinib (daily oral doses of up to 500 mg) proved disappointing in these patients (28). The invasive potential of glioma causes widespread proliferation of high-grade glioma cells outside regions of the tumor core and inside areas of otherwise normal brain, where the function of the BBB is intact. In addition, ABC transporters can be expressed in tumor cells as well and may confer intrinsic chemoresistance, thus forming an additional hurdle against treatment (5,7). Clinical trials are therefore necessary to assess whether antitumor response rates can be improved using ABCB1/ABCG2 inhibition strategies.
CONCLUSION
We tested the efficacy of elacridar and high-dose erlotinib to increase the brain distribution of the dual ABCB1/ABCG2 substrate 11C-erlotinib in nonhuman primates as a model of the human BBB. Both compounds resulted in significant increases in brain distribution of 11C-erlotinib. We report for the first time the target plasma concentrations at which these two transporter inhibitors effectively inhibited ABCB1 and ABCG2 at the BBB. Our findings suggest that the use of dual ABCB1/ABCG2 inhibition at the BBB to improve brain delivery of TKIs may become clinically feasible.
DISCLOSURE
Sylvain Auvity and Michael Soussan received a public grant overseen by the French National Research Agency (ANR) as part of the “Investissement d’Avenir” program, through the “Lidex-PIM” project funded by the IDEX Paris-Saclay, ANR-11-IDEX-0003-02. Oliver Langer received grants from the Lower Austria Corporation for Research and Education (NFB) (LS12-006) and from the Austrian Science Fund (FWF) (F 3513-B20, KLI 480-B30). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Aug. 4, 2016.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication May 27, 2016.
- Accepted for publication July 12, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Impact of P-Glycoprotein-Mediated Active Efflux on Drug Distribution into Lumbar Cerebrospinal Fluid in Nonhuman Primates
- A Proof-of-Concept Study to Inhibit ABCG2- and ABCB1-Mediated Efflux Transport at the Human Blood-Brain Barrier
- P-Glycoprotein (ABCB1) Inhibits the Influx and Increases the Efflux of 11C-Metoclopramide Across the Blood-Brain Barrier: A PET Study on Nonhuman Primates
- Molecular Imaging of ABCB1 and ABCG2 Inhibition at the Human Blood-Brain Barrier Using Elacridar and 11C-Erlotinib PET
- Elucidation of the Impact of P-glycoprotein and Breast Cancer Resistance Protein on the Brain Distribution of Catechol-O-Methyltransferase Inhibitors