


Abstract
Positive allosteric modulators (PAM) of metabotropic glutamate receptor 2 (mGluR2) are a potential therapy for anxiety, schizophrenia, and addiction. Aside from pathophysiologic imaging studies, an mGluR2 PET tracer would enable confirmation of sufficient central target engagement and assist dose selection for proof-of-concept studies of PAM compounds. 11C-JNJ-42491293, a novel high-affinity radioligand (human 50% inhibitory concentration = 9.6 nM) for the PAM site of mGluR2, was evaluated as a selective mGluR2 PAM PET tracer. Methods: In vitro and ex vivo autoradiography binding experiments in Wistar and in mGluR2 knockout and wildtype rats as well as in vivo biodistribution and brain PET imaging studies in wildtype and mGluR2 knockout rats in a primate and in humans were performed. Results: In vitro binding studies and in vivo imaging studies in Wistar rats showed moderate brain uptake, with a distribution pattern fully consistent with the reported intracerebral distribution of mGluR2. Given these promising findings, biodistribution, dosimetry, and brain kinetic modeling of 11C-JNJ-42491293 were determined in humans. Because of an unexpected high myocardial retention, additional 11C-JNJ-42491293 imaging studies were performed in recently available mGluR2 knockout and wildtype rats and in a monkey using a structurally distinct mGluR2 PAM ligand with affinity for the same site, demonstrating off-target binding in vivo that could not have been anticipated from previous in vitro experiments. To date, the target of this non-mGluR2 tracer binding remains unknown. Conclusion: On the basis of in vivo selectivity issues suggested by human distribution and demonstrated in knockout rat models, 11C-JNJ-42491293 was considered unsuitable as a specific PET ligand for in vivo imaging of mGluR2. These results emphasize the importance of elaborated in vitro/in vivo comparative studies and, when available, validation with knockout animal models or structurally distinct ligands with affinity for the same site, in radiotracer development.
Glutamate is the major excitatory neurotransmitter in the central nervous system of vertebrates that elicits and modulates synaptic responses by activating 2 families of receptors, the ligand-gated cation channels, termed ionotropic glutamate receptors, and G-protein–coupled glutamate receptors, termed metabotropic glutamate receptors (mGluRs) (1). By their modulating effects on neurotransmission, mGluRs are involved in multiple normal and pathologic brain processes. In particular, mGluR2 is primarily expressed presynaptically where it reduces glutamate release and protects neurons from excitotoxicity, modulates dopaminergic and adrenergic neurotransmission, and participates in synaptic plasticity. As such, mGluR2 has been implicated in the pathogenesis of several neurologic and psychiatric disorders including schizophrenia (2), addiction (3), anxiety (4), major depression (5), and neurodegeneration (6,7). The cerebral distribution of mGluR2 messenger RNA and protein has been described in rodents (8,9) and humans (10,11), with expression throughout the cortex, striatum, thalamus, and cerebellum. Augmenting mGluR2 function either by a direct orthosteric agonist or by a positive allosteric modulator (PAM) is expected to reduce activity-dependent glutamate release. Several orthosteric agonists for mGluR2 have been identified and have shown beneficial effects in preclinical behavioral models and in clinical conditions including schizophrenia (2), anxiety (4), and addiction (12) but show narrow therapeutic margins. mGluR2 PAMs are potentially more selective and give a more nuanced approach to enhancing mGluR2 function (13).
PET neuroimaging is a valuable tool for assessing the neurobiology of brain disorders by its unique ability to visualize and quantify molecular processes in vivo (14). The development of PET ligands targeting mGluR binding sites has been an active research field over the last decades. The first efforts included radiolabeling of derivatives of the mGluR2/3 orthosteric agonist LY354740. However, these compounds proved difficult to label and demonstrated negligible brain uptake. Exploratory studies with tritiated LY341495, an antagonist derivative, showed modest brain uptake but no evidence of specific binding in competition studies (15). A principal limitation of this source of lead compounds was that they target the orthosteric binding site for endogenous glutamate. This binding site is highly conserved, and the structural requirements for pharmacophores that occupy only the mGluR2 binding pocket are quite restrictive. For instance, the recently developed PET radioligand 11C-CMGDE (16) lacks specificity for mGluR2 as it also binds to mGluR3. Drugs and imaging ligands targeting allosteric binding sites on mGluRs have been more tractable, and negative allosteric modulator PET ligands have been developed for mGluR1 (17–19) and mGluR5 (20). A radioligand that enables selective quantitative assessment of mGluR2 would be a major step forward in understanding the role of this receptor in the neurobiology of multiple brain disorders and a valuable tool for testing target engagement and dose occupancy of clinical drug leads with affinity for the same receptor site (5,21).
Our group published the radiosynthesis and preliminary characterization in rats of 11C-JNJ-42491293 (Fig. 1) (22). In vitro, 11C-JNJ-42491293 binds to an allosteric modulator site of the human mGluR2 with 9.6 ± 3.8 nM binding affinity and reversible kinetics and is displaceable by the highly potent mGluR2 PAM JNJ-42153605 (50% inhibitory concentration [IC50] = 15 nM) (Fig. 1) (23). Extensive screening in vitro against cerebral targets (receptors/ion channels) revealed no significant off-target affinities. Initial preclinical in vivo evaluation, involving biodistribution, plasma, and brain radiometabolite analysis, and preliminary small-animal PET studies in Wistar rats suggested that 11C-JNJ-42491293 would be a promising PET radioligand for in vivo brain imaging of mGluR2 (22). The purpose of the present study was to further evaluate this potential of 11C-JNJ-42491293 in both animals and humans.
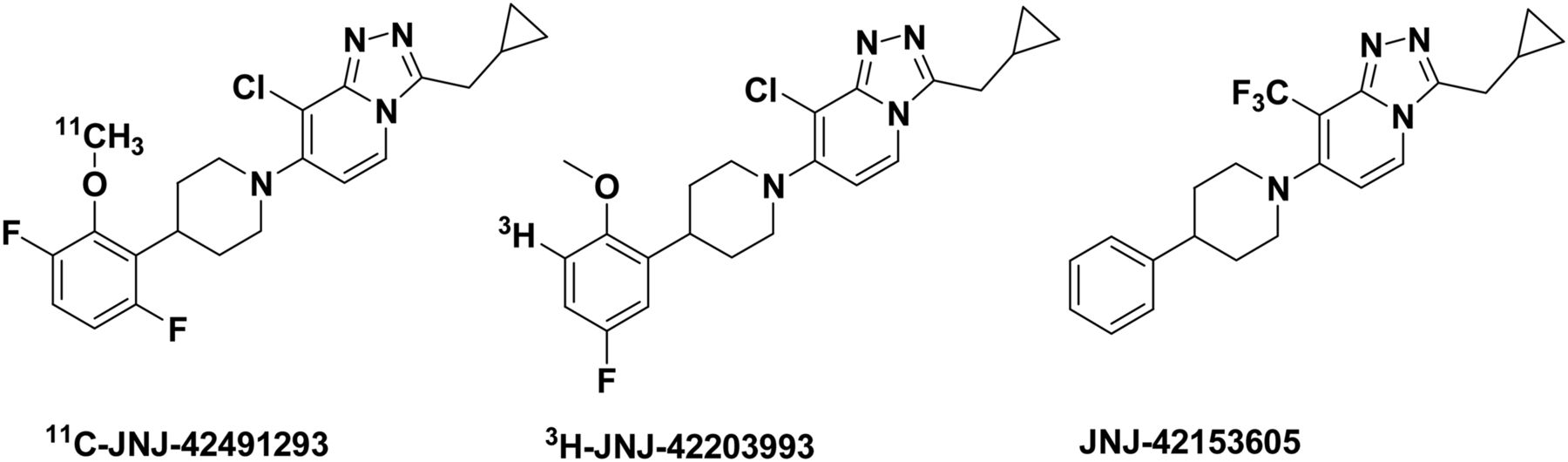
Chemical structure of PET tracer 11C-JNJ-42491293, its tritiated analog 3H-JNJ-42203993, and mGluR2 PAM JNJ-42153605 used in the blocking and displacement studies.
MATERIALS AND METHODS
Chemistry
The synthesis and radiolabeling of JNJ-42491293 were performed according to the method described by Andrés et al. (22). For autoradiography studies in mGluR2 knockout (KO) and wildtype (WT) rats, 3H-JNJ-42203993 (22), a tritiated analog of JNJ-42491293, was used (Fig. 1). The mGluR2 PAMs JNJ-42153605 (23) (Fig. 1) and JNJ-46356479 (Andrés et al., unpublished, data, 2011) used for blocking and displacement experiments were synthesized by Janssen Research and Development. JNJ-46356479 belongs to the same 1,2,4-triazolo[4,3-a]pyridine chemical series as JNJ-42491293, JNJ-42153605, and JNJ-42203993, but with a different substitution pattern. One of the main structural differences between JNJ-46356479 and the other 3 compounds, which are shown in Figure 1, is that JNJ-46356479 has a basic nitrogen whereas the piperidine nitrogen of the other 3 derivatives is not basic.
Animals
Rodent studies were conducted in healthy Wistar rats and in mGluR2 KO rats and their WT littermates (weight, 200–600 g). The Wistar rats (RjHan:WI or Crl:WI (Han)) were provided by Janvier or Charles River, and the mGluR2 KO and WT rats were provided by Janssen Research and Development. A macaque monkey was provided by the Laboratory for Neuro- and Psychophysiology (KU Leuven). All animal studies were approved by the local Animal Ethics Committee and were performed according to European Ethics Committee guidelines (decree 86/609/EEC).
Autoradiography Binding Studies
In Vitro Autoradiography with 11C-JNJ-42491293
Before decapitation, brain removal, and slice preparation, Wistar rats were anesthetized (2.5% isoflurane in O2 at 1 L/min flow rate). Twenty-micrometer-thick horizontal and sagittal brain sections were preincubated for 2 × 10 min at room temperature in 50 mM Tris-HCl buffer, pH 7.0, in the presence of 2 mM MgCl2 and 2 mM CaCl2. After being dried, the sections were incubated with 11C-JNJ-42491293 (110 kBq/mL), in the absence or presence of 10 μM JNJ-42153605 (in MeOH), for 30 min at room temperature. The sections were then rinsed 3 × 5 min with 50 mM Tris-HCl buffer, pH 7.0, in the presence of 2 mM MgCl2, 2 mM CaCl2, and 0.3% bovine serum albumin at 4°C. After a quick dip in ice-cold distilled water, the slides were dried and exposed to a high-performance phosphor storage screen (superresolution screen; Perkin Elmer) for 30 min. The screens were read using a Cyclone Plus system (Perkin Elmer) and analyzed using Optiquant software (Perkin Elmer).
Ex Vivo and In Vitro Autoradiography with 3H-JNJ-42203993
Male mGlu2 KO (n = 4) and WT (n = 4) rats were treated by intravenous injection of 3H-JNJ-42203993 (370 kBq) 30 min before sacrifice (without anesthesia). Brains were removed and frozen in dry-ice-cooled 2-methylbutane (−40°C). Twenty-micrometer-thick sections were cut and loaded in a β-imager (Biospace) for 20 h, and radioactivity emerging from delineated brain area was quantified using the β-vision program (Biospace). In vitro autoradiography binding studies were performed using adjacent sections from the same animals. After being thawed, sections were dried under a stream of cold air and washed 2 × 10 min in preincubation buffer (Tris-HCl 50 mM, pH 7.4, containing 2 mM MgCl2 and 2 mM CaCl2). After incubation with 3 nM 3H-JNJ-42203993, the excess of radioligand was washed off 3 × 5 min in ice-cold incubation buffer and rinsed in distilled water. Nonspecific binding was measured on adjacent sections, in the presence of 10 μM JNJ-42341806 (another mGluR2 PAM). After being dried, slides were placed in light-tight cassettes and exposed to Fujix IP plates for 7 d. Notably, the in vivo binding of 3H-JNJ-42203993 did not interfere with the signal related to the in vitro mGlu2 binding, because it was too weak to be detected with Fujix IP plates. Fujix IP plates were scanned using FLA-7000 (Life Science).
Perfused Rat Heart Radiometabolite Analysis
The same procedure as previously described for perfused brain radiometabolite analysis was applied (22).
Animal PET Imaging Studies
The small-animal PET acquisition protocol has been described previously (22). During all scan sessions, the rats were kept under gas anesthesia (2.5% isoflurane in O2 at 1 L/min flow rate). The monkey was sedated with ketamine (Ketalar; Pfizer Ltd.) and xylazine (Rompun; Bayer Ltd.). Rats and the monkey received an intravenous slow bolus injection of 40–54 and 201–215 MBq of 11C-JNJ-42491293, respectively. For rats, pretreatment studies were performed by subcutaneous injection with 20 mg/kg of unlabeled JNJ-42491293 at 60 min before radiotracer injection. Displacement studies were performed by intravenous injection of JNJ-42153605 (0.1 mg/kg), JNJ-46356479 (4 mg/kg), and ritanserin (0.3 mg/kg) during scanning at 30 min after radiotracer injection. The monkey used in the baseline study was also scanned after pretreatment with JNJ-46356479 (1 mg/kg, intravenously) at 36 min before tracer injection.
Small-animal PET data were analyzed as previously described (22). For the monkey, the 112RM-SL T1-weighted macaque atlas was used (24). Regional time–activity curves are expressed using SUVs as SUV = activity in volume of interest (Bq/cm3) × body weight (g)/injected activity (Bq). For kinetic modeling using the metabolite-corrected input function, a 1-tissue-compartment model, 2-tissue-compartment model, and Logan plot were fitted to the volume-of-interest PET data.
Input Function and Radiometabolite Analysis
Before Wistar rats were scanned (n = 5), the femoral artery was cannulated under isoflurane anesthesia using polyethylene tubing. After radiotracer injection, 3 droplets of arterial blood were collected each 15 s for the first 3 min, and larger samples (100–200 μL) were taken at 4, 6, 8, 10, 15, 20, 25, 35, 40, 50, 60, and 85 min. Plasma was counted for radioactivity in a γ-counter. Plasma samples collected at 4, 10, 20, 40, 60, and 85 min after injection were additionally analyzed for radiometabolites following the procedure of Andrés et al. (22).
Imaging Studies in Humans
Subjects
Six healthy male volunteers (age range, 19–36 y) were recruited by advertisements in University Hospital and community newspapers. All subjects had normal physical examination; blood and urine tests, including urine toxicology; and high-resolution T1-weighted brain magnetization-prepared rapid gradient-echo sequence MRI (Philips Ingenia, Health care), and had no history of neurologic or psychiatric disease. Three subjects participated in a biodistribution or dosimetry part and 3 in a brain kinetic modeling part. The study was approved by the local Ethics Committee, and all subjects gave written informed consent. The clinical trial registration number was NCT01359852 (http://www.clinicaltrials.gov/).
PET Imaging
Scans were acquired on a HiRez PET/CT camera (Siemens) after an intravenous bolus injection of 323 ± 29 MBq of 11C-JNJ-42491293. Acquisition and analysis procedures for the biodistribution or dosimetry (25) and brain imaging (26) studies have been described previously.
For brain kinetic modeling, the automated anatomic labeling atlas was used for volume-of-interest definition. Using arterial radiometabolite-corrected input function, we derived the 11C-JNJ-42491293 total volume of distribution (VT) from 1-tissue- and 2-tissue-compartmental models and from the Logan approach using PMOD software (version 3.1; PMOD).
RESULTS
In Vitro Autoradiography Binding Studies in Wistar Rat
11C-JNJ-42491293 binding was evaluated in vitro using healthy rat brain sections. Figure 2A shows the anatomic distribution of binding sites with high uptake in the cortex, cerebellum, caudate–putamen, hippocampus, and thalamus, consistent with the reported brain localization of mGluR2 in rodents (9). Addition of the mGluR2 PAM ligand JNJ-42153605 effectively blocked binding (Fig. 2B), further suggesting the specificity of 11C-JNJ-42491293 for mGluR2. A similar binding pattern was obtained when rat brain sections were incubated with 3H-JNJ-42203993 and with 3H-LY341495 (not shown).
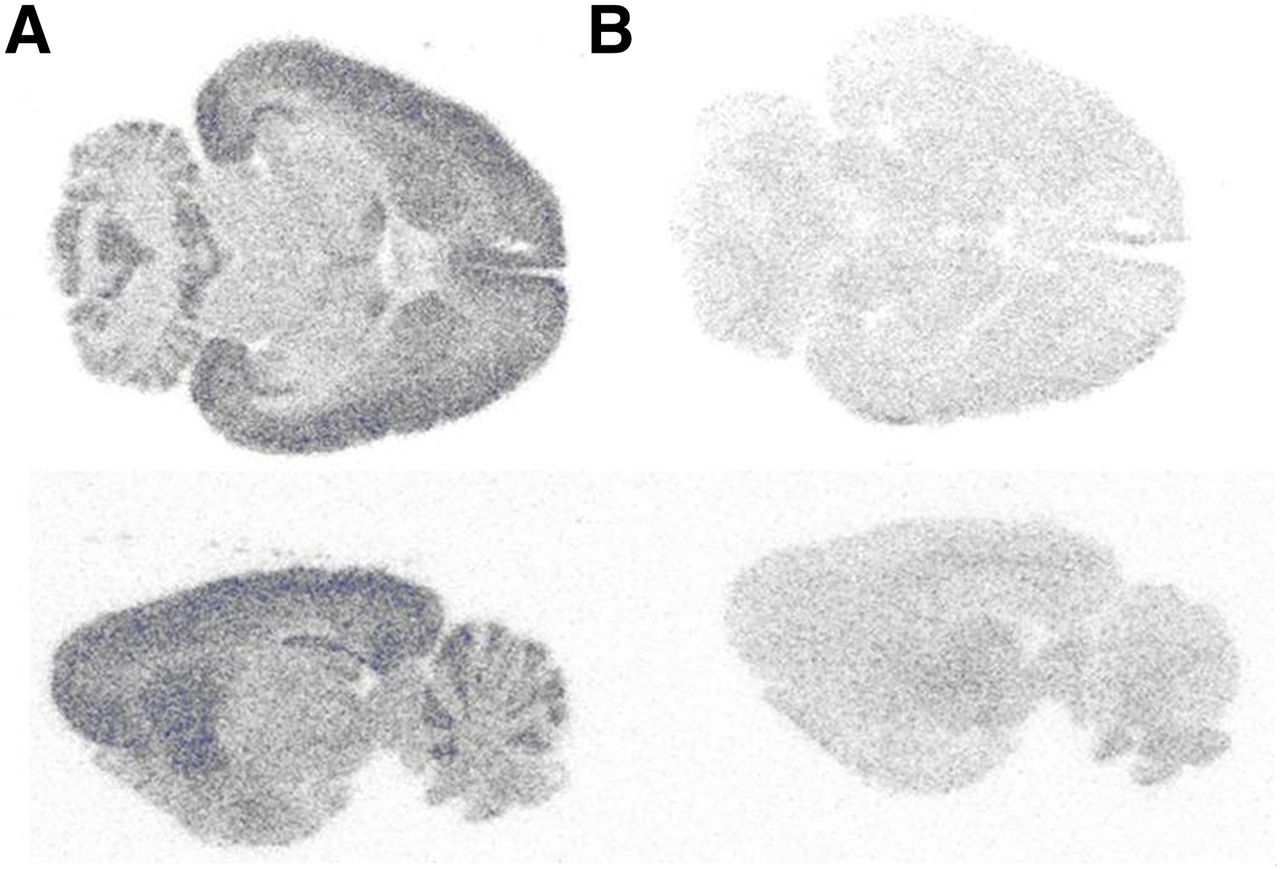
In vitro autoradiographic distribution of 11C-JNJ-42491293 bound to normal rat brain. Horizontal and sagittal sections (20 μm) of rat brain incubated with 11C-JNJ-42491293 (110 kBq/mL) for 30 min. Specific binding was assessed by incubating adjacent sections without (A, total tracer binding) and with (B, nonspecific binding) JNJ-42153605 (10 μM).
Brain Small-Animal PET Imaging in Wistar Rats
Baseline and Displacement Studies
As previously reported (22), baseline 11C-JNJ-42491293 brain uptake occurred diffusely in the cortex, cerebellum, and subcortical regions in accordance to the known mGluR2 distribution in rats, with highest uptake in the caudate–putamen and cerebellum. Dynamic imaging showed rapid gray matter uptake, peaking at 10–20 min after tracer injection, then slowly clearing from the brain during the next hour. To determine whether the observed 11C-JNJ-42491293 binding was reversible, imaging studies were repeated with an intravenous bolus administration of the high-affinity mGluR2 PAM JNJ-42153605, injected 30 min after 11C-JNJ-42491293. Administration of JNJ-42153605 (0.1 mg/kg) resulted in a 62% reduction in SUVmean (Fig. 3). Also, to test potential cross selectivity, displacement studies were performed with the 5HT2A receptor antagonist ritanserin (0.3 mg/kg, intravenously, 30 min after tracer injection, n = 2). Ritanserin failed to produce any change in radiotracer regional uptake (Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org), indicating no shared affinity of 11C-JNJ-42491293 for the 5HT2A receptor, unlike another mGluR2 PAM compound (27).

In vivo 11C-JNJ-42491293 small-animal PET displacement study with JNJ-42153605 (0.1 mg/kg, intravenous, 30 min after tracer injection) in normal rat brain. (A) Average coronal image through basal ganglia and cerebellum before (12.5–27.5 min after radiotracer injection) and after (67.5–87.5 min) administration of JNJ-42153605. Color scale indicates SUV units and images are in radiologic orientation. (B) Corresponding 11C-JNJ-42491293 time–activity curves. Data are corrected for physical decay of 11C.
Kinetic Modeling
The 1-tissue-compartment model and Logan plot provided stable fitting for all baseline studies in rats, and a good correlation was observed between the VT calculated with the Logan approach and the 1-tissue-compartment model (Supplemental Fig. 2).
From these preclinical studies in healthy Wistar rats, it was concluded that 11C-JNJ-42491293 behaves as a selective PET ligand for the mGluR2 PAM site and has competitive binding with JNJ-42153605 in rodents. 11C-JNJ-42491293 was therefore considered promising for mGluR2 PET imaging in humans.
In Vivo Human PET Imaging
Biodistribution and Dosimetry
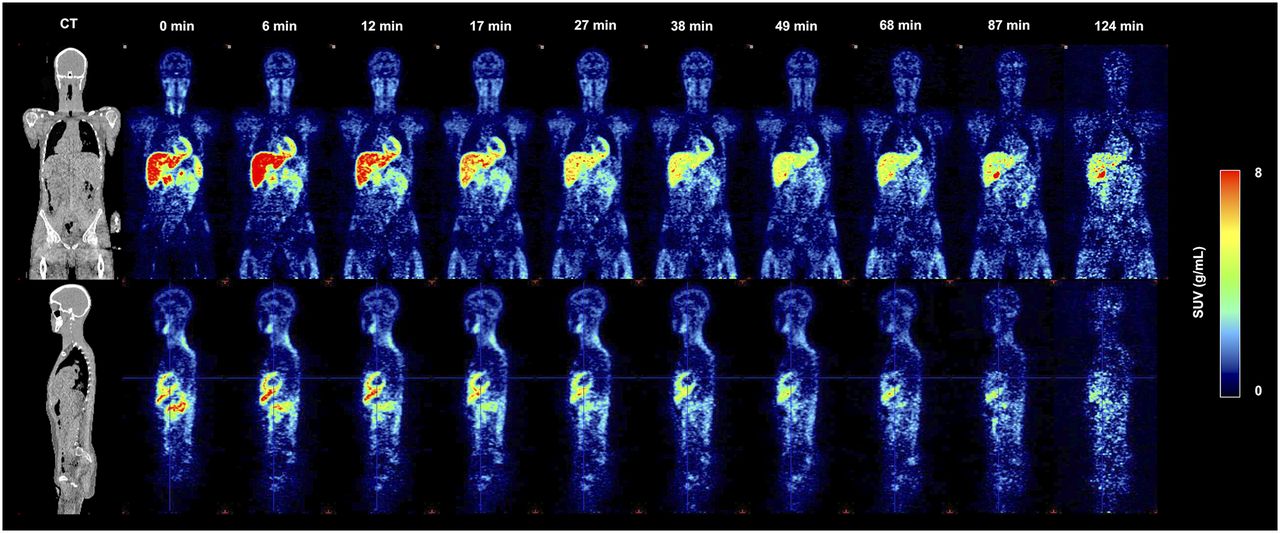
Dynamic whole-body imaging after tracer injection showed rapid brain uptake, high retention in the cardiac wall, and a predominant hepatobiliary clearance route (Fig. 4). The effective radiation dose determined in 3 healthy male volunteers was 4.53 ± 0.73 μSv/MBq. The organ-absorbed doses are presented in Supplemental Table 1. For 300 MBq of injected activity, 11C-JNJ-42491293 would result in 1.4 mSv effective dose.

Time–activity biodistribution of 11C-JNJ-42491293 in a human body. Coronal and parasagittal decay-corrected SUV images at 10 time points after tracer injection. Notice high uptake in myocardium. For anatomic reference, corresponding low-dose CT scan is shown on left.
Brain Imaging
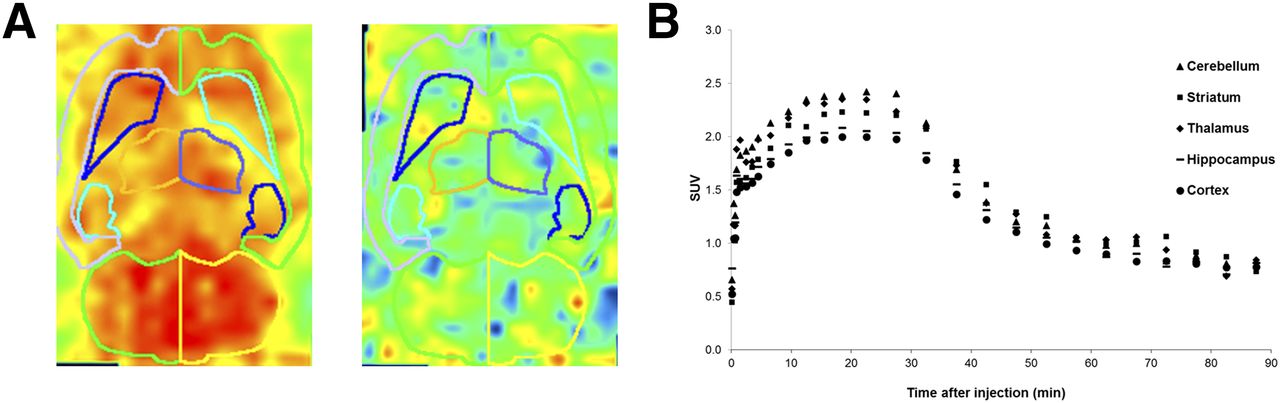
11C-JNJ-42491293 exhibited reversible and relatively fast kinetics, with brain uptake reaching a maximum at approximately 30 min after bolus injection and a gradual brain washout over the next hour (Fig. 5A). When a radiometabolite-corrected arterial input function was used, both a 2-tissue-compartmental model and a Logan plot provided good fit to the data. The highest VT values were observed in the cerebellum, striatum, thalamus, and cortex, whereas intermediate values were seen in the pons and hippocampus, and the lowest detected in white matter (Fig. 5B).

Typical 11C-JNJ-42491293 time–activity curves (A) and VT (B) in human brain.
Radiometabolite Analysis
The metabolism of 11C-JNJ-42491293 in arterial plasma is shown in Supplemental Figure 3. At 15 min after injection, 52% ± 9% of the total radioactivity in plasma corresponded to 11C-JNJ-42491293, declining to 24% ± 7% at 90 min.
Safety
11C-JNJ-42491293 was well tolerated at an injected activity and mass of 321 ± 18 MBq (range, 396–345 MBq) and 0.75 ± 0.38 μg (range, 0.40–1.00 μg), respectively. All reported adverse events were mild in intensity and were not considered to be related to the tracer. No significant changes in vital signs, electrocardiograms, or laboratory studies arose.
Despite these promising clinical results, the unexpected high cardiac retention observed in the human distribution study prompted us to further examine the specificity of tracer binding by extending preclinical research using both a novel mGluR2 KO rat model and a structurally distinct specific mGluR2 PAM ligand (JNJ-46356479), which were previously not available, and in Wistar rats for heart uptake.
Heart Imaging Studies in Wistar Rats
To determine whether the observed retention of 11C-JNJ-42491293 in the human heart was reversible and specific, dynamic small-animal PET imaging studies were conducted at baseline (n = 2), after pretreatment with cold JNJ-42491293 (self-block, n = 1), and in a displacement study with JNJ-42153605 (n = 2) (Supplemental Fig. 4). These experiments revealed reversible and specific binding of 11C-JNJ-42491293 to the normal rat heart. Heart radiometabolite analysis showed an absence of radiometabolites, confirming that only intact tracer was responsible for the observed heart uptake.
Brain Imaging Studies in WT and mGluR2 KO Rats
Baseline small-animal PET imaging studies showed similar 11C-JNJ-42491293 regional brain uptake in WT and mGluR2 KO rats (Fig. 6A). Moreover, the displacement study using mGluR2 PAM JNJ-46356479 failed to show any tracer displacement in both WT and KO rats (Fig. 6B). In vivo, the lack of difference between WT and mGluR2 KO rats for specific binding of 11C-JNJ-42491293 suggests that brain retention of 11C-JNJ-42491293 is dominated by binding to a non-mGluR2 target. JNJ-42153605 also has affinity for this so far unidentified binding site, whereas JNJ-46356479 does not seem to display occupancy of this site in view of the lack of tracer displacement in vivo.
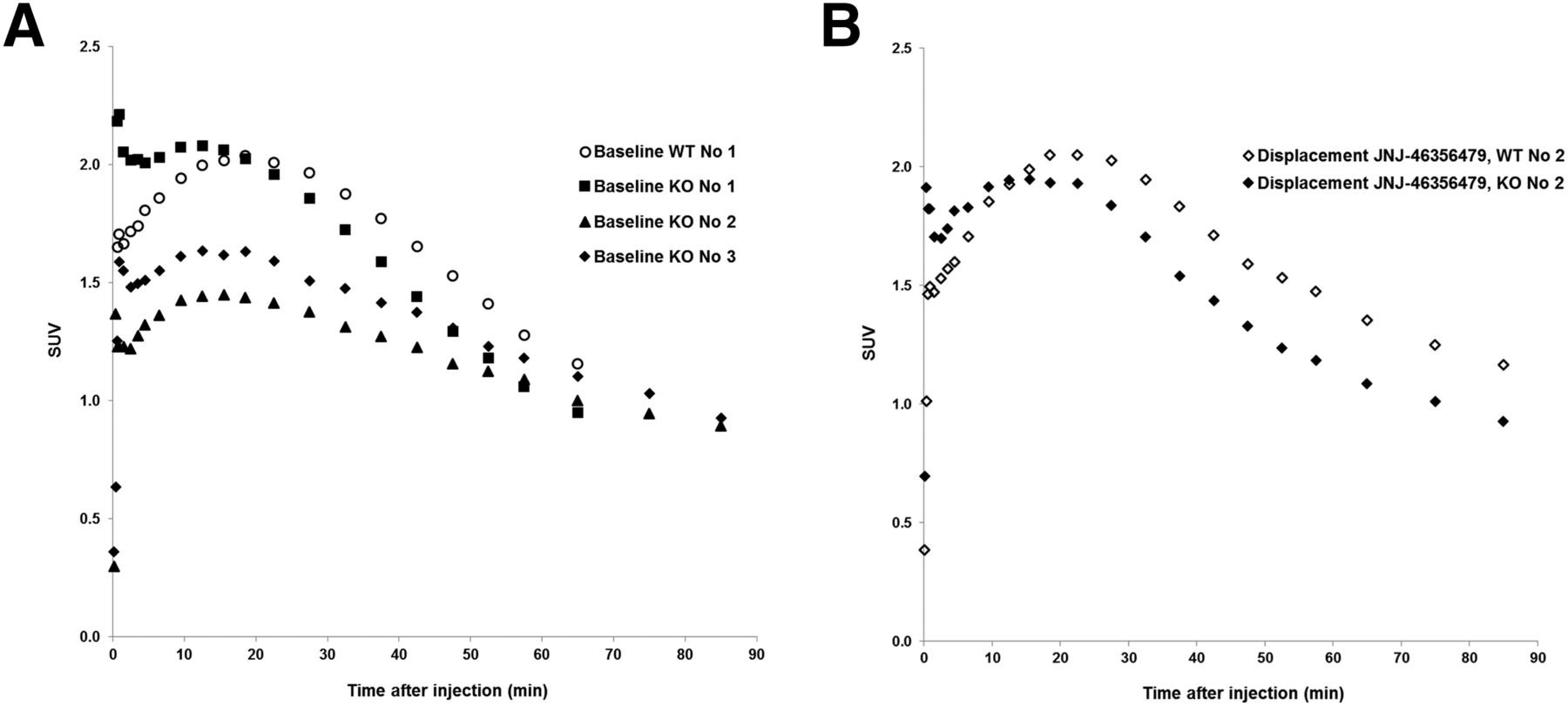
Total rat brain time–activity curves of 11C-JNJ-42491293 in WT and mGluR2 KO rats at baseline (A) and in displacement study with JNJ-46356479 (4 mg/kg, intravenous, 30 min after tracer injection) (B).
Ex Vivo and In Vitro Autoradiography Binding Studies in WT and mGluR2 KO Rats
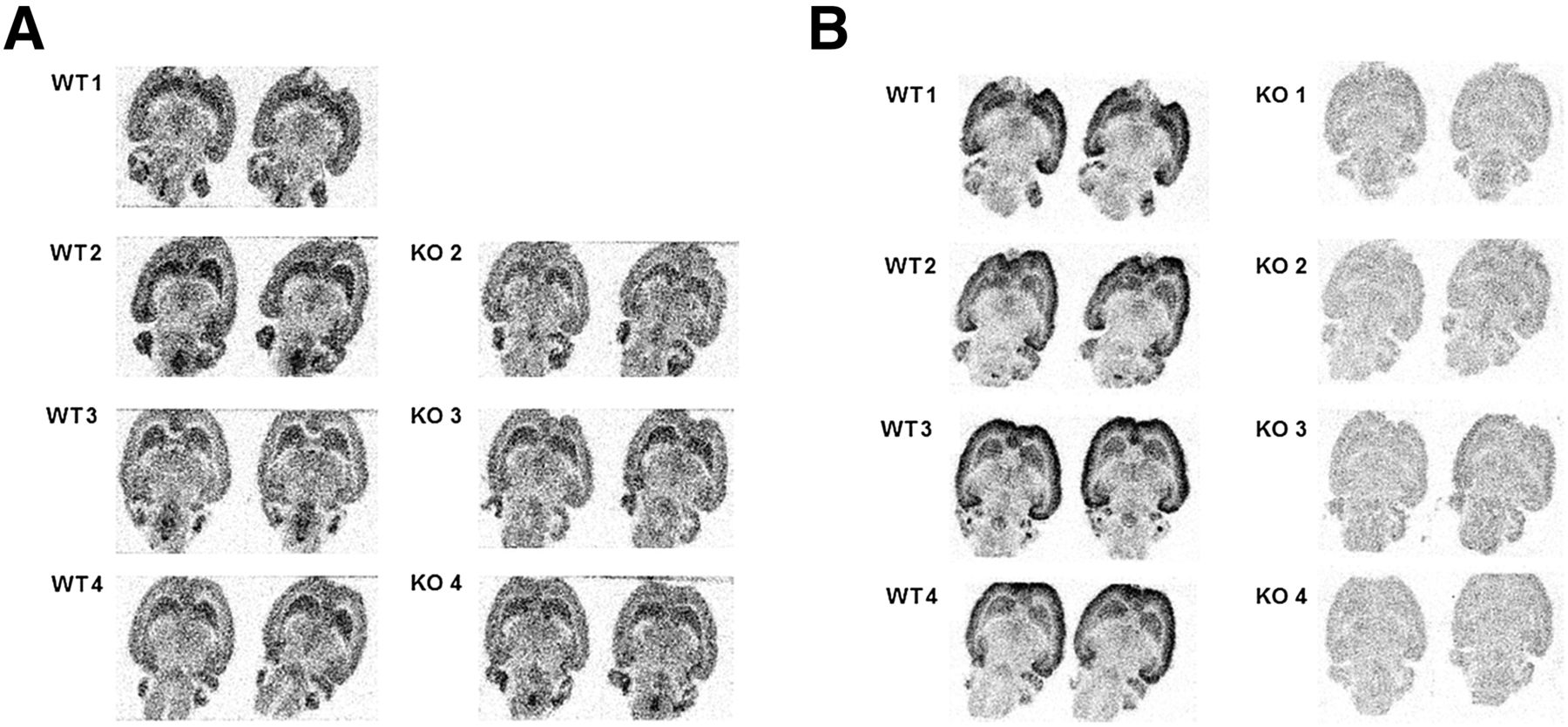
Ex vivo binding studies (intravenous injection of the tritiated ligand followed by sacrification of the animal, isolation, and cryosectioning of the brain and visualization of brain retention with autoradiography) were performed with 3H-JNJ-42203993. Brain binding consistent with specific binding was, as expected, observed in WT rats. However, similar in vivo binding was also observed in the brains of mGluR2 KO rats (Fig. 7A), further suggesting that brain binding of this ligand in vivo is dominated by mGluR2-independent interactions with an unidentified binding site.

In vivo (A) and in vitro (B) autoradiographic distribution of bound 3H-JNJ-42203993, a tritiated analog of JNJ-42491293, in WT (n = 4) and mGluR2 KO rats (n = 4, 1 rat, KO 1, failed intravenous injection). Adjacent sections from same rats were used in both experiments. In vitro specific binding to WT rat brain sections only is observed, whereas in vivo tracer binding in both WT and mGluR2 KO rat brains is observed.
Repeating the previous experiment in vitro (incubation of adjacent brain slices of same animals with the tritiated ligand followed by autoradiography) showed binding, consistent with the reported cerebral mGluR2 distribution, to WT rat brain but not to KO rat brain (Fig. 7B), suggesting mGluR2-specific and selective binding of 3H-JNJ-42203993 in vitro.
In Vitro Binding Studies in Wistar Rat Cortical Membranes
To further substantiate the use of the structurally distinct PAM ligand JNJ-46356479 as chase or block compound in the rat and monkey PET studies, additional in vitro binding experiments with 3H-JNJ-42203993 (the tritiated analog of JNJ-42491293) on Wistar rat cortical membranes were performed, showing full displacement of the tritiated ligand by JNJ-46356479 (IC50 = 107 nM; n = 2; Supplemental Fig. 5). Full displacement of 3H-JNJ-42203993 by JNJ-4635479 demonstrates competitive binding of both compounds to the same mGluR2 PAM site in vitro.
In Vivo Brain PET Imaging in Rhesus Monkey
Baseline dynamic 11C-JNJ-42491293 imaging in the monkey showed regional tracer distribution similar to human brain uptake (Supplemental Fig. 6). Akin to the in vivo imaging studies in WT and mGluR2 KO rats, pretreatment with the structurally distinct mGluR2 PAM JNJ-46356479 did not affect tracer binding in the brain, indicating that, also in primates, JNJ-46356479 does not bind to the binding site that dominates brain retention of 11C-JNJ-42491293 in vivo.
DISCUSSION
When in vitro and in vivo imaging studies in rats and in vivo imaging studies in primate and humans were combined, the present findings demonstrated that 11C-JNJ-42491293 specifically and selectively bound to the mGluR2 PAM site in vitro, but that in vivo off-target binding was majorly responsible for the observed tracer brain retention. Experiments using mGluR2 KO rats helped us to disentangle this curious in vitro mGluR2 PAM specific to in vivo mGluR2 unspecific binding switch. Yet to be identified, this off-target binding site is shared with the JNJ-42494293–related PAM compound JNJ-42153605 and is also present in the myocardium. Importantly, JNJ-46356479, a structurally distinct mGluR2 PAM ligand, did not exhibit this in vivo off-target binding and was therefore valuable in further evaluating the in vivo specificity of 11C-JNJ-42491293 in rats and especially in a monkey for which KO models do not exist.
The development of PET radioligands for neuroreceptor imaging is a demanding task, and although in vitro binding has been demonstrated for many radioligands, only a few are eventually suitable as PET tracers for quantitative in vivo occupancy studies. Previously, a series of 6 11C-labeled triazolopyridines, targeting an mGluR2 allosteric binding site, were radiolabeled and preliminarily evaluated in vivo by our group (22). From this investigation, 11C-JNJ-42491293 was the most promising ligand for mGluR2 PET imaging and was therefore selected for further preclinical and clinical evaluation. The postmortem regional distribution of brain mGluR2 has been well characterized in both rodents (8,9) and humans (10,11), with good overall interspecies concordance except for the mesencephalon, in which mGluR2 was expressed in humans but absent or expressed at low levels in rodents. The initial findings of in vitro autoradiography binding studies and in vivo small-animal PET imaging studies using 11C-JNJ-42491293 in healthy Wistar rats were in qualitative agreement with these data, suggesting the suitability of 11C-JNJ-42491293 for imaging mGluR2 in the brain in vivo. Also in humans, 11C-JNJ-42491293 brain uptake was consistent with the known cerebral distribution of mGluR2, and initial kinetic modeling showed that 11C-JNJ-42491293 behaved with favorable characteristics. These results initially suggested this radioligand to be the first valuable tool for determining mGluR2 drug occupancy and studying regional receptor availability in the human brain under physiologic and several neuropathologic conditions.
However, because the human heart is known to be devoid of mGluR2 (28), the high 11C-JNJ-42491293 heart accumulation seen in the whole-body human PET imaging was unexpected and fostered more extended preclinical evaluation to further study the specificity of tracer binding. Focusing on the heart in Wistar rats, additional small-animal PET imaging studies including blocking and displacement experiments suggested specific tracer binding to the normal rat heart in vivo. The additional heart radiometabolite analysis confirmed that intact tracer was responsible for the observed myocardial binding. Although present, mGluR2 expression in the rat heart was low and predominated in the atrium (28), further challenging the specificity of 11C-JNJ-42491293 for mGluR2 in vivo.
At that point in time, an mGluR2 KO rat model became available. Extensive preclinical research combining in vitro and ex vivo autoradiography binding studies and in vivo small-animal PET baseline, blocking, and displacement studies in this mGluR2 KO rat model revealed that 11C-JNJ-42491293 did specifically bind to mGluR2 in vitro, whereas in vivo an off-target (non-mGluR2) binding site was visualized. JNJ-46356479, which displaced 3H-JNJ-42203993 in vitro (Supplemental Fig. 5), did not block tracer binding in vivo, suggesting that—in contrast to 11C-JNJ-42491293, 3H-JNJ-42203993, and JNJ-42153605—JNJ-46356479 may specifically bind to mGluR2 without in vivo off-target affinity. To further investigate the contribution of an in vivo off-target binding in higher species, imaging studies were performed in a monkey. Because no mGluR2 KO models were available for monkeys, a pretreatment imaging study with the structurally distinct mGluR2 PAM JNJ-46356479 was performed, showing no blocking of 11C-JNJ-42491293 brain binding. This suggested that also in monkeys, and presumably also in humans, the observed in vivo tracer binding of 11C-JNJ-42491293 predominantly reflects specific binding to a non-mGluR2 target. Collectively, these data demonstrate that mGluR2 binding of 11C-JNJ-42491293 is strong and specific in vitro, whereas it becomes low (if any) in vivo. In contrast, although not seen in vitro, a non-mGluR2 binding dominated in vivo, highlighting the difficulty in recognizing off-target binding in the initial (in vitro) characterization steps. We hypothesized the presence of an in vivo mediator-enhanced binding mechanism resulting in non-mGluR2 binding to a target that has high affinity and sufficient capacity in vivo, but not in vitro. The identity of this off-target binding site remains yet to be discovered.
Two limitations of this study should be mentioned. First, autoradiography studies in WT and KO rats were not performed with 11C-JNJ-42491293 but with 3H-JNJ-42203993, an analog of the 11C-labeled tracer (Fig. 1). However, JNJ-42203993 and JNJ-42491293 bind to the same mGluR2 PAM site with comparable potency in vitro (IC50 = 9.2 and 11.2 nM, respectively) and have similar pharmacokinetics in vivo when labeled with 11C (22), supporting the validity of the present findings. Second, a potential confounding effect of anesthesia procedures cannot be excluded. Yet, autoradiography experiments in WT and KO rats, which were performed without prior anesthesia, showed good binding agreement with autoradiography and imaging studies in rats performed under anesthesia. Furthermore, the similarity in the brain time–activity curves of 11C-JNJ-42491293 in humans and monkey suggest that the use of ketamine/xylazine did not significantly affect tracer binding. Collectively, this suggests that the impact of anesthesia procedures on the present findings was, if present, minimal and did not threaten the validity of the conclusions.
CONCLUSION
Despite promising preclinical and normal-behaving clinical imaging studies, 11C-JNJ-42491293 does not specifically bind to mGluR2 in vivo. This translational research highlights the possible discrepancy between in vitro and in vivo findings and shows the importance of using KO animal models or structurally distinct ligands with affinity for the same target (especially important when KO models are not available) in studying binding specificity at early stages of PET radioligand and drug development. When specific binding is established in vitro, additional off-target affinity screening using in vivo settings is essential.
DISCLOSURE
The study was sponsored by Johnson & Johnson Pharmaceutical Research & Development, L.L.C. Xavier Langlois, Jose-Ignacio Andrés, Paula te Riele, Jesús Alcázar, and Mark E. Schmidt are employees of Janssen Research & Development. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank the team of PET radiopharmacy UZ Leuven for their skilled work on this study and Kwinten Porters, Julie Cornelis, Ivan Sannen, Peter Vermaelen, Ann Van Santvoort, and Mieke Steukers for their excellent help in preclinical and clinical data acquisition.
Footnotes
↵* Contributed equally to this work.
Published online Jul. 28, 2016.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 11, 2016.
- Accepted for publication July 11, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Design, synthesis, and characterization of [18F]mG2P026 as a high contrast PET imaging ligand for metabotropic glutamate receptor 2
- PET imaging studies to investigate functional expression of mGluR2 using [11C]mG2P001
- Synthesis and characterization of 5-(2-fluoro-4-[11C]methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carboxamide as a PET imaging ligand for metabotropic glutamate receptor 2
- Differential Pharmacology and Binding of mGlu2 Receptor Allosteric Modulators