


Abstract
Radioactive ligands for the prostate-specific membrane antigen (PSMA) are under development for therapy of metastasized prostate cancer. Since PSMA expression is also found in the kidneys, renal tracer uptake can be dose-limiting. Because kidney kinetics differ from tumor kinetics, serial application of PSMA inhibitors such as 2-(phosphonomethyl)pentanedioic acid (PMPA) may improve the kidney-to-tumor ratio. In this study, we evaluated the effect of PMPA on the biodistribution of 2 promising PSMA ligands. Methods: Human prostate cancer xenografts (LNCaP) were transplanted subcutaneously into mice. After injection of 125I-MIP1095, a 16-h latency period was allowed for tracer clearance from the blood and renal calices. After baseline scintigraphy, PMPA was injected in doses of 0.2–50 mg/kg (n = 3 per dose, 5 controls), followed by scans at 2, 4, 6, and 24 h after PMPA injection. Kidney and tumor displacement was determined as a percentage of baseline. A shortened but similar design was used to evaluate the PSMA ligand MIP1404, which contains a chelate for 99mTc/rhenium. Results: PMPA injection 16 h after MIP1095 translated into a rapid and quantitative relevant displacement of renal activity. Tumor uptake was reduced to a significantly lesser extent in a dose-dependent manner. PMPA doses of 0.2–1 mg/kg appear optimal for sustaining nearly complete tumor uptake while simultaneously achieving near-total blocking of specific renal PSMA binding. The effect was successfully validated with the PSMA ligand MIP1404. Conclusion: PSMA-targeted radionuclide therapy can benefit from serial PMPA comedication by reducing off-target radiation to the kidneys. These data will be used for a first approximation in clinical translation, although in patients an optimization of the dose and time schedule may be necessary.
Intense overexpression of the cell-surface peptidase prostate-specific membrane antigen (PSMA) in most primary and metastatic prostate cancers, as well as a positive correlation between PSMA and traditional adverse prognostic factors, has given rise to growing interest in the evaluation of this target (1–3). Initially, monoclonal antibodies (mAb) targeting PSMA have been used for prostate cancer imaging and therapeutic approaches. The 111In-labeled mAb capromab (ProstaScint; EUSA Pharma) targeted to a cytoplasmic domain of PSMA was even approved in the United States. However, the diagnostic value of intracellular targeting was rather limited (4). The mAb J591 targets the extracellular domain of PSMA, but like most full-length mAbs it is characterized by a slow accumulation in tumor and a long circulation time in blood. Thus, diagnostic mAb tracers require prolonged imaging—even days after injection (5). In the context of radionuclide therapy, mAbs commonly translate into an unfavorable dosimetry with pronounced hematotoxicity (6). Recently, the development of the Glu-urea–based high-affinity small-molecule PSMA inhibitors MIP1072 and MIP1095, either labeled with 123I for imaging or 131I for targeted radionuclide therapy, rendered rapid tumor uptake possible (7–9). Since PSMA is internalized through clathrin-coated pits (10), either spontaneously or after binding of an antibody or an inhibitor, it is also an excellent target for systemic radionuclide therapy. In this respect, the accumulation of the tracer in normal tissues has to be considered in order to prevent or diminish the extent of side effects. In a PET-based dosimetry study of 124I-MIP1095 in patients with castration-refractory prostate cancer, doses of up to 300 Gy in lymph nodes and bone metastases were assessed. The organs with the highest off-target radiation doses were salivary glands (3.8 mSv/MBq), liver (1.7 mSv/MBq), and kidneys (1.4 mSv/MBq); red marrow was 0.37 mSv/MBq (11). A 23-Gy tolerance limit was suggested for kidneys during conventional radiotherapy (12), and the biologic effective dose in systemic radionuclide therapy, which depends on the half-life of the used radionuclide, can markedly vary in both directions (13). Therefore, the kidneys may be the limiting factor for the maximum cumulative activity that can be administered safely. However, minimizing kidney uptake without losing tumor dose is a real challenge because PSMA is physiologically expressed in the kidney tubules (1). The pharmacokinetics of MIP1072 versus MIP1095 in animals have already been evaluated in detail (7,14). The authors of the mentioned studies reported a similar accumulation of both compounds in PSMA-expressing LNCaP xenografts but with very different pharmacokinetic profiles. MIP1072 clears rapidly from target and nontarget tissues. In contrast, MIP1095 has a longer biologic half-life in tumor but not in kidneys, thus corresponding to a higher fraction of ligand-induced receptor internalization in tumor cells.
For the development of PSMA ligands, the structurally unrelated PSMA inhibitor 2-(phosphonomethyl)pentanedioic acid (PMPA) is commonly used as a competitor in blocking studies to demonstrate the specific binding of the molecule of interest. In this respect, simultaneous coinjection of a 50 mg/kg dose of PMPA resulted in complete blocking of MIP1095 binding sites in tumor and kidneys (7).
The thesis for this study was that, after internalization of Glu-urea–based radiolabeled PSMA ligands into the tumor cells, subsequent administration of high-dose PSMA competitor PMPA may no longer block tumor uptake but can still displace the “non-” or “not yet” internalized PSMA ligand from the renal tubular cells, thus improving the projected tumor-to-kidney dose ratio.
MATERIALS AND METHODS
Inoculation of Mice with LNCaP Xenografts
All animal studies were approved by the Institutional Animal Care and Use Committee in accordance with the Guidelines for Care and Use of Laboratory Animals. Mice were housed under standard conditions in approved facilities with 12-h light/dark cycles and given food and water ad libitum. BALB/c nude mice were implanted behind the left shoulder with 5 × 106 LNCaP cells (BD Biosciences) suspended in Matrigel (BD Biosciences). The mice were used for our study after 8–12 wk, when the tumors reached approximately 1.5 cm in diameter.
Radiolabeling of MIP1095 and MIP1404
The synthesis of MIP1095 (S)-2-(3-((S)-1-carboxy-5-(3-(4-iodophenyl)ureido)pentyl)ureido)pentanedioic acid, the synthesis of the radiolabeling precursor trimethyltin-MIP-1095, and the subsequent radiolabeling with a variety of iodine isotopes were described previously (14,15). MIP1404 was provided by Molecular Insight Pharmaceuticals and labeled with 99mTc eluted from a clinical European Medicines Agency (EMA)–approved generator (Elumatic III; IBA). The synthesis of MIP1404 has also been described previously (16).
Workflow of MIP-1095 and MIP-1404 Animal Studies
Mice bearing LNCaP tumors of 1.5-cm diameter were injected via the tail vein with 37 MBq of 125I-MIP1095 in 100 μL of saline at a specific activity of more than 37,000 MBq/μmol. The mice were imaged at 16 h after injection (baseline), giving enough time for tumor uptake and internalization and tracer clearance from the bloodstream and kidney calices. Immediately after the baseline scan, PMPA in 100 μL of physiologic saline solution was injected via the tail vein in doses of 50, 10, 1, or 0.2 mg/kg (n = 3 per dose), or the animals served as controls (n = 5). Planar scans were performed 2, 4, 6, and 24 h after PMPA injection (Fig. 1A).

Chart demonstrating experimental setup with first image at 16 h after injection. (A) Immediately after scintigraphy, saline (control group) or different doses of PMPA were given and additional imaging was done at the indicated times after injection. (B) Example images of controls and each PMPA group.
MIP1404 was evaluated after labeling with 99mTc in LNCaP-bearing BALB/c nude mice. Baseline scintigraphy was done 1 h after injection of 9.3–11 MBq of 99mTc-MIP1404 in 100 μL of saline, directly followed by the injection of a 50 mg/kg dose of PMPA in 50 μL of saline or injection of 50 μL of saline alone (n = 3 each). Planar scans were done 1 and 3 h after the subsequent PMPA or saline injection. The flow chart of the experiment is presented in Figure 2A. Quantification was done with kidney regions of interest to obtain counts per minute, which were then converted to percentage-of-baseline values. The same protocol was used to evaluate kidney uptake of 10.3–11.6 MBq of 99mTc-MIP1404 in 100 μL of saline, directly followed by the injection of a 50, 10, and 1 mg/kg dose of PMPA in 50 μL of saline or injection of 50 μL of saline alone (n = 4 each) in non–tumor-bearing NMRI mice (Supplemental Fig. 1A; supplemental materials are available at http://jnm.snmjournals.org). In a pilot experiment, planar scans after injection of the salivary gland tracer 99mTc-pertechnetate were acquired, and the diagnostic value of the images was judged visually.

Chart demonstrating experimental setup with first image at 1 h after injection. (A) Immediately after scintigraphy, saline (control group) or 50 mg/kg dose of PMPA was given and additional imaging was done at the indicated times after injection. (B) Scintigraphic images of control and PMPA-treated animals.
Technique for Small-Animal Scintigraphy
During imaging, mice were anesthetized using 1% isoflurane gas in oxygen flowing at 0.6 L/min. Serial planar scans were obtained with a Gamma Imager-sct (Biospace labs). Tumor and kidney uptake was quantified by determining the counts per minute in the target region by region-of-interest technique. The baseline scan before administration of PMPA served as a reference, and the subsequent images are reported as a percentage of the baseline value.
RESULTS
MIP1095
Injection of PMPA 16 h after MIP1095 translated into a rapid and quantitatively significant displacement of renal activity for all PMPA doses tested. Scintigraphy scans from one example of each group are presented in Figure 1B. The course of residual activity of 125I-MIP1095 measured at the particular time points after subsequent administration of a 0.2, 1, 10, or 50 mg/kg dose of PMPA or controls is presented in Figure 3A for kidney and Figure 3B for tumor regions of interest. Even at a very low dose of 0.2 mg/kg, there was a highly significant (P < 0.01) displacement from the kidneys. The effect was more pronounced at higher doses and showed a dose dependency, with 1 mg/kg revealing an even higher kidney washout than the 0.2 mg/kg dose (P < 0.01). However, doses greater than 1 mg/kg did not result in further improvement of the kidney-protective effect (differences between 1, 10, and 50 mg/kg PMPA groups were nonsignificant). In tumor tissue, the uptake of 125I-MIP1095 was displaced by subsequent injection of PMPA, and this effect appeared to be dose-dependent: residual uptake 2 h after PMPA was 98% with 0.2 mg/kg, 93% with 1 mg/kg, 77% with 10 mg/kg, and 75% with 50 mg/kg. However, because of the small size of the group (n = 3), none of the differences was statistically significant.
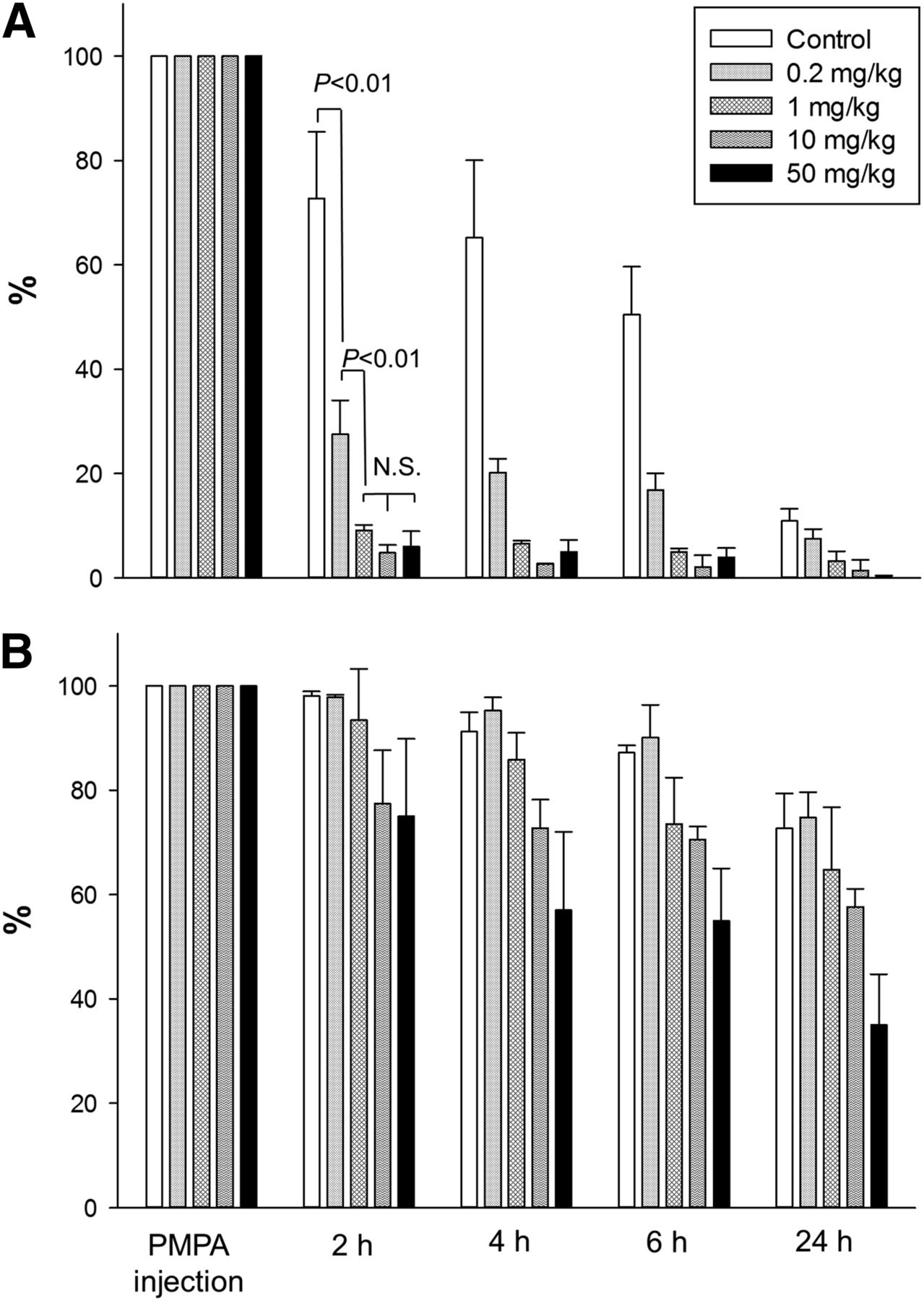
Time course of residual activity in kidney (A) and tumor (B) expressed as percentage of value at PMPA or saline injection.
MIP1404
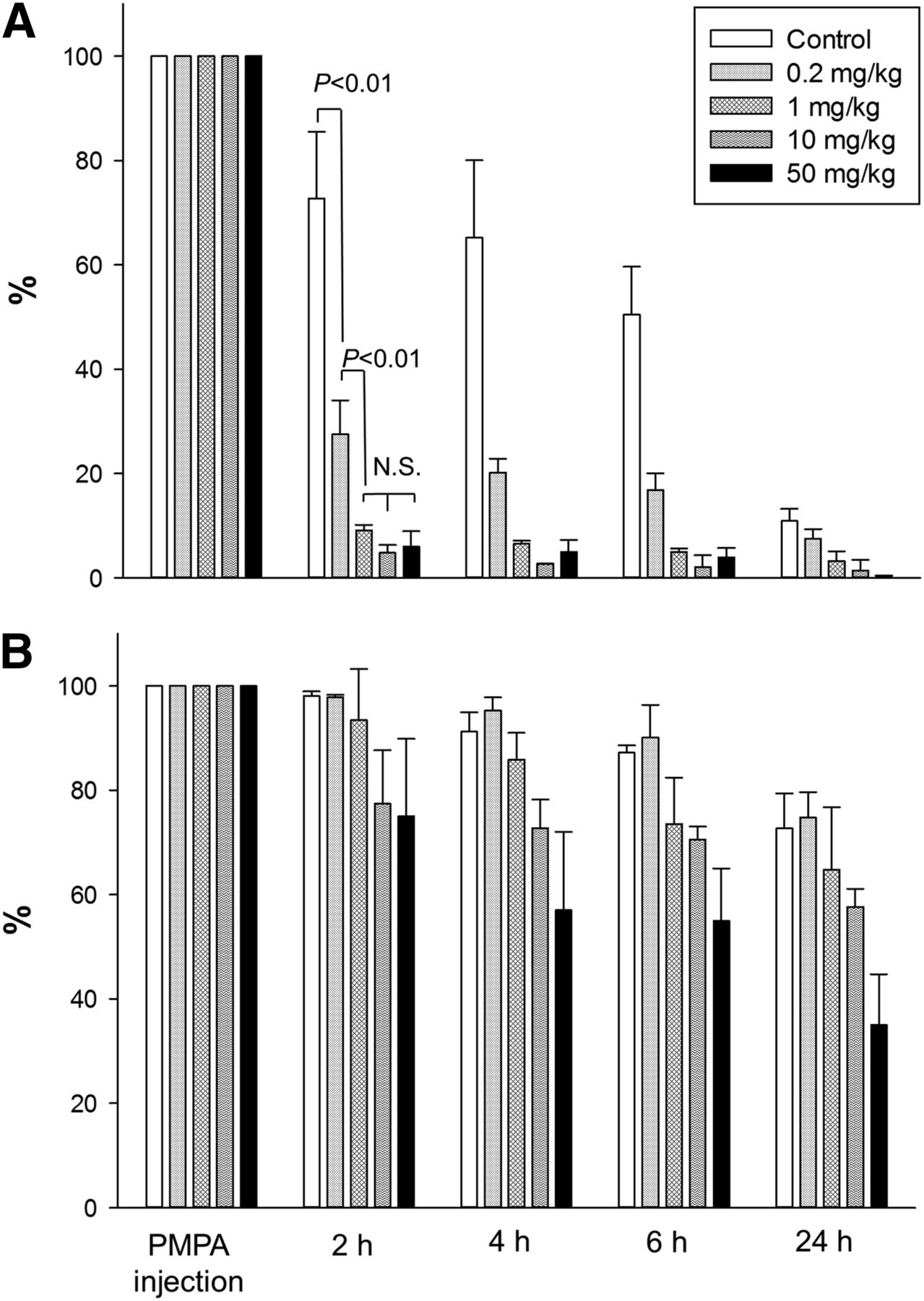
Normalized to tumor uptake 1 h after injection, washout of MIP1404 from the kidneys was fast, even without intervention. When a 50 mg/kg dose of PMPA was administered directly after the baseline scan, the washout was significantly (P < 0.01) enhanced at 2 and 4 h (Figs. 2B and 4A). Compared with controls, the difference in tumor uptake measured in the PMPA group was already nonsignificant with the 50 mg/kg PMPA dose (Fig. 4B). Because MIP1404 seemed rather robust against tumor displacement even after high-dose PMPA, the dilution series with 50, 10, and 1 mg/kg and saline alone were done in non–tumor-bearing NMRI mice. Respective residual kidney uptake at 2 and 4 h was 36% and 15% in controls, 13% and 9% after a 1 mg/kg dose of PMPA, 15% and 10% after 10 mg/kg, and 11% and 7% after 50 mg/kg (Supplemental Fig. 1B). This pattern is consistent with the larger MIP1095 series, demonstrating optimal kidney displacement even at 1 mg/kg and no benefit from higher PMPA doses.
In a pilot study, planar scans of the mice were acquired after injection of 99mTc-pertechnetate, which is known to accumulate in the salivary glands, but because of the limited spatial resolution of the camera we were not able to sufficiently delineate these small structures in mice (Supplemental Fig. 2). Thus, we concluded that it is not feasible to further evaluate potential displacement of MIP1404 out of the salivary glands with this study design.
Time course of residual activity in kidney (A) and tumor (B) expressed as percentage of value at PMPA or saline injection.
DISCUSSION
PSMA is an emerging target for imaging and radionuclide therapy of metastasized prostate cancer. With regard to radionuclide therapy, MIP1095 and MIP1404 are promising PSMA ligands because these small molecules enable fast tumor targeting, have fast clearance from untargeted organs, and have sufficient residency times in tumor because of ligand-induced cellular internalization (7,11). MIP1404 is of particular interest because its single-amino-acid chelator can be labeled with either 99mTc for diagnostic applications or 186Re or 188Re for therapeutic applications (16,17). Technetium and rhenium are chemically related and have structural and reactive similarities. 99mTc is available worldwide from a variety of regionally approved generator systems and can be imaged with the numerous Anger cameras already installed. 186Re (half-life, 3.7 d; maximum energy of β emission, 1.07 MeV; 11% coemission of 137-keV γ emission for imaging) presents an attractive “matched pair” for therapy. 188Re (half-life, 17 h; maximum energy of β emission, 2.12 MeV; 15% coemission of 155 keV γ emission for imaging) can be obtained from a 188W/188Re generator (Oak Ridge National Laboratory) that was Food and Drug Administration–approved and would be suitable for clinical application (18). MIP1095 can be tagged with different isotopes of iodine. Until now, most of the clinical experience has been with this compound. Labeled with 124I, it has already been used for PET-based dosimetry, and labeled with 131I, for therapy of patients with treatment-refractory advanced prostate cancer (11). For human beings, 123I offers good imaging probabilities (159-keV γ emission; 12-h half-life) for conventional scintigraphy (7). Labeled with 125I (half-life, 60 d; γ emission, 27–35 keV), MIP1095 presents the best characteristics for small-animal imaging, and therefore 125I-MIP1095 was chosen for our main experiment.
After the subsequent injection of PMPA, relevant kidney displacement was observed with both radiotracers whether the evaluated compound included a chelate (MIP1404) or did not (MIP1095). Thus, in addition to some nonspecific kidney uptake due to tubular reabsorption—which might be further improved by modifications affecting the chelate or linker of the tracer molecule—a relevant part of kidney uptake seems to be related to specific PSMA binding.
It is common practice to show specific binding of PSMA ligands by simultaneous coinjection of a 50 mg/kg dose of PMPA, which results in complete blocking of PSMA binding sites in tumor and kidneys (7,14,16). Our findings now imply that a subsequent injection of this competitor cannot displace the endosome-fixed ligand from the tumor; by contrast, it can still displace the noninternalized ligands from the kidneys.
PSMA was found to be equivalent to the enzyme glutamate carboxypeptidase II (GCP II), and today the two terms are used synonymously. In 1997, immunostaining had already revealed a physiologic expression of PSMA in the kidney tubules (1). Glutamate carboxypeptidase III (GCP III), a GCP II homolog with 67% amino acid identity, was cloned and evaluated on RNA level in 2007, but because of the lack of a GCP III–specific antibody, the effect of different amounts of GCP II and III on the cell surface of different tissues could not be assessed on protein level until now (19). A third enzyme highly similar to GCP II is NAALADase L (20). Therefore, it is possible that the PSMA expressed by the renal tubular cells represents simply a different isoform of the molecule with a lower internalization rate.
Even if the kidney expresses GCP II, there potentially are other mechanisms that could account for the different tracer kinetics. In one report, binding of PSMA to filamin A reduced its internalization rate by 50% (21). Glycosylation can also regulate GCP II enzyme activity via redistribution of the protein from the plasma membrane to intracellular loci, as well as by affecting its half-life (22). Furthermore, the sensitivity of PSMA to treatment with different glycosidases was different in different prostate carcinoma cell lines, indicating that these may be composed of different types of sugar structures (23). Thus, a different expression of PSMA cofactors or different glycosylation patterns in kidney and tumor would also be a sufficient explanation for our observations.
Preclinical animal studies are associated with some limitations. In our study design, the PSMA expressed by the LNCaP xenografts represents the human form of GCP II, whereas the kidney PSMA represents the ortholog of mice. Rovenská et al. characterized human PSMA with its rat and pig orthologs and found the GCP II of these species suitable to approximate human GCP II enzyme activity (24). However, variants of PSMA among different species should always be considered when using animal models and may complicate the extrapolation of preclinical observations to clinical application.
The salivary glands are another organ that show high uptake of diagnostic PSMA tracers (8) and 131I-MIP1095; vice versa, this effect was found to translate into xerostomia as a side effect in PSMA-targeted radionuclide therapy (11). Different reports exist concerning PSMA expression in salivary glands. Silver et al. (1) and Mhawech-Fauceglia et al. (25) found no expression in the parotids. However, PSMA RNA has been found in salivary glands (26), in tissue extracts (27), and by immunohistochemistry (28). A variety of mAbs binding to different epitopes of the extracellular domain of PSMA presented with different stainings of normal tissues (28)—a possible indicator that different splicing variants of PSMA may exist. However, the 3 antibodies evaluated in the cited study showed no binding to the kidneys, raising some doubt about the reliability of the specificity and binding affinity of the used antibodies. Therefore, the situation in salivary glands is rather unclear. If PSMA expression is responsible for tracer accumulation in the salivary glands, PMPA could also be suitable for displacing radioactive PSMA ligands from these organs. However, in our small-animal imaging experiments, no sufficient delineation of this target structure was possible—not even in explicit salivary gland scintigraphy with 99mTc-pertechnetate. Therefore, we cannot presently conclude whether PMPA is also suitable for decreasing the risk of xerostomia as a potential side effect of PSMA-targeted radionuclide therapy.
With regard to the kidneys, the optimal dose of PMPA ranged from 0.2 to 1.0 mg/kg. A 1.0 mg/kg dose translates into near-total renal displacement with only a minimal (<10%) effect on tumor uptake. With 0.2 mg/kg, the decrease in tumor uptake was less than 5%, but still a relevant improvement of kidney uptake could be observed. Nevertheless, renal activity was only subtotally displaced with the lower dose. Doing a conservative, body surface–based, extrapolation from the 0.2 mg/kg mouse dose to humans, an 80-kg patient should receive about 60 mg of PMPA.
The orally bioavailable PSMA inhibitor 2-(3-mercaptopropyl)pentanedioic acid, which is nearly as potent as 2-PMPA (inhibitory concentration of 50%, 85 nM vs. 30 nM (29,30)), has already been evaluated, in 25 healthy subjects with a mean body weight of 71 kg. Doses of up to 750 mg (i.e., ∼10 mg/kg) were found to be safe and generally well tolerated (31). This finding indicates an approximately 10-fold safety factor in comparison to our recommended starting dose for the evaluation of PMPA-based kidney protection during PSMA-targeted radionuclide therapy in humans. In animal studies even much higher doses of PMPA have also been found tolerable (32). Preliminary data also suggest that selective PSMA inhibitors such as 2-MPPA (2-(3-mercaptopropyl)pentanedioic acid) or 2-PMPA may have inherent potential against prostate cancer and may simultaneously attenuate docetaxel-induced neuropathy (33).
It is difficult to predict how the displacement effect observed in this investigation will translate into the dose to kidney in prostate cancer patients because the metabolic turnover of PSMA inhibitors is much faster in rodents than in humans. The biologic half-life of MIP1095 in the mouse kidney of our control group was approximately 6 h (Fig. 3A). In contrast, it was 60–72 h in patients evaluated with 124I-MIP1095 (11). Therefore, it is not yet reasonable to simulate the dosimetry effect in humans on the basis of the currently available data. Further studies are needed to quantify the kidney dose reduction that can actually be achieved with the presented concept in clinical application.
CONCLUSION
Our data demonstrate that it is possible to lower the dose-limiting kidney uptake in PSMA-targeted radionuclide therapy with the subsequent injection of the small-molecule PSMA inhibitor PMPA without significantly diminishing tumor uptake and retention. Taking into account the potential benefits versus the residual risks for the unapproved administration of PMPA, we consider it ethically reasonable to offer subsequent PMPA comedication as a kidney protective during PSMA-targeted radionuclide therapy. However, further studies are required to optimize the schedule for the subsequent application of PMPA after radiolabeled PSMA ligands and to extrapolate from animals to humans.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported initially by Molecular Insight Pharmaceutical, Inc., and later by Progenics Pharmaceuticals Inc., in providing the precursors of MIP1095 and MIP1404, respectively. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Ursula Schierbaum for her help in inoculation of the LNCaP xenografts and Susanne Kraemer for execution of the radiolabeling process. We also thank Maria Rius Montraveta for creating the figure graphics.
Footnotes
↵* Contributed equally to this work.
Published online Jan. 22, 2015.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Prostate-Specific Membrane Antigen Radioligand Therapy Using 177Lu-PSMA I&T and 177Lu-PSMA-617 in Patients with Metastatic Castration-Resistant Prostate Cancer: Comparison of Safety, Biodistribution, and Dosimetry
- Normal-Tissue Tolerance to Radiopharmaceutical Therapies, the Knowns and the Unknowns
- The Impact of Monosodium Glutamate on 68Ga-PSMA-11 Biodistribution in Men with Prostate Cancer: A Prospective Randomized, Controlled Imaging Study
- 177Lu-Labeled Albumin-Binder-Conjugated PSMA-Targeting Agents with Extremely High Tumor Uptake and Enhanced Tumor-to-Kidney Absorbed Dose Ratio
- The Effects of Monosodium Glutamate on PSMA Radiotracer Uptake in Men with Recurrent Prostate Cancer: A Prospective, Randomized, Double-Blind, Placebo-Controlled Intraindividual Imaging Study
- JHU-2545 Selectively Shields Salivary Glands and Kidneys during PSMA-Targeted Radiotherapy
- Development of Novel PSMA Ligands for Imaging and Therapy with Copper Isotopes
- 177Lu-PSMA-617 Radioligand Therapy in Metastatic Castration-Resistant Prostate Cancer Patients with a Single Functioning Kidney
- Effect of External Cooling on 177Lu-PSMA Uptake by the Parotid Glands
- First Clinicopathologic Evidence of a Non-PSMA-Related Uptake Mechanism for 68Ga-PSMA-11 in Salivary Glands
- Evaluation of 111In-DOTA-5D3, a Surrogate SPECT Imaging Agent for Radioimmunotherapy of Prostate-Specific Membrane Antigen
- Monosodium Glutamate Reduces 68Ga-PSMA-11 Uptake in Salivary Glands and Kidneys in a Preclinical Prostate Cancer Model
- Salivary Gland Toxicity of PSMA Radioligand Therapy: Relevance and Preventive Strategies
- Targeted {alpha}-Therapy of Metastatic Castration-Resistant Prostate Cancer with 225Ac-PSMA-617: Swimmer-Plot Analysis Suggests Efficacy Regarding Duration of Tumor Control
- PSMA-Targeted Radionuclide Therapy and Salivary Gland Toxicity: Why Does It Matter?
- Dual-Target Binding Ligands with Modulated Pharmacokinetics for Endoradiotherapy of Prostate Cancer
- PSMA-Targeted Radionuclide Therapy of Metastatic Castration-Resistant Prostate Cancer with 177Lu-Labeled PSMA-617
- 177Lu-Labeled Prostate-Specific Membrane Antigen Radioligand Therapy of Metastatic Castration-Resistant Prostate Cancer: Safety and Efficacy
- New Strategies in Prostate Cancer: Prostate-Specific Membrane Antigen (PSMA) Ligands for Diagnosis and Therapy