


Abstract
Radiolabeled antibodies have become indispensable tools in nuclear medicine. However, the natural roles of antibodies within the immune system mean that they have several intrinsic limitations as a platform for radiopharmaceuticals. In recent years, the field has increasingly turned to antibody engineering to circumvent these issues while retaining the manifold benefits of the immunoglobulin framework. In this “Focus on Molecular Imaging” review, we cover recent advances in the application of antibody engineering to immunoPET, immunoSPECT, and radioimmunotherapy. Specifically, we address how antibody engineering has been used to improve radioimmunoconjugates on four fronts: optimizing pharmacokinetics, facilitating site-specific bioconjugation, modulating Fc interactions, and creating bispecific constructs.
Since the pioneering work of Pressman and Korngold over half a century ago, the field of nuclear medicine has been captivated by the promise of monoclonal antibodies (mAbs) as tools for the delivery of radionuclides to tumors (1,2). Decades of advancements in this area have culminated in an array of promising clinical results in radioimmunotherapy with 131I-, 225Ac-, and 177Lu-labeled radioimmunoconjugates as well as immunoPET with an ever-expanding array of 89Zr-labeled mAbs (Fig. 1). Yet even a cursory look at the immunoPET, immunoSPECT, and radioimmunotherapy literature reveals a common thread: the development of effective radioimmunoconjugates often requires addressing the intrinsic limitations of antibodies themselves (3). Nature, after all, did not design immunoglobulins as simple vehicles for the delivery of cargoes to target cells. Rather, antibodies evolved as multifunctional components of complex immune systems. Turning to nature for a radiopharmaceutical vector is thus a double-edged sword. On the one hand, radiolabeled antibodies offer exquisite specificity and affinity for cancer antigens, tremendous in vivo stability, and high levels of tumoral accretion. On the other hand, they are difficult to synthesize in a well-defined and homogeneous manner, can have problematically long biological residence times, and can produce elevated uptake in healthy tissues.

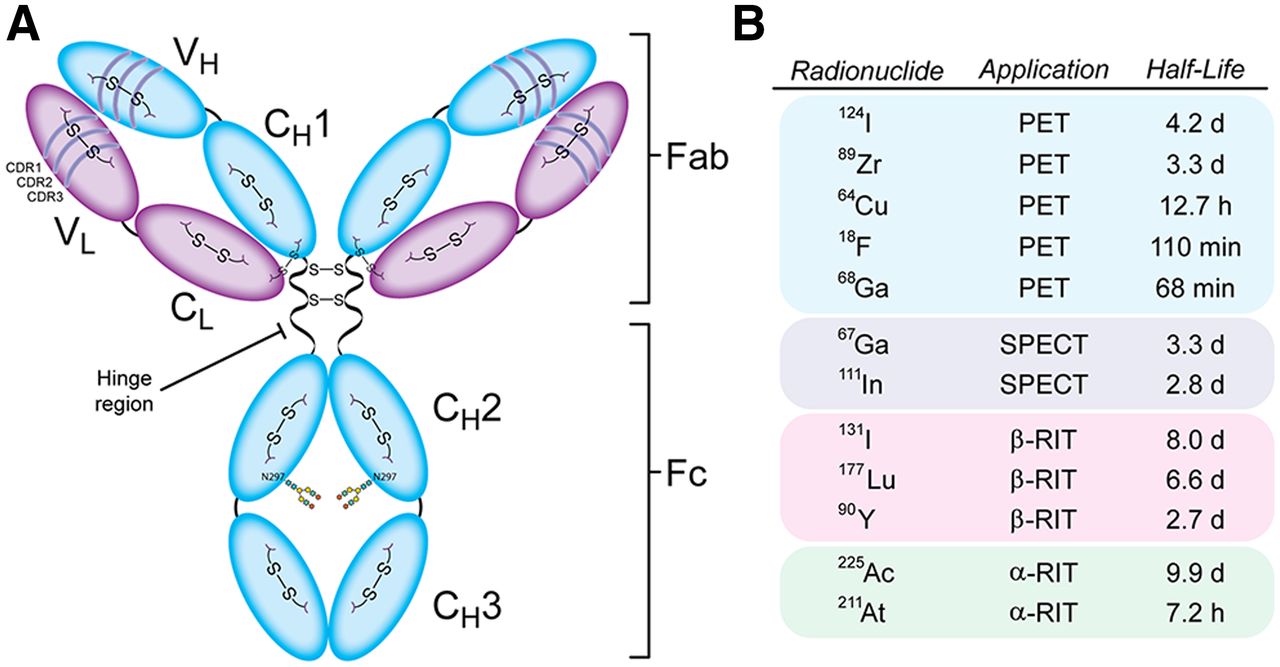
Anatomy of IgG1 antibody (A) and radionuclides discussed in this review (B). RIT = radioimmunotherapy.
The innate advantages of antibodies as radiopharmaceutical vectors have fueled a great deal of research dedicated to circumventing their limitations and optimizing their performance as diagnostics, theranostics, and therapeutics. This work has generally followed three different veins. In one approach—in vivo pretargeting—the antibody is decoupled from the radioactivity, the former is injected days before the latter, and the two components combine at the target site via a highly selective ligation (4). This strategy alleviates the dosimetric challenges associated with traditional radioimmunoconjugates but inevitably introduces significant scientific and logistical complexity. Others have sought to create radiopharmaceuticals based on antibody mimetics, synthetic biomolecules such as Affibody molecules and designed ankyrin repeat proteins that are inspired by the structure and function of immunoglobulins (5,6). These constructs frequently offer improved pharmacokinetic profiles compared to radiolabeled antibodies but lose other benefits associated with IgG-based scaffolds, including bivalency, high tumoral uptake, and in vivo stability.
In this installment of the “Focus on Molecular Imaging” series, we will discuss the third approach: antibody engineering. In the broadest sense, antibody engineering is predicated on using molecular biology to make modifications—large or small—to the immunoglobulin framework to improve the performance of antibodies or immunoconjugates. The best-known medical application of antibody engineering is easily the humanization of antibodies, and the wide array of methods used for antibody engineering have been skillfully reviewed elsewhere (7,8). In the specific context of nuclear medicine, antibody engineering offers an opportunity to optimize the performance of radioimmunoconjugates without the inherent complexity of pretargeting or the attendant sacrifices of antibody mimetics. These efforts generally seek to achieve one of four objectives: shortening the pharmacokinetic profile of full-length antibodies, facilitating site-specific bioconjugation, modulating Fc interactions, or creating constructs capable of binding multiple targets.
OPTIMIZING PHARMACOKINETICS
The neonatal Fc receptor (FcRn)–mediated recycling and size of full-length IgG gives traditional radioimmunoconjugates elongated, multiday serum half-lives. This pharmacokinetic behavior often manifests in high uptake in tumor lesions. However, it can also produce elevated activity concentrations in the blood and other healthy tissues, a phenomenon that can lower image contrast and reduce therapeutic indices (3). Antibody fragments offer an alternative platform with more rapid pharmacokinetic profiles, as they retain the binding properties of full-length mAbs but come in a smaller package. A variety of fragments have been harnessed for nuclear imaging and therapy, including F(ab′)2 fragments (∼110 kDa), (scFv)2-Fc fragments (∼100 kDa), minibodies (∼75 kDa), diabodies (∼60 kDa), F(ab) fragments (∼50 kDa), single-chain variable fragments (scFv, ∼27 kDa), and single-domain antibodies (sdAb, ∼15 kDa; also known as VHH and Nanobodies [Ablynx]) (Fig. 2A). The more rapid pharmacokinetic profiles of antibody fragments provide an opportunity to replace the long-lived radionuclides typically used with full-length IgG (i.e., 89Zr, 131I, 225Ac, and 177Lu) with isotopes bearing shorter physical half-lives (e.g., 64Cu, 18F, 68Ga, and 211At), a switch that only enhances the dosimetric and logistical benefits of fragments. The improved pharmacokinetic profiles of fragments often come with a price, however. Fragment-based radioimmunoconjugates generally produce lower uptake in tumor tissue than their full-length analogs, and some fragments accumulate at very high levels in the kidneys during renal elimination.
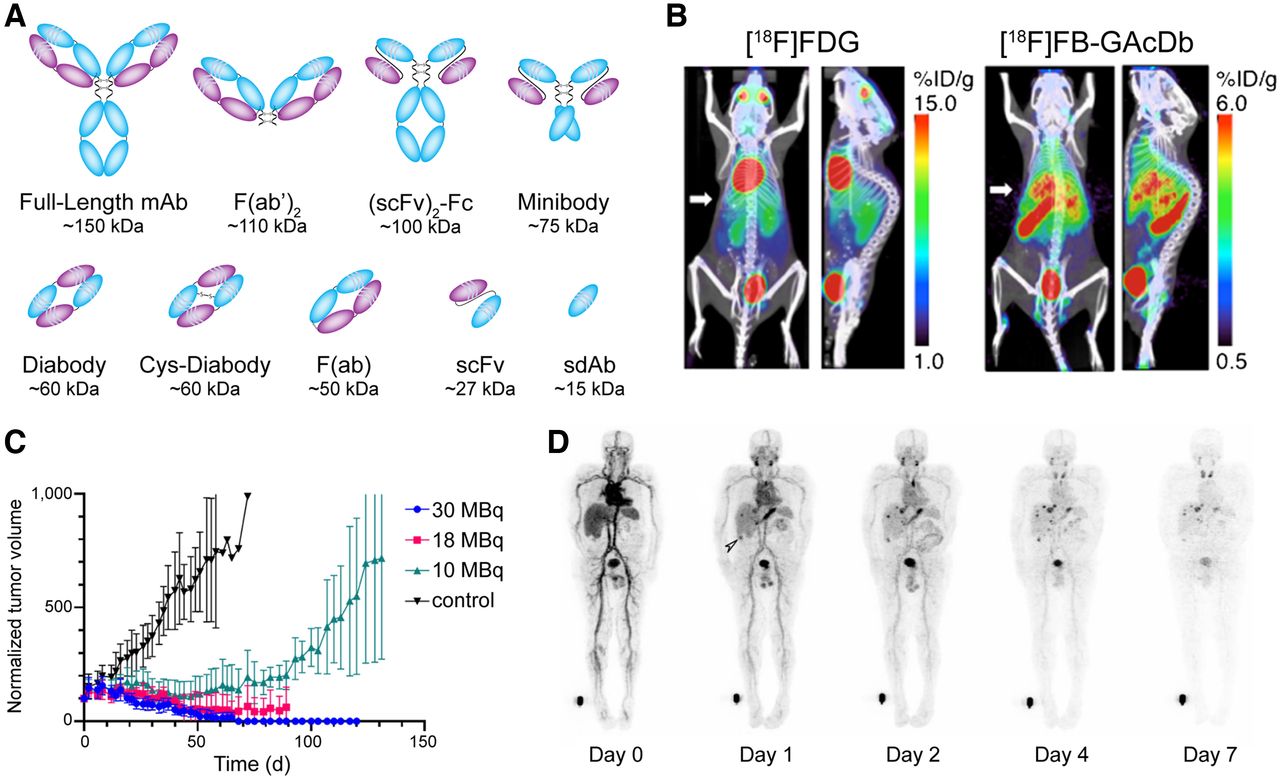
(A) Commonly used antibody fragments. (B) Uptake of cys-diabody—[18F]FB-GAcDb—in liver metastases in a transgenic murine model of B-cell lymphoma. (Reprinted with permission of (10).) (C) Normalized tumor volumes observed after the treatment of mice bearing HER2-positive breast cancer xenografts with iso-[131I]SGMIB-VHH_1028. (Reprinted from (12).) (D) Whole-body PET images of a patient injected with TAG-72–targeted pegylated diabody, [124I]I-PEG-AVP0458. (Reprinted from (17).) %ID/g = percent injected dose per gram.
Several promising preclinical imaging and therapy studies featuring antibody fragments have emerged in the last half-decade. In 2018, for example, Seo et al. reported the use of a 64Cu-labeled anti-CD8 cys-diabody—dubbed 169cDb—to image CD8-positive T-cell density in FVB, BALB/c, and C57BL/6 mice (9). The tracer proved capable of delineating and quantifying CD8-positive T-cell populations in lymphoid organs, suggesting that a similar approach to imaging could be used in the clinic to quantify T-cell density and monitor the response of patients to immunotherapy. A year later, a team at UCLA described the development of a CD20-targeting cys-diabody for the same-day immunoPET of B-cell lymphoma. The authors used transgenic mice that expressed human CD20 on mature B cells to demonstrate the ability of an 18F-labeled diabody, [18F]FB-GAcDb, to detect both endogenous compartments expressing human CD20 and B-cell lymphoma liver metastases (Fig. 2B) (10). Shifting to radioimmunotherapy, Pruszynski et al. evaluated the biodistribution of a 225Ac-labeled human epidermal growth factor receptor 2 (HER2)–targeting sdAb in murine models of HER2-positive ovarian cancer and HER2-negative triple-negative breast cancer. The radioimmunoconjugate produced 15-fold higher uptake in HER2-positive than in HER2-negative tumors at 48 h after injection, as well as producing promising HER2-positive tumor–to–healthy-tissue activity concentration ratios at the same time point (>100 for blood; >40 for muscle; >8 for bone), suggesting that sdAbs may be suitable vectors for targeted radionuclide therapy with α-emitting isotopes (11). In a separate study, Feng et al. used a HER2-targted sdAb bearing a residualizing 131I-labeled prosthetic group for radioimmunotherapy in murine models of HER2-positive breast and ovarian cancer. In mice bearing BT474 xenografts, the radioimmunoconjugate—iso-[131I]SGMIB-VHH_1028—yielded a tumor-to-kidney therapeutic index of more than 8.5 and produced nearly complete tumor growth inhibition after doses of 18 and 30 MBq (Fig. 2C) (12). The same team recently reported preliminary data demonstrating the efficacy of HER2-targeted radioimmunotherapy with a sdAb bearing a similar residualizing prosthetic group labeled with the α-emitting radiohalogen 211At (13).
Even more exciting than these preclinical results are a handful of recent clinical trials. In 2016, Pandit-Taskar et al. reported the first-in-human application of [89Zr]Zr-IAB2M—a89Zr-labeled minibody engineered from the prostate-specific membrane antigen (PSMA)–targeting mAb J591—for the imaging of patients with metastatic prostate cancer (14). This initial study verified that the radioimmunoconjugate was safe, exhibited a favorable pharmacokinetic profile, and provided good lesion visualization 48 h after injection. Subsequent trials have explored the utility of [89Zr]Zr-IAB2M in PSMA-positive localized prostate cancer and have revealed that the radioimmunoconjugate exhibits performance comparable to that of [68Ga]Ga-PSMA-11 (15). Yet another minibody-based probe—the CD8-targeting [89Zr]Zr-IAB22M2C—was recently translated to the clinic for the imaging of CD8-positive T-cells. A first-in-human study in 2020 demonstrated that [89Zr]Zr-IAB22M2C was safe, well tolerated, and successfully targeted CD8-positive T-cell–rich regions such as the lymph nodes and the spleen (16). Finally, in 2020, Scott et al. reported on the use of a 124I-labeled diabody that had been pegylated to reduce first-pass renal clearance and thus increase circulation time ([124I]I-PEG-AVP0458) for PET in patients with ovarian cancer, metastatic prostate cancer, or primary prostate cancer (17). Whole-body PET scans acquired as early as 1 d after injection clearly demonstrate the promise of the radiotracer as a tool for delineating TAG-72–expressing cancers (Fig. 2D).
SITE-SPECIFIC BIOCONJUGATION
An abiding paradox in the study and use of radioimmunoconjugates is that these tools that are supposed to enable precision medicine are themselves synthesized in a surprisingly imprecise way. The overwhelming majority of radioimmunoconjugates are created via the stochastic ligation of amine-reactive prosthetic groups—most often chelators but occasionally radiohalogenated moieties—to lysines within the mAb. This approach, though facile, produces heterogeneous constructs and can interfere with the ability of the mAb to bind its antigen. A wide range of site-specific and site-selective approaches to bioconjugation have been developed to circumvent these issues, and the data consistently show that site-specifically modified radioimmunoconjugates exhibit better in vivo performance than randomly synthesized analogs (18). Precision is especially critical in the synthesis of fragment-based radioimmunoconjugates, as their smaller size increases the odds that cargoes will inadvertently be attached within their antigen-binding domains. The most frequently used approach to site-specific bioconjugation relies on ligations between thiol-reactive probes (e.g., maleimides) and cysteines generated via the reduction of the antibody’s interchain disulfide linkages. This strategy is an improvement over traditional methods, but the maleimide–thiol reaction is reversible under physiologic conditions, and the formation of 4–8 free cysteine residues in full-length IgG (depending on the reduction conditions) means that some heterogeneity remains inevitable. Not surprisingly, the field has increasingly turned to antibody engineering for more robust and reliable approaches to site-specific bioconjugation.
The incorporation of amino acids—either natural or unnatural—at unique sites within the antibody for bioconjugation has proven a promising strategy, particularly in the context of fragment-based probes. For example, Chigoho et al. used an hPD-L1–targeting sdAb with a C-terminal cysteine residue and a maleimide-bearing variant of NOTA to create [68Ga]Ga-NOTA-mal-hPD-L1, a radiotracer that displayed promising in vivo performance (19). Sharma et al. followed a slightly different approach to create a [89Zr]Zr-desferrioxamine (DFO)-labeled probe for the visualization of delta-like ligand 3 expression. In this case, the authors did not incorporate new cysteines into the mAb. Rather, the heavy-chain cysteines within the upper hinge region that would normally form disulfide linkages were mutated to serine residues, leaving a pair of free cysteines on the light chains that the team specifically modified with phenyloxadiazolyl methylsulfone–bearing chelators (Fig. 3A) (20). Genetic code expansion to incorporate unnatural amino acids with orthogonally reactive moieties offers a more sophisticated variation on this theme with unrivaled site specificity. In 2020, Ahn et al. leveraged this technology to create a variant of trastuzumab containing a quartet of p-azido-methyl-phenylalanine residues in the Fc region. The authors used the strain-promoted azide-alkyne cycloaddition reaction to couple dibenzocyclooctyne-bearing variants of DFO and DO3A to the mAb and subsequently labeled the site-specifically modified immunoconjugates with 89Zr and 111In, respectively (Fig. 3B) (21).

(A–C) Schematics illustrating the creation of free cysteine residues via a pair of C220S mutations (A), the site-specific modification of unnatural pAMF via strain-promoted azide-alkyne cycloaddition reaction (B), and the sortase-mediated bioconjugation of glycine-bearing chelator to Fab fragment bearing C-terminal LPETG tag (C). (D) Serial PET images of mice bearing A431 xenografts obtained after the administration of an EGFR-targeting Fab that has been site-specifically labeled with 64Cu. (Reprinted with permission of (22).)
Antibody engineering has also been used to build peptide recognition sites into immunoglobulins for chemoenzymatic modifications. In 2021, for example, Rudd et al. used the transpeptidase sortase A to catalyze the attachment of glycine-bearing chelators to an epidermal growth factor receptor–targeting Fab bearing a C-terminal LPETG recognition sequence, ultimately creating both 64Cu- and 89Zr-labeled radioimmunoconjugates that displayed excellent in vivo performance in a murine model of epidermal growth factor receptor–positive epidermoid carcinoma (Figs. 3C and 3D) (22). Bridoux et al. used a similar approach to append a variant of NOTA with a GGGYK tag to an hPD-L1–targeting sdAb bearing a C-terminal LPETG recognition element. Critically, the authors demonstrated that the site-specifically modified radioimmunoconjugate—[68Ga]Ga-NOTA-(hPD-L1)—outperformed a randomly labeled analog in vivo (23). The Fc region of full-length IgG has also attracted interest for engineering-driven bioconjugation. To wit, Jeger et al. developed a variant of the L1CAM-targeted mAb chCE7 with a N297Q mutation that eliminated glycosylation and allowed for the transglutaminase-mediated attachment of DFO at the Q297 and Q295 sites of each heavy chain (24). The resulting radioimmunoconjugate—[67Ga]Ga-DFO4-chCE7agl—displayed markedly better in vivo behavior than an analogous randomly conjugated variant.
MODULATING FC INTERACTIONS
The interactions between the Fc domains of immunoglobulins and Fc receptors are instrumental in determining the in vivo behavior of mAbs. Although FcRn is responsible for the recycling of immunoglobulins, a variety of Fcγ receptors—including receptors I, II, IIIA, and IIIB—mediate the interplay between antibodies, immunocomplexes, and the immune system. In light of this, it is not surprising that the field has turned to the engineering of the Fc region as a route to improved radioimmunoconjugates.
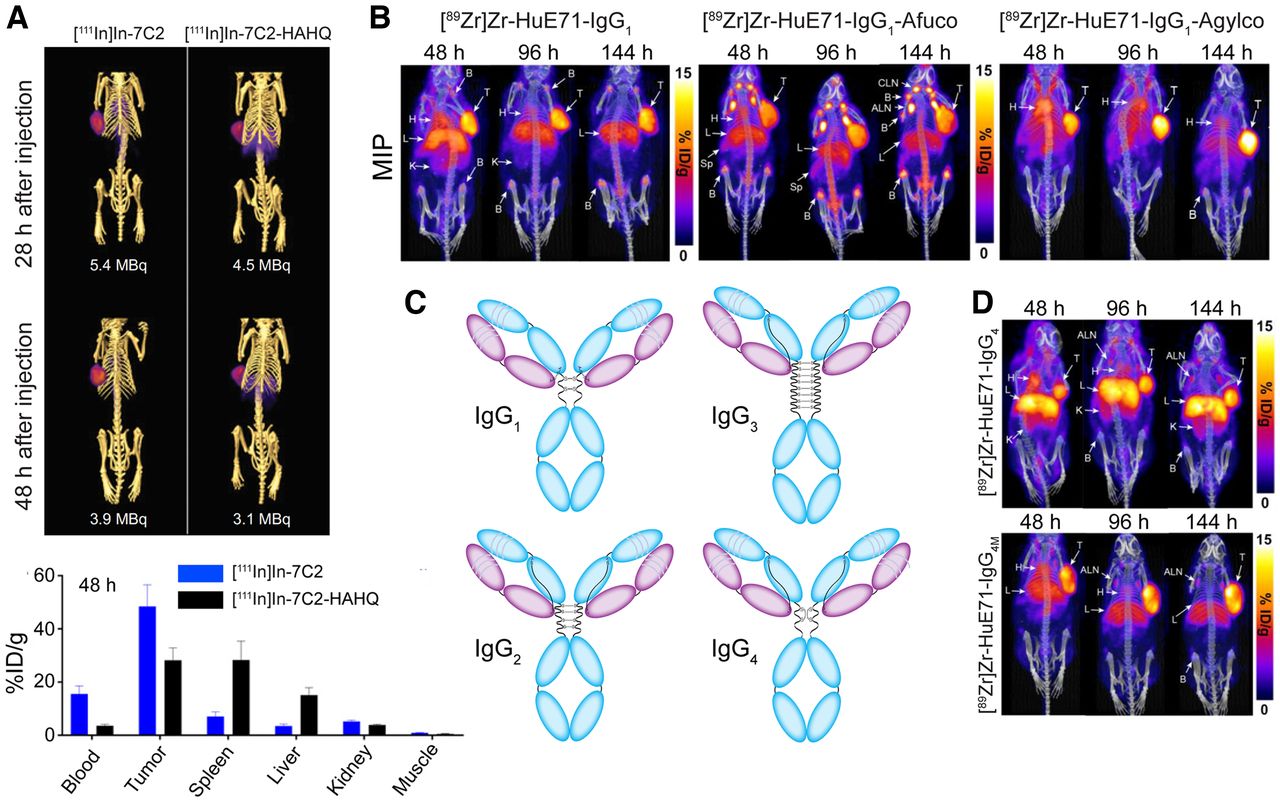
Several laboratories have sought to improve the behavior of mAb-based radioimmunoconjugates by attenuating or enhancing Fc receptor binding. Nazarova et al. explored immunoSPECT imaging using a HER2-targeting 111In-labeled mAb bearing a pair of histidine mutations—H310A and H435Q—that abrogated FcRn binding (25). The double mutant displayed a more rapid pharmacokinetic profile and greater tumor-to-blood activity concentration ratios than its wild-type parent but at the cost of reduced tumoral uptake and lower tumor-to-spleen and tumor-to-liver contrast (Fig. 4A). Burvenich et al. made similar observations with 111In- and 177Lu-labeled anti-Lewis-Y mAbs with two mutations (I253A and H310A) that reduce FcRn binding (26). The critical role of the heavy-chain glycans in modulating the Fcγ receptor engagement of mAb has led others to pursue glycoengineered radioimmunoconjugates. In 2019, Vivier et al. demonstrated that the enzymatic deglycosylation of [89Zr]Zr-DFO-trastuzumab attenuates FcγRI binding and reduces uptake in healthy tissues in tumor-bearing NSG and huNSG mice compared with a fully glycosylated analog (27). More recently, Sharma et al. demonstrated the power of this approach using two glycoengineered variants of the L1CAM-targeting radioimmunoconjugate [89Zr]Zr-DFO-HuE71 (Fig. 4B) (28). An afucosylated variant with enhanced FcγRIIIA binding exhibited reduced tumoral uptake and enhanced accretion in the liver and lymphoid tissues compared with the parent radioimmunoconjugate. In contrast, an aglycosylated variant with abrogated Fcγ receptor binding yielded dramatically reduced accretion in the bone and lymph nodes compared to parent [89Zr]Zr-DFO-HuE71. Finally, Bensch et al. recently reported a clinical trial focused on immunoPET with [89Zr]Zr-lumretuzumab, a HER3-targeting mAb glycoengineered to exhibit enhanced FcγRIIIA engagement and thus antibody-dependent cellular cytotoxicity (29).

(A) SPECT images (top) and biodistribution data (bottom) collected 28 and 48 h after the administration of a HER2-targeting radioimmunoconjugate ([111In]In-7C2) or a mutant with abrogated FcRn binding ([111In]In-7C2-HAHQ) to mice bearing KPL-4 xenografts. (Reprinted with permission of (25).) (B) Serial PET images obtained using wild-type, afucosylated, and aglycosylated [89Zr]Zr-IgG1 in mice bearing L1CAM-expressing SKOV3 xenografts. (Reprinted with permission of (28).) (C) Four subclasses of IgG. (D) Serial PET images obtained using wild-type IgG4-based [89Zr]Zr-mAb ([89Zr]Zr-HuE71-IgG4) and an analog mutated to have IgG1-like interchain disulfide linkages ([89Zr]Zr-HuE71-IgG4M). (Reprinted from (28).)
The differing Fc interactions of the four subclasses of IgG—IgG1, IgG2, IgG3, and IgG4—have also been leveraged to create better radioimmunoconjugates (Fig. 4C). In 2020, Bicak et al. created an IgG3-based radioimmunoconjugate of the hexokinase 2–targeting mAb hu11B6 for the 225Ac-radioimmunotherapy of prostate cancer (30). The investigators hypothesized that the enhanced complement activation and Fcγ receptor engagement of the IgG3 scaffold would lead to “immunotherapeutically enhanced” radioimmunotherapy, but in practice neither [225Ac]Ac-hu11B6-IgG3 nor an analog bearing a R435H mutation to rescue FcRn binding produced improved therapeutic efficacy over [225Ac]Ac-hu11B6-IgG1. In the study from Sharma et al. mentioned above, the authors also explored 89Zr-immunoPET with two L1CAM-targeting IgG4-based radioimmunoconjugates, as this subclass has attracted attention in the immunotherapeutics community because it elicits reduced effector functions relative to other subclasses (Fig. 4D) (28). Although a radioimmunoconjugate based on the wild-type IgG4 framework produced high levels of nonspecific uptake in the kidneys, this behavior was eliminated in a variant with S228P mutations in the hinge region that facilitated the formation of IgG1-like interchain disulfides and thus mitigated in vivo Fab arm exchange. Finally, Man et al. moved beyond the IgG isotype, using immunoSPECT to study the in vivo behavior of an IgE-based anti-CSPG4 antibody designed to elicit a more potent immune response (31). In the absence of FcRn-mediated recycling, the [111In]In-IgE was cleared from the blood of tumor-bearing mice much more rapidly than its [111In]In-IgG counterpart. Furthermore, the [111In]In-IgE produced low activity concentrations in tumor tissue alongside high accretion in the liver, but its very low uptake in the blood yielded tumor-to-blood activity concentration ratios comparable to the analogous IgG.
Radioimmunoconjugates based on fusion proteins that combine antigen-binding fragments and Fc domains lie at the intersection of two threads of our discussion. These single-chain constructs offer many characteristics of full-length mAbs—that is, large size, multivalency, and control over FcRn engagement—in a package that is easier to produce given that there is no need for the coexpression of both heavy and light chains. In 2014, Rochefort et al. developed an effective CA19-9–targeting 124I-labeled (scFv)2-Fc that included an H310A mutation that abrogated FcRn binding (32). More recently, Delage et al. described a 177Lu-labeled anti-TEM-1 (scFv)2-Fc that produced moderate uptake in TEM-1–positive xenografts, but the authors made no mention of Fc engagement or mutations (33).
BINDING MULTIPLE TARGETS
Antibody engineering has also been harnessed to create radioimmunoconjugates based on bispecific antibodies (BsAbs), immunoglobulins designed to target more than one antigen. BsAbs offer several advantages over their monospecific cousins, as the ability to bind a second antigen can be harnessed to elicit synergistic antitumor effects, circumvent drug resistance, recruit immune cells, or block protumor signaling pathways. Generally speaking, full-length BsAbs consist of a heavy-chain/light-chain pair from one mAb and a second from another mAb (Fig. 5A). However, a wide variety of bi- and trispecific constructs have been developed using fragment-based frameworks.

(A) Examples of BsAb. (B) ImmunoPET images obtained from mice bearing HuT tumors 6 d after the administration of bispecific [89Zr]Zr-REGN4018 (left), [89Zr]Zr-REGN4018 plus unlabeled mucin 16-binding mAb (middle), and [89Zr]Zr-REGN4018 plus unlabeled CD3-binding mAb (right). Spleen is indicated by yellow arrow; lymph nodes by green arrow; and tumor by red arrow. (Reprinted with permission of (35).) (C) Pretargeted immunoPET with TF2 and [68Ga]Ga-IMP288 in a patient with HER2-negative breast cancer produces high uptake in liver lesion (arrow, top) which was not seen by [18F]FDG PET (bottom). (Reprinted from (39).)
BsAbs, like mAbs, have increasingly been used as companion imaging agents for therapeutic counterparts. Along these lines, a clinical trial focused on immunoPET with a 89Zr-labeled T-cell–engaging BsAb capable of binding CD3 and carcinoembryonic antigen in patients with gastrointestinal adenocarcinoma (34). The CD3/carcinoembryonic antigen–targeting BsAb accumulated in both lymphoid organs and tumor lesions, thus providing data on the heterogeneity of antigen expression that could aid in the selection of patients likely to respond to therapy and in planning personalized drug dosing schedules. In 2019, Crawford et al. evaluated the in vivo behavior of another T-cell–engaging BsAb that targets CD3 and mucin 16 in a murine model of ovarian cancer and found the highest activity concentrations in the tumor tissue and lymphoid organs (35). A blocking study elegantly illustrated the role of the probe’s dual specificity in this behavior: blocking with a CD3-specific mAb selectively reduced uptake in the lymphoid tissues, whereas blocking with a mucin 16–specific mAb did the same for the tumor (Fig. 5B).
The field has also turned to BsAbs to create radioimmunoconjugates capable of crossing the blood–brain barrier. The size and polarity of mAbs generally precludes their transport across the blood–brain barrier, thus curtailing the use of radiolabeled antibodies for neuroimaging and therapy. However, the incorporation of a transferrin-binding Fab fragment in a BsAb has been shown to facilitate the transferrin receptor–mediated transcytosis of the bispecific construct across the blood–brain barrier. In 2017, Syvänen et al. used this technique to create a 124I-labeled tribody for the visualization of amyloid-β protofibrils—protein aggregates implicated in Alzheimer disease—in the brain (36).
A third application of BsAbs in nuclear medicine lies in pretargeted imaging and therapy (4,37). In this context, the BsAb is designed to bind both a cancer antigen and an exogeneous radiolabeled hapten. The unlabeled BsAb is administered first and allowed to accumulate at the tumor and clear from the blood. Once the BsAb has reached this optimal biodistribution, the radiolabeled hapten is administered, and the high affinity and selectivity of the BsAb for the hapten facilitates the in vivo ligation between the two components. This strategy enables the use of short-lived radionuclides that are not normally compatible with mAb-based vectors and reduces radiation dose rates to healthy tissues. Two recent clinical studies have explored the use of pretargeted PET with a carcinoembryonic antigen–targeted BsAb (TF2) and a 68Ga-labeled peptidic hapten ([68Ga]Ga-IMP288) in patients with breast cancer and colon cancer and have found that this approach not only was safe but also provided better sensitivity and specificity than [18F]FDG (Fig. 5C) (38,39). Importantly, a second approach to in vivo pretargeting predicated on BsAbs capable of binding tumor biomarkers as well as DOTA-based haptens has also shown promise for both imaging and radioimmunotherapy in murine models of disease (40).
CONCLUSION
The next few years will undoubtedly be critical for work at the intersection of nuclear medicine and antibody engineering. It is undeniably easy to get excited about the preclinical data produced by many of these innovations, including sdAb-based radioimmunotherapeutics, new approaches to site-specific bioconjugation, and novel strategies for pretargeted imaging and therapy. Yet the key step for many of these technologies will be the move from the laboratory to the clinic. This is, admittedly, easier said than done, but their impact on patient care will ultimately hinge on clinical outcomes, so generating clinical data as soon as possible is imperative. There is more to do in the laboratory, too. For example, the preliminary work on the interplay between radioimmunoconjugates and Fc receptors is intriguing, but it behooves the field to explore these relationships in murine models that better recapitulate the human immune system. Finally, we hope to see even more bridges built between the nuclear medicine community and those studying immunotherapy and antibody–drug conjugates, as the cross-pollination of ideas between these fields can only lead to improved technologies for the clinic.
DISCLOSURE
Support was received from the NIH (R01CA240963, U01CA221046, R01CA204167, R21EB030275, and R01CA244327 to Brian Zeglis) and the Academy of Finland (331659 to Outi Keinänen). No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jul. 21, 2022.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 15, 2022.
- Revision received July 7, 2022.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}