


Abstract
The application of radiopharmaceutical therapy for the treatment of certain diseases is well established, and the field is expanding. New therapeutic radiopharmaceuticals have been developed in recent years, and more are in the research pipeline. Concurrently, there is growing interest in the use of internal dosimetry as a means of personalizing, and potentially optimizing, such therapy for patients. Internal dosimetry is multifaceted, and the current state of the art is discussed in this continuing education article. Topics include the context of dosimetry, internal dosimetry methods, the advantages and disadvantages of incorporating dosimetry calculations in radiopharmaceutical therapy, a description of the workflow for implementing patient-specific dosimetry, and future prospects in the field.
Currently, there is intense interest in radiopharmaceutical therapy (RPT), particularly in terms of a theranostic paradigm that incorporates both diagnostic and therapeutic elements. Ideally, this consists of matched pairs of radiopharmaceuticals: a diagnostic partner (labeled with a positron-emitting or single-photon–emitting radionuclide) that provides information about disease extent and phenotype and serves to predict the utility of a therapeutic partner (usually labeled with a β- or α-particle–emitting radionuclide) that delivers targeted radiation. A key issue in protocol design is the choice of therapeutic activity and the time schedule for its administration. These may be based on patient-specific information or a simpler one-size-fits-all (i.e., population-averaged) approach.
The RPT strategy should align with the therapeutic objective: curative or palliative. A treatment designed to maximize the likelihood of cure would aim to deliver a high therapeutic dose over a relatively short time. The scope for future retreatment would be limited, similar to the case of external-beam radiotherapy (XRT). In contrast, a treatment designed to maximize the duration of disease control would entail a less aggressive delivery of therapy over an extended period, ideally retaining the option for future retreatment. Most current RPT designs for systemic treatment conform with the latter approach. Examples of clinical applications of RPT are provided in the supplemental material (available at http://jnm.snmjournals.org) (1–23).
Traditionally, in medicine, dose refers to the mass amount (e.g., mg) of drug administered. For radiopharmaceuticals, the analogous quantity is activity (e.g., MBq). Prescribing treatment in terms of mass dose is rational for nonradioactive drugs because there is little way of knowing how much localizes in target and nontarget tissues and how this localization varies among patients. In contrast, most radiopharmaceuticals permit quantification of their biodistribution and enable the administered activity to be modified on the basis of patient-specific factors via the metric of absorbed dose, the radiation energy deposited per unit mass (expressed as grays).
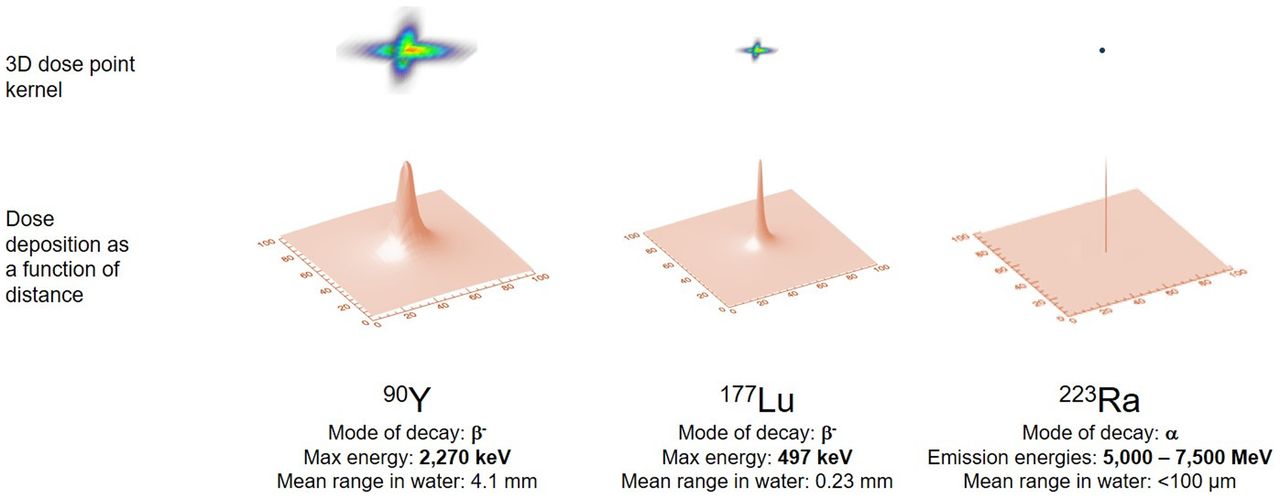
The aim of dosimetry is to estimate the absorbed dose to normal tissues and tumors and thereby anticipate the biologic effects of radiation. Calculation of absorbed dose takes into account (patient-dependent) anatomy and radiopharmaceutical biodistribution and (patient-independent) radionuclide properties. For RPT, the absorbed dose is calculated as the area under the dose rate–versus–time curve for a given target volume and generally requires multiple point estimates of dose rate. The dose rate in a tissue is proportional to the activity concentration in that tissue (self-dose) and in other tissues within range of the radionuclide’s emissions (cross-dose). For radionuclides typically used for therapy, most of the energy is emitted as short-range particles, and for those tissues that receive the highest absorbed doses, self-dose is the dominant contribution (Fig. 1; Table 1).
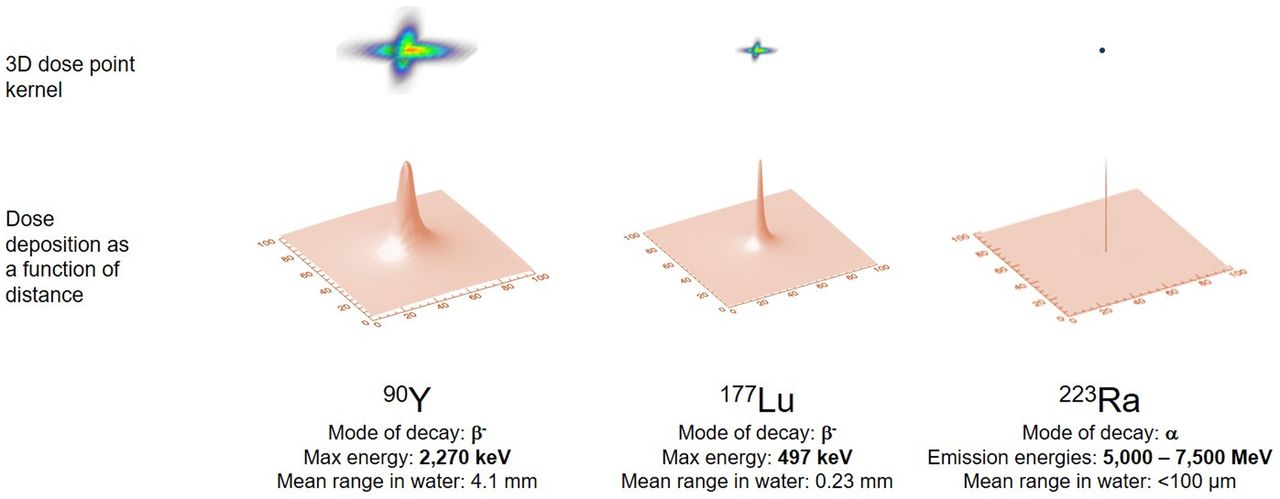
Graphical representation of dose deposition ranges delivered by different radionuclides having varying modes of decay. Top row shows relative geometric dose deposition delivered by point source of activity. Bottom row shows same data represented with 2-dimensional curve, illustrated as point spread function. Figure illustrates variable dose deposition properties and is not to scale.
Summary Characteristics of Commonly Used Radionuclides in RPT
The key physical determinants of biologic response are the radiation quantity (absorbed dose) and quality (linear energy transfer [LET]). XRT and brachytherapy treatments are prescribed, and normal-tissue tolerances defined, in terms of absorbed dose. However, for RPT, patient-specific dosimetry remains controversial. Important, recently introduced therapies featuring 223RaCl2, 177Lu-DOTATATE, and 177Lu-PSMA-617 are prescribed on the basis of activity, not absorbed dose. Several factors contribute to this current practice. First, protocol design and pivotal clinical studies are increasingly dictated by industrial sponsors, for whom it is advantageous to minimize logistical complexity and maximize throughput. As more RPTs are approved, simplified treatment delivery will become even more desirable. Second, a dosimetry-based protocol requires reliable tumor and normal-tissue dose–response information. However, RPT dose–response data remain largely anecdotal. There is a chicken-and-egg element to this: dosimetry is not performed because dose–response data are lacking, and dose–response data are lacking because dosimetry is not performed. Each radiopharmaceutical has a range of administered activity that results in at least some clinical responses without excessive adverse effects, generally identified in chemotherapy-like dose-escalation (i.e., phase 1 and 2) trials. Third, dosimetric and clinical factors in RPT make direct comparisons with XRT problematic. Absorbed dose distributions are driven by biology in RPT but by the geometry of intersecting radiation beams in XRT. XRT dose distributions are uniform at the microscopic level, but in RPT they are nonuniform. Clinically, RPT is a systemic treatment in which targets may differ in size, location, and phenotype. Tumors too small to be imaged cannot be treated with XRT but are valid targets for RPT. Further, RPT patients have typically received prior therapies, complicating normal-organ radiation response.
Prescribing RPT on the basis of a one-size-fits-all activity ignores patient-specific differences and sets limits on treatment that are defined by the most susceptible patients (24,25). A dosimetry-based approach is predicated on the assumption that better clinical results can be achieved using individualized absorbed dose estimates rather than fixed activities. However, this hypothesis must be tested clinically. Recent approvals of new therapeutic radiopharmaceuticals were based on the results of randomized controlled clinical trials (3,12,26). For dosimetry, too, clinical trials will be required to determine whether it can improve RPT outcomes.
DOSE PRESCRIPTION ALGORITHMS
There are 3 prescription algorithms for RPT: fixed administered activity (e.g., MBq, MBq/kg of body mass, and MBq/m2 of body surface area), maximum tolerated absorbed dose (MTAD), and prescribed tumor-absorbed dose (PTAD).
The approach using a fixed administered activity is patient-independent and does not require any patient measurements, apart from possibly mass and height. Treatment activities are based on chemotherapylike dose-escalation phase 1 and 2 clinical trials. For example, 177Lu-DOTATATE (Lutathera; Advanced Accelerator Applications) treatment of somatostatin receptor–expressing neuroendocrine tumors is generally delivered in 4 cycles of 7.4 GBq at 8-wk intervals (27). For 223RaCl2 (Xofigo; Bayer), 6 administrations of 55 kBq/kg are given at 4-wk intervals (28). A fixed administered activity is the simplest, most convenient, and least expensive approach. Inevitably, however, some patients could safely have received higher (and presumably more therapeutically effective) activities and were thus underdosed. Conversely, other patients receiving the same fixed activity may have experienced excessive normal-tissue side effects and were therefore overdosed (24).
The MTAD and PTAD approaches are both patient-specific and involve absorbed-dose projections. These approaches typically require a series of measurements, either performed in advance of the therapy or during the first administration of a multiadministration treatment. The objective is to predict the activity to administer to achieve a specified absorbed dose either to the dose-limiting normal tissue or to the tumor.
In the MTAD approach, it is likely that only a small number of normal tissues will receive absorbed doses approaching tolerance limits. For 131I-iodide treatment of metastatic thyroid cancer, one approach is to prescribe a therapeutic activity that is calculated to deliver 2 Gy to blood (29). For renal toxicity, an MTAD of 23 Gy is often used as a guideline, based on XRT data, and is reasonably consistent with RPT experience with 90Y-DOTA-octreotide after appropriate radiobiologic corrections for dose rate (30). However, it may be an inappropriately low threshold for 177Lu-DOTATATE (31). Organ MTAD likely depends on the type of emissions, uniformity of the activity/dose distribution, dose rate, prior treatment, and life expectancy (32).
Treating patients according to PTAD is a concept extended from XRT practice. However, there are few dose–response data available for RPT on which to base treatment prescription. In a small series of postthyroidectomy thyroid cancer patients, Maxon et al. (33) successfully treated lymph node metastases in 74% of patients with thyroid remnants and in 86% of athyrotic patients with a single administration of 131I calculated to deliver lesion-absorbed doses of 85 and 140 Gy, respectively. Dewaraja et al. (34) found that for 90Y-microsphere radioembolic therapy of liver tumors, the mean absorbed dose and biologically effective dose (an absorbed dose-based metric that takes account of radiobiologic features) that yielded a 50% tumor control probability were 292 and 441 Gy, respectively (34). The current state of knowledge of tumor dose response was recently summarized (35). However, only macroscopic, imageable tumors are amenable to the PTAD approach.
PARADIGM FOR PATIENT-SPECIFIC DOSIMETRY
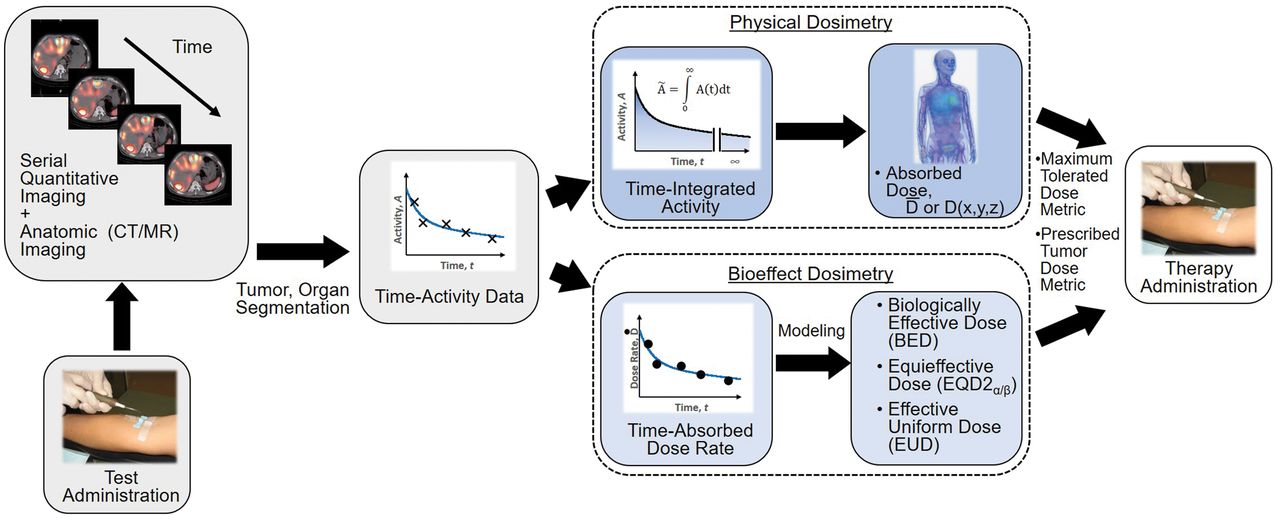
The paradigm for patient-specific dosimetry for RPT (Fig. 2) is as follows: administration of a test activity of either the therapeutic or a surrogate radiopharmaceutical; measurement—by serial imaging and possibly blood and whole-body counting—of its time-dependent biodistribution; definition of the pertinent anatomy by high-resolution structural imaging (CT, MRI); derivation of time-dependent activity concentration or absorbed dose rate, with appropriate adjustment for differences in half-life between the therapeutic and surrogate radionuclides; integration of time–activity data to yield region- or voxel-specific time-integrated activity coefficients (alternatively, time–dose-rate data can be integrated directly to yield absorbed dose); calculation of absorbed dose coefficients for organ at risk or tumor for the therapeutic radiopharmaceutical (optional modifications for radiobiologic modeling can be incorporated at this step); and prescription of the activity to deliver the intended absorbed dose to the organ at risk or tumor.
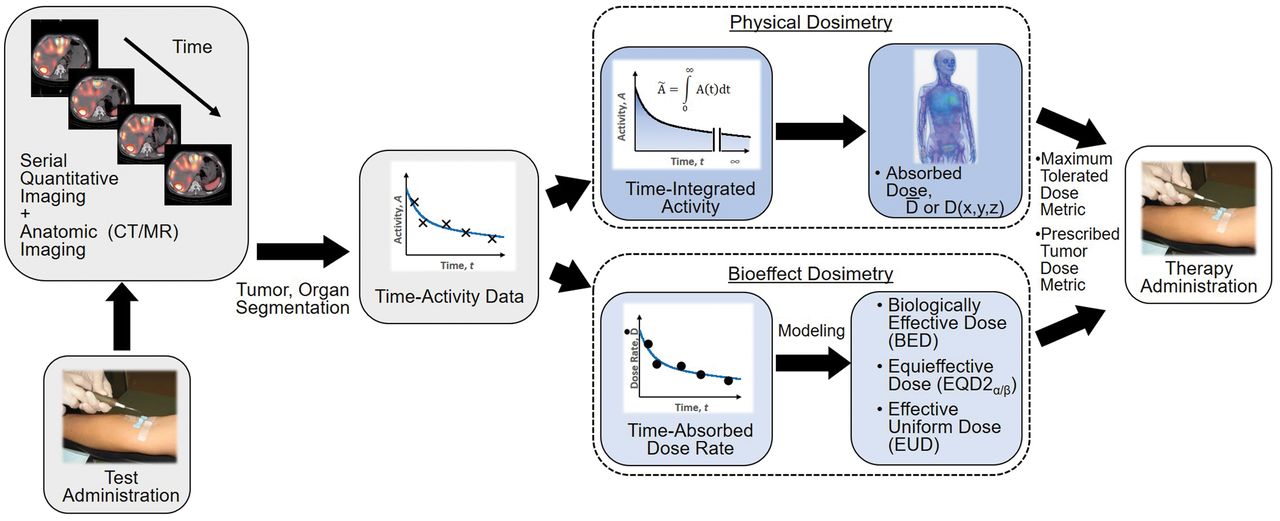
General workflow for RPT dosimetry. Process begins with test administration (may be either pretherapy administration or first cycle of multidose therapy regimen). Serial quantitation measurements can then support calculation of absorbed doses, either in terms of tumor and organ mean doses (D) or dose distributions (D[x,y,z]). Dose estimation per unit of administered activity can then be used to tailor treatment. The term dose metric may refer to absorbed dose (for physical dosimetry) or biologically effective dose (BED), equieffective dose (EQD2α/β), or effective uniform dose (EUD) (for bioeffect dosimetry).
Implicit in this paradigm is that the absorbed dose coefficients for the full RPT will be the same as those projected on the basis of the test study, and this is more likely to be true when the test and therapeutic radiopharmaceutical are chemically identical. If the test and therapeutic radiopharmaceuticals are different or if target tissue uptake depends nonlinearly on administered mass or activity (36), this approach may be less reliable. Changes in the patient’s condition between test and therapy administrations, such as thyroid stunning (37), may also undermine this approach.
The time and effort required for the dosimetry paradigm may be considerable. Preparation and assay of the radiopharmaceutical may take 10–20 min; its administration may take less than a minute for a bolus injection to as long as 1–2 h for a slow infusion. The imaging time per point ranges from 2–5 min for a single static image to 20–40 min for a whole-body scan or single-bed-position SPECT/CT study to 1–2 h for a multiple-bed-position SPECT/CT study. A single imaging time point may be sufficient for reasonably accurate dosimetry, greatly reducing the time commitment. Segmentation (i.e., contouring) of normal organs and tumors can be particularly time-consuming—several hours—if done manually. Automated and semiautomated segmentation procedures can accelerate this process, and ultimately, artificial intelligence (AI)–based routines may make segmentation fully automated and rapid. Subsequent steps in the workflow—fitting or integrating mathematic functions to measured data and calculating absorbed doses or dose distributions—are computer-intensive but largely automated. Individuals performing clinical dosimetry calculations must have appropriate training and a full understanding of the process. Recent international guidance suggests allotting 1.1 d of a medical physicist’s time to perform calculations per case (38).
MEASUREMENT OF ACTIVITY AND TIME–ACTIVITY DATA
Radiopharmaceutical activity is routinely measured with a dose calibrator with uncertainties of ±5% or less. However, for isotopes with complex decay schemes—with nonequilibrium progeny such as some α-particle emitters, pure β-particle emitters (e.g., 90Y), and nonstandard source geometries—dose calibrator uncertainties can be significant (39). For such isotopes, reference standard sources traceable to a national agency should be used to verify accuracy. Any uncertainties associated with activity measurements will be propagated through the entire dosimetry analysis (40).
Therapeutic radiopharmaceuticals are often single-photon emitters, and their time-dependent activities or activity concentrations may be measured by serial planar γ-camera imaging (i.e., the conjugate-view method (41)), SPECT/CT (42), or a combination of planar and SPECT/CT imaging (the hybrid method (42)). Subject to corrections for collimator–detector response, scatter, attenuation, and partial-volume effects, the count rate per voxel in reconstructed tomographic images is proportional to the local activity concentration. The corrected count rate (cps) per voxel is divided by a measured system calibration factor [(cps/voxel)/(kBq/mL)] to yield activity concentrations:
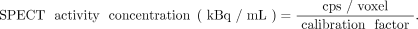 Eq. 1
Eq. 1
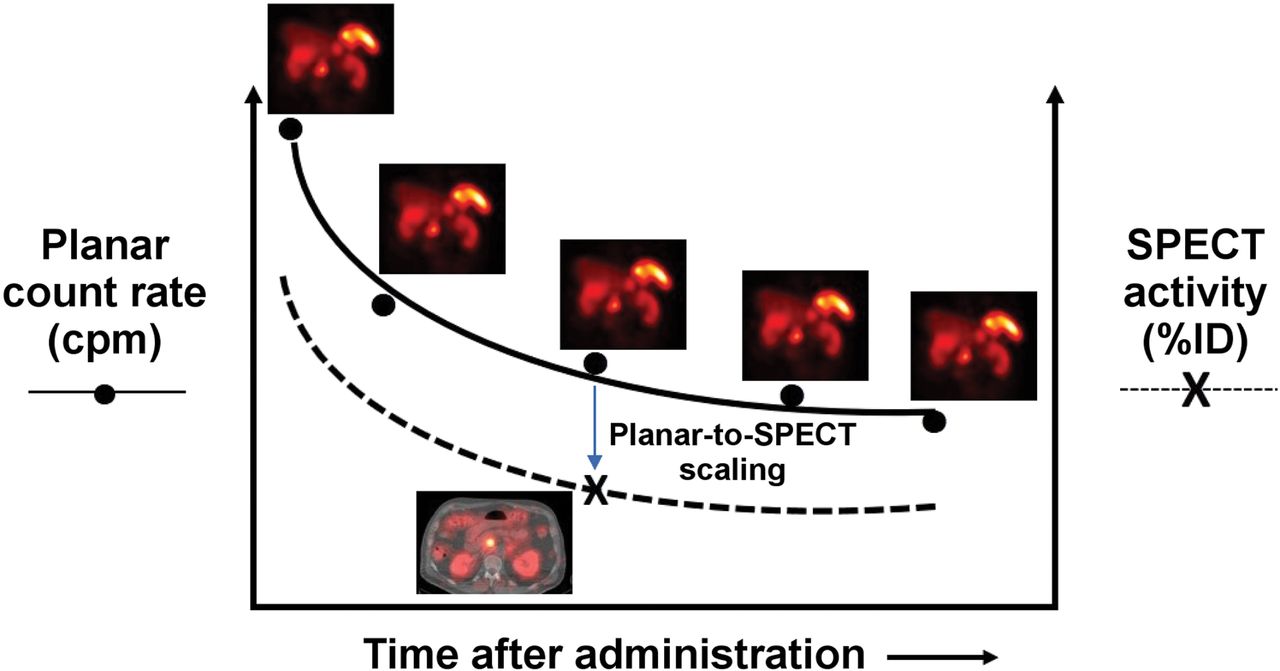
SPECT/CT imaging is relatively time-consuming (15–30 min per bed position). A practical alternative is hybrid SPECT/planar imaging, in which both SPECT/CT and planar scans are acquired at a single time point and only the more rapid planar scans are acquired at the remaining time points (Fig. 3) (42). The multiple planar scans provide the shapes of the source-region time–activity curves (i.e., the kinetics), and the single SPECT/CT study provides a (more accurate) point estimate of activity. Comparison of the contemporaneous planar and SPECT/CT scans provides a SPECT/CT-to-planar scaling factor.
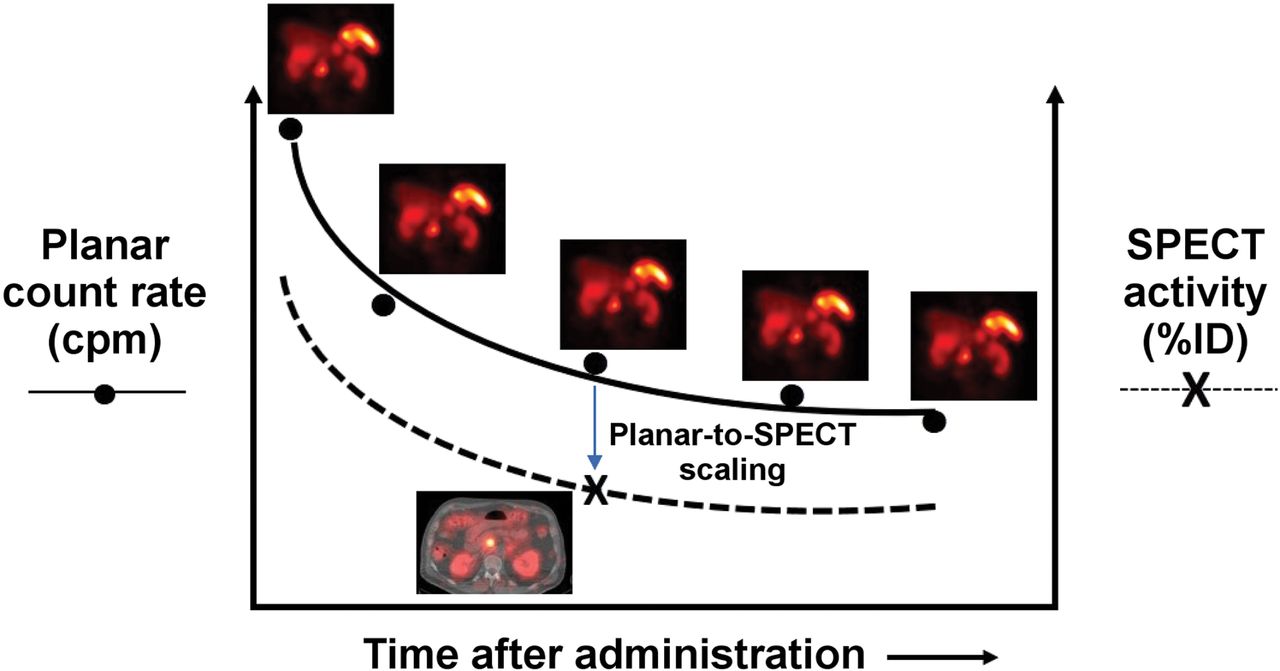
Hybrid SPECT/planar imaging approach to imaging-based measurement of time–activity data (55).
Quantitative PET remains more mature than quantitative SPECT, but with rare exceptions (10), positron-emitting radionuclides are not used for RPT. Positron emitter–labeled surrogates may, however, be used to provide time–activity data for therapeutic radiopharmaceuticals (43), such as the 124I/131I PET/therapeutic radionuclide pair in metastatic thyroid cancer. The PET and therapeutic radionuclides must be well matched in terms of physical half-life for serial PET scans to be performed over a sufficiently long total time frame to yield reliable estimates of the time–activity data for the therapeutic radionuclide. 124I (physical half-life, 4.18 d) and 131I (physical half-life, 8.04 d) satisfy this criterion. In contrast, 68Ga-DOTATATE (physical half-life, 67.7 min) is too short-lived to estimate later tissue activities of 177Lu-DOTATATE (physical half-life, 6.65 d).
For radiopharmaceuticals with well-characterized kinetics that exhibit little variability among patients, population-averaged normal-organ time–activity curves may be scaled by image-derived, patient-specific organ activities measured at a judiciously selected single time point (44,45). The utility of this method has been demonstrated for 90Y-DOTATOC (46) and 177Lu-DOTATATE/DOTATOC (47) for kidney dosimetry. The reliability of single-time-point imaging for planning RPT requires further validation and may be less applicable to tumors.
The hematopoietic bone marrow is radiosensitive and is often dose-limiting for RPT (48). However, quantifying activity in red marrow for dosimetry is especially challenging as it is a widely distributed source region with regional variations in activity concentration. One practical approach is based on counting weighed samples of peripheral blood in a scintillation well counter. For radiopharmaceuticals that do not localize to blood or marrow cells, the activity concentration in plasma has been estimated as equal to that in the red marrow extracellular fluid (∼20% of the marrow by volume) at equilibrium (49,50). Alternatively, red-marrow activity concentration may be estimated by scintigraphic imaging of vertebrae (51,52).
Whole-body clearance kinetics may be measured by serial conjugate-view whole-body scans or probe-based counts beginning shortly after radiopharmaceutical administration but before the patient’s first postadministration void or bowel movement. The initial net (background-subtracted) geometric-mean whole-body or probe count rate corresponds to 100% of the administered activity. The values at each subsequent time point, normalized to the 100% count-rate value, yield whole-body activity (as a percentage of the administered activity).
CALCULATION OF ABSORBED DOSE
Calculation of absorbed doses requires estimating the source region time-integrated activity coefficients, calculated as areas under curves of activity/activity concentration or dose rate. These data may be fitted by mathematic functions (typically sums of exponentials) and integrated analytically to infinity. Alternatively, numeric methods (e.g., trapezoidal integration) may be used to integrate to the last measured point with an additional contribution to account for terminal behavior. Operationally, the terminal contribution may be taken to correspond to physical decay or apparent clearance derived from the last 2 measurements. Although it has been recommended that the last measurement be performed no earlier after administration than twice the radionuclide’s physical half-life (53), this is rarely done for radionuclides with relatively long half-lives (e.g., 131I [8.0 d] and 177Lu [6.7 d]). Areas under curves can also be deduced by compartmental modeling (54).
There are 3 approaches to calculating absorbed dose from internal radionuclides: dose factor–based calculation (such as the MIRD formalism), dose point kernel convolution, and Monte Carlo (MC) radiation transport simulation (55).
In the organ-level time-independent formulation of the MIRD schema(56), the absorbed dose coefficient 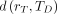 (mGy/MBq) is defined, for a target region
(mGy/MBq) is defined, for a target region 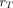 irradiated over a time period
irradiated over a time period 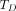 , as the absorbed dose (mGy) normalized to the administered activity (MBq):
, as the absorbed dose (mGy) normalized to the administered activity (MBq):
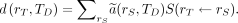 Eq. 2
Eq. 2
Here, 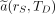 is the time-integrated activity coefficient, and
is the time-integrated activity coefficient, and 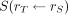 is known as the S value (or S coefficient), the absorbed dose to
is known as the S value (or S coefficient), the absorbed dose to 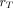 per unit time-integrated activity in source region
per unit time-integrated activity in source region  . S values have been tabulated for a large number of radionuclides and source-region–target-region pairs in several reference anatomic models from newborns to adult men and women (57). Self-irradiation dose factors for tumors, modeled as unit-density spheres, are also available (58,59). Several computer programs for organ-level dosimetry have been developed; these include OLINDA (approved by the U.S. Food and Drug Administration) (58), MIRDOSE (its predecessor) (60), IDAC-Dose 2.1 (61), and MIRDcalc (59); the latter two are freely available.
. S values have been tabulated for a large number of radionuclides and source-region–target-region pairs in several reference anatomic models from newborns to adult men and women (57). Self-irradiation dose factors for tumors, modeled as unit-density spheres, are also available (58,59). Several computer programs for organ-level dosimetry have been developed; these include OLINDA (approved by the U.S. Food and Drug Administration) (58), MIRDOSE (its predecessor) (60), IDAC-Dose 2.1 (61), and MIRDcalc (59); the latter two are freely available.
Suborgan and subtumor dosimetry, or voxel-level dosimetry, is addressable by MC radiation transport simulation (62,63), dose point kernel convolution (64,65), or voxel S values (66). MC simulation has the advantages of applicability to inhomogeneous media, complex 3-dimensional geometries, and conditions in which charged-particle equilibrium is not achieved (e.g., tissue interfaces). A historical drawback of MC was its large computational burden, but technologic advances have made it increasingly practical. Dose point kernel has also been adapted to heterogenous media by applying relatively simple multiplicative scaling factors to those in water-equivalent media, yielding results that closely approximate those of MC with reduced computational overhead.
A software tool that has been developed, MIRDcell, adapts the MIRD formalism to cellular and subcellular dosimetry (67). This freely downloadable applet models the radiation dose to the cellular and subcellular compartments (i.e., the cell membrane, cytoplasm, and nucleus modeled as concentric unit-density spheres) for both isolated cells and collections of cells using cellular S values (68). It also models the responses of the labeled and unlabeled cell populations as a function of the fraction of cells radiolabeled.
UNCERTAINTIES IN DOSE ESTIMATION
Sources of uncertainty in radiopharmaceutical dosimetry include assay of administered activity, determination of organ and tumor volumes or masses, measurement of time-dependent activity distributions, estimation of time-integrated activities, and translation of activity/time-integrated activity and anatomic data to dose-rate/absorbed dose. The European Association of Nuclear Medicine has published guidelines on uncertainty analysis for RPT absorbed-dose calculations (69). Error propagation in RPT results in net uncertainties of 10%–15% for absorbed dose estimates to the major organs and much higher values for small lesions. Efforts to determine and minimize the quantitative uncertainties in SPECT/CT activity quantification have been reviewed (70). Harmonization of calibration procedures, acquisition protocols, and reconstruction techniques will be required to achieve, in multicenter trials, the precision needed to build robust dose–response data.
BIOEFFECTS MODELING
Factors other than absorbed dose can impact the outcome of RPT. Historically, the linear-quadratic model has been used to describe normal-tissue and tumor responses to radiation (71,72):
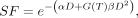 Eq. 3
where SF is the surviving fraction (i.e., the fraction of irradiated cells that has not undergone reproductive failure), D is the absorbed dose (in Gy), α is the linear sensitivity coefficient (in Gy−1), β is the quadratic sensitivity coefficient (in Gy−2), and G(T) is a modifier that, for RPT, depends on the dose-rate curve and the time constant for repair (73).
Eq. 3
where SF is the surviving fraction (i.e., the fraction of irradiated cells that has not undergone reproductive failure), D is the absorbed dose (in Gy), α is the linear sensitivity coefficient (in Gy−1), β is the quadratic sensitivity coefficient (in Gy−2), and G(T) is a modifier that, for RPT, depends on the dose-rate curve and the time constant for repair (73).
The modulation of biologic response due to differences in dose rate or fraction size has led to the concept of biologically effective dose (71,74). This is the absorbed dose (Gy) projected to cause some biologic effect if it were delivered at the mathematic limit of infinitely low dose rate. The equieffective dose (EQDX, in Gy), like biologically effective dose, is dependent on the α/β ratio and is usually written as EQDXα/β (75). Typically, X = 2 Gy is taken as the reference dose because of its common use in conventionally fractionated XRT, yielding EQD2α/β. Using this notation, the biologically effective dose could be expressed as EQD0α/β (i.e., a reference dose per fraction of 0 Gy, corresponding to radiation treatment delivered by an infinite number of infinitesimally small fractions or at an infinitesimally low dose rate).
Tumor therapeutic response and normal-tissue toxicity may not correlate with mean absorbed doses because of spatial nonuniformity in the dose distribution. The equivalent uniform dose is the single value of absorbed dose that, if distributed uniformly, would achieve the same overall survival fraction as a nonuniform dose distribution (76,77). The equivalent uniform dose has been formulated as the “equivalent uniform biological effective dose” (77). Several studies have shown a better response correlation with equivalent uniform dose than with tumor mean absorbed dose (78–80).
RPT WITH α-EMITTERS
The radiobiologic advantages of high-LET radiation include intense ionization density along particle tracks that produce difficult-to-repair DNA damage and a reduced dependency on dose rate and local oxygen tension (81). In addition to high-LET radiobiologic advantages, the short range of α-particles (40–90 μm) can produce highly localized dose delivery (82) with the possibility of beneficial normal-tissue sparing if parts of critical organs lie beyond their emission range (e.g., marrow stem cells from radium deposition on bone surface). Other high-LET radiations include Auger electrons, emitted from some radionuclides. However, high-LET effects from Auger electron emission have only nanometer ranges and are not currently of clinical significance (83).
Alpha-particle–emitting radionuclides that have been used in clinical trials include 213Bi (84,85), 211At (86,87), 212Pb (88), 223Ra (89), 225Ac (90,91), and 227Th (92), listed in Table 1. These radionuclides may be separated into those that emit a single α-particle (213Bi, 211At, and 212Pb) and those that undergo multiple decays with up to 4 (223Ra and 225Ac) or 5 (227Th) α-particle emissions.
For 213Bi, the combination of a short 46-min half-life and absence of α-particle–emitting progeny allows for the administration of relatively high, imageable (440-keV γ-ray) activities (∼37 MBq) (84). 211At has a longer 7.2-h half-life and emits characteristic x-rays (77–92 keV) suitable for quantitative imaging (93). 212Pb (10.6-h half-life) decays via β-particle emission, but its progeny emit, on average, one α-particle either via 212Bi (36%) or 212Po (64%). It has 2 disadvantages: the first is that one of its progeny, 208Tl, emits a very-high-energy γ-ray (2.6 MeV; 36%) that complicates radiation protection; the second is that about 40% of 212Pb β-transitions are accompanied by nuclear deexcitation by internal conversion, producing an Auger-electron cascade and charge-neutralization effect that can lead to molecular fragmentation and release of 212Bi (94,95). However, 212Pb has the advantage that it forms a theranostic pair with 203Pb for imaging (52-h half-life; 279-keV γ-ray) and for patient selection and dosimetry (96,97).
Imaging 223Ra, 225Ac, or 227Th poses challenges for accurate activity quantification. Because of their long half-lives and multiple α-particle–emitting progeny, trials with these radionuclides use only kBq/kg activities, compared with MBq/kg activities for 213Bi and GBq activities for 177Lu. The resulting low-count images are noisy. Another challenge is determining the fate of the radioactive progeny dissociated from the radiopharmaceutical, as α-particle decay results in a high (100 keV) nuclear recoil energy that disrupts chemical bonds. Radioactive progeny can thus potentially translocate from the site of the parent decay, as with bismuth translocation to the kidney after 225Ac decay in blood (98). Although current γ-cameras are ill-equipped for imaging many α-particle–emitting radionuclides, scanners with improved energy resolution may distinguish the imaging signals from multiple progeny (99).
Xofigo is the first, and thus far only, α-particle–emitting radiopharmaceutical approved by the Food and Drug Administration, for the treatment of patients with castration-resistant prostate cancer metastatic to bone with no visceral component. Initial studies in Europe and the United States included imaging and dosimetry, but this is not required for current protocols. The low administered activities of 223Ra (55 kBq/kg per treatment) produce noisy images (100) in which bone lesions are often inconspicuous, and a 99mTc-diphosphonate or 18F-fluoride bone scan is required for definitive identification. The partial-volume effect reduces lesion contrast further, and even though uptake in bony lesions is stable, the lack of lesion mass information makes accurate dose estimation problematic.
Dosimetry of α-particle emitters is additionally challenging with respect to imaging because of their short emission range, 2 orders of magnitude smaller than γ-camera pixel dimensions. Even if γ-camera images of α-particle–emitting radionuclides can provide biodistribution data (100), subvoxel microscopic nonuniformities in dose distributions may produce different biologic effects depending on the association of the agent to tumor cells or normal tissue structures. Obtaining source microdistribution within patients is currently not possible except in limited cases from biopsy or surgical samples (101). One possibility may be to infer this information from preclinical studies in tumor-bearing animals by autoradiographic methods. Using α-particle MC codes, digitized histologic images, and radiobiologic modeling, cell survival fractions may thus be deduced (67,102,103).
Currently, there is considerable interest in RPT with α-particle–emitting radionuclides, at least partly because of reports of remarkable clinical responses in some patients with macroscopic disease, often after a limited clinical response to RPT with β-particle–emitting radionuclides (104–106). From a purely dosimetric perspective, α-particles have a limited range (several cell diameters), and optimal target sizes would be expected to be of submillimeter dimensions. In addition, the adverse effects of nonuniform radiopharmaceutical uptake in macroscopic disease would be expected to be severe. Taken together, this suggests that α-RPT would be optimally used in the adjuvant or neoadjuvant setting, specifically addressing subclinical microscopic disease. The unanticipated clinical effectiveness of α-RPT for macroscopic disease may be related to immunologic or abscopal factors or to absorbed dose contributions from diffusible α-particle–emitting progeny. Despite our lack of understanding of the biologic mechanisms involved, clinical implementation of α-RPT is accelerating. However, its full potential may not be realized unless rigorous dosimetric analyses are performed (107). It is a field that warrants proceeding cautiously since many unknowns remain.
FUTURE PERSPECTIVES
RPT has emerged as a major new treatment modality spurred by the recent approval of Lutathera, 177Lu-vipivotide tetraxetan (Pluvicto; Novartis), and Xofigo and the anticipation of new agents to follow. At present, the ability to perform accurate 3-dimensional dosimetry for RPT is clinically achievable, though requiring specialized software and technical capabilities and a significant commitment of time by the treating facility and patient. The available dose–response data, though still sparse, suggest that patient-specific dosimetry may help to improve RPT by minimizing toxicity or maximizing efficacy. However, additional dose–response data are still required and should remain a priority of future studies. Constructing a tumor dose–based prescription will be challenging, as at least some of the targets will be either phenotypically diverse or too small to be imaged. The use of RPT for the treatment of bulky macroscopic disease is likely transitional, and in the future, this type of disease conformation may be better treated by the addition of a supplementary XRT component, as part of a combined-modality therapy. RTP can selectively target and treat subclinical microscopic disease, even if imaging is not possible.
Practical dosimetry is rapidly advancing, progressing beyond historical obstacles. New imaging hardware has recently been introduced as well: γ-cameras and SPECT scanners with solid-state detector technologies that allow better energy resolution, SPECT scanners with full-ring detector geometries (making whole-body SPECT faster and more feasible), and whole-body PET scanners allowing whole-body dynamic imaging and reliable imaging of much lower administered activities than those currently used. Advancements in commercial software and regulatory approval of tools that facilitate clinical implementation will provide new opportunities for standardization of methods across centers. Artificial intelligence–assisted workflows that may reduce dosimetry time and effort and improve standardization are also being developed.
RPT dosimetry remains a work in progress. Specialized centers will continue to refine dosimetric methodology, introduce novel radiopharmaceuticals, investigate combined-modality therapies, and elucidate issues such as patient-specific susceptibility to radiation injury, interactions with the immune system, and abscopal effects of radiation. Work to standardize and validate dosimetry calculations and simplify the dosimetry process must continue, bearing in mind Einstein’s dictum of “…as simple as possible but no simpler.” In particular, scenarios in which single-time point imaging provides adequate dosimetric accuracy will need to be identified. The opposing considerations of minimizing complexity and maximizing throughput on the one hand and optimizing treatment for individual patients on the other need to be recognized and reconciled. This will be especially important as the field expands—the recent approval of 177Lu-PSMA-617 therapy will have a major impact on patient load, and it is likely that an increasing number of RPT agents will become available. If dosimetry is to become more than an academic exercise, we need to show that it makes a significant difference to clinical outcomes with RPT. Ultimately, the only acceptable way of achieving this is through multicenter randomized controlled clinical trials comparing dosimetry-based prescriptions with one-size-fits-all activity-based prescriptions.
ACKNOWLEDGMENT
We thank Dr. Lukas Carter for his contribution to Figure 2.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- 1.↵
- 2.
- 3.↵
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.↵
- 11.
- 12.↵
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.
- 106.↵
- 107.↵
- Received for publication December 14, 2021.
- Accepted for publication July 14, 2022.
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Computational Nuclear Oncology Toward Precision Radiopharmaceutical Therapies: Current Tools, Techniques, and Uncharted Territories
- Dosimetry of [177Lu]Lu-DOTATATE in Patients with Advanced Midgut Neuroendocrine Tumors: Results from a Substudy of the Phase III NETTER-1 Trial
- The MIRD Schema for Radiopharmaceutical Dosimetry: A Review
- Reply: Dosimetry in Radiopharmaceutical Therapy
- Dosimetry in Radiopharmaceutical Therapy