


Neurodegenerative disorders are common brain afflictions with increasing prevalence in countries with aging populations. This is true for the two most common neurodegenerative disorders, Alzheimer disease (AD) and Parkinson disease (PD), both demonstrating increases in prevalence over the last decade, with more than 6 million diagnosed with AD and 1 million with PD in the United States (1,2). The impact on the individual patient, the burden on families, and the societal and financial costs are becoming more urgent as these epidemiologic trends play out (3).
Recently, progress has been made in understanding the etiology of neurodegenerative disorders at the molecular level. We know that these diseases represent a series of pathologic brain conditions characterized by protein misfolding and the progressive accumulation of cytotoxic fibrils and oligomers that are believed to result in selective neurologic and functional impairment (4). The unrelenting progression of symptoms and disability represents ongoing neuronal degeneration resulting from dissemination of these cytotoxic proteins in the brain. In AD, there are PET biomarkers targeting the 2 aberrant proteins, β-amyloid contained in neuritic plaques and tau found in neurofibrillary tangles, the pathology described originally by Alois Alzheimer.
These imaging biomarkers have already been deployed to support the development of therapeutics that target amyloid (5). The equivalent protein in PD is α-synuclein (a-syn), found in Lewy bodies. Currently, there is no available PET agent for interrogating a-syn deposition and no easy way to track in vivo brain changes mechanistically relevant to clinical progression (6).
WHY A-SYN PET IN PD?
Dopamine transporter (DaT) imaging using SPECT and 123I-tropane imaging agents such as 123I-ioflupane demonstrate significant reductions in striatal binding ratios in PD patients enrolled in longitudinal studies of 1–2 y or more. DaT SPECT is also an important tool for identifying DaT deficits in premotor and at-risk individuals (7,8). Yet DaT imaging interrogates one piece of the pathology of PD, presumably the consequence of a-syn deposition in the substantia nigra. DaT SPECT does not directly assess a-syn, which represents an upstream process against which disease-modifying treatments are targeted. Hence, the 2 imaging biomarker targets, DaT and a-syn, are theoretically complementary.
A-syn is a 140-amino-acid protein highly prevalent in the brain, representing 1% of the cytosolic protein and thought to be localized to the presynaptic terminal, where it facilitates the release of neurotransmitter into the synapse. The protein undergoes extensive posttranslational modification and is natively unstructured, rather taking its conformation from its local milieu. Within cells, fibrils form oligomers, thought to be the cytotoxic form of a-syn. Different conformational states characterize the different synucleinopathies (9,10).
A-syn has now emerged as a key target for development of a PET biomarker in the evaluation of the pathophysiology and putative treatments for modifying the course of PD. Why? First, the primary brain pathologies in PD are Lewy neurites and Lewy bodies, which are composed of aggregated a-syn resulting from misfolding of protein and seeding and whose pattern of brain spread is suggested by postmortem brain examination of Lewy body distribution (11,12). These studies indicate a discrete pattern of Lewy body formation that describes a pathway of spread going from the brain stem systematically to the midbrain (including nigrostriatal projections) and finally to higher cortical regions. A-syn is found in many body tissues outside the brain, including the skin, salivary glands, and gut. Biopsy of tissue in PD can demonstrate this and has been proposed as another biomarker in clinical therapeutic trials. Interestingly, recent work suggests that the gastrointestinal tract may have a role in the etiology of PD. Alterations in the microenvironment of the gut biota can lead to inflammatory changes and conditions that favor the formation of a-syn fibrils (13). These may then travel into the central nervous system via the vagus nerve or through vascular pathways. Once seeded in the lower brain stem, fibrils are believed to cross the synapse to affect adjacent connected neurons (Fig. 1). The presence of these alien fibrils creates conditions that enhance the formation of additional fibrils and aggregates. As a-syn spreads superiorly, involvement in midbrain structures may result in some premotor symptoms associated with PD, including rapid-eye-movement sleep behavior disorder and hyposmia. Both of these have been used for cohort enrichment for enrollment of at-risk or premotor individuals (14).
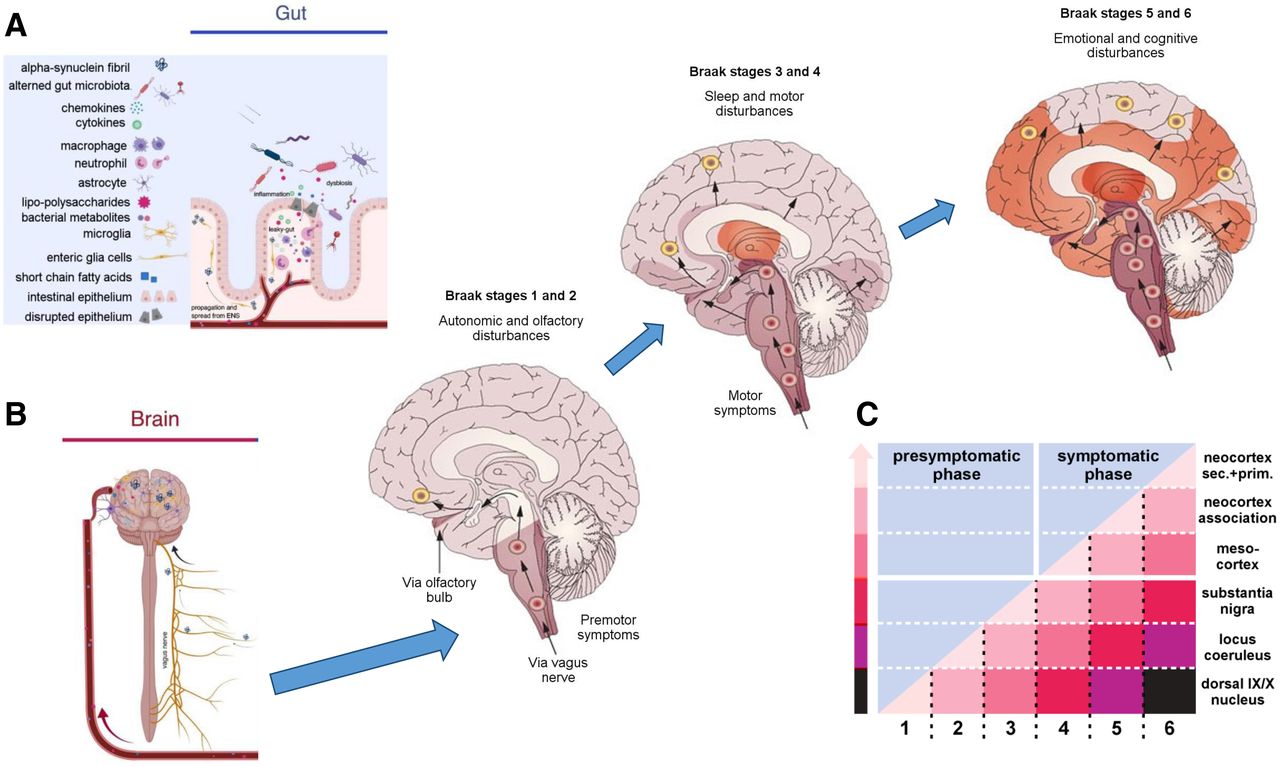
Etiologic model of PD and other synucleinopathies. (A) Proposed model for development of Lewy body diseases such as PD suggests gut may be locus of initial production of a-syn fibrils. This takes place in context of inflammatory changes associated with disruption of integrity of intestinal epithelium, as occurs in different microbiota environments. (B) These fibrils are taken up by vagus nerve and transported to lower brain stem. (C) Through process of cell-to-cell spread following along network lines, additional cells become affected, systematically moving up through brain stem to midbrain and cortex and resulting in progressive symptoms reflective of involved region. Motor symptoms begin at stage 3, when substantia nigra becomes involved. ENS = enteric nervous system. (Adapted from (11,13).)
Another reason for interest in a-syn in PD comes from genetic studies. Genomewide association studies have shown a strong association of variations in the a-syn gene with PD (15,16). In addition, mutations in the a-syn gene promote formation of a-syn aggregates and fibrils. These findings support a-syn as an important target of disease-modifying treatments and potentially a path to better understanding of the onset and longitudinal course of disease across the synucleinopathies: PD, dementia with Lewy bodies, and multiple-system atrophy. Further, knowing how differences in a-syn conformational structure affect pathophysiologic manifestations of disease may offer additional clues to treatment, clarify phenotype with regard to differential diagnosis and prognosis, and offer needed tools—such as at-risk screening, proof of target engagement, and assessment of drug efficacy—needed to conduct therapeutic trials.
The enthusiasm for a PET biomarker of a-syn is underscored by the sponsorship of the Michael J. Fox Foundation, which has offered a $2 million prize to the first team that develops a viable selective a-syn PET tracer and agrees to make that tracer available broadly. The ability to image a-syn deposition in the brain was described in the program announcement as “a game-changing achievement for the Parkinson’s disease field.” Efforts by both industry and academic groups are under way in this and other a-syn biomarker initiatives.
NEW ROLES FOR PET IMAGING IN PD
Up to now, treatment for PD has been symptomatic, rather than disease-modifying. Designing a clinical trial for a drug that actually slows, stops, or reverses clinical symptoms and improves function is exceedingly difficult. Questions arise as to which participants to enroll, what metric to use for measuring treatment efficacy, and how many subjects are needed to power a potentially small reduction in a process that changes relatively slowly as PD progresses.
Medications taken for managing symptoms, such as l-DOPA, can be another confounder in determining off-medication clinical status. To get true off-medication assessments, several weeks of withdrawal from the symptomatic medications may be necessary. This is not easily done or ethical given the amount of increased morbidity experienced by patients off medications for that period (17). Another issue regarding medications is whether the putative disease-modifying agent has any direct symptomatic effect, making it harder to tease out symptom relief from true efficacy in altering the mechanisms of disease (18).
Questions about when in the course of illness to recruit the test cohort are extremely important since clinical manifestations occur only after years of silent, abnormal pathologic processes. How does one diagnose and treat a disease that is not clinically manifest? This presymptomatic phase of illness is when a disease-modifying intervention might be most effective, rather than later when there is less salvageable tissue. Recruiting from this cohort also gets around the medication issues mentioned above. The length of this window between initiation of pathology and subsequent clinical manifestation is on the order of years. As an example, use of the DaT agent 123I-ioflupane and SPECT imaging in the Parkinson Progression Marker Initiative de novo PD cohort (19), which was serially scanned over 4 y (baseline and years 1, 2, and 4), allows back-extrapolation of striatal specific binding ratio curves to the point of normalcy, permitting a rough estimate of the duration of the clinically silent course of progressive change in the brain. This duration turns out to be about 13 y in the most affected brain regions, thus suggesting the important roles that a-syn PET might play in the arena of clinical trials. For example, a-syn might provide early confirmation of disease pathology in at-risk individuals, serve as a screening tool to ensure the diagnostic integrity of the cohort, offer a biomarker directly related to the mechanisms of disease progression, provide evidence of target engagement, and assess the efficacy of an intervention designed to slow this progression.
AD DRUG DEVELOPMENT AND TAU PET: A MODEL FOR A-SYN PET?
The recent development and application of imaging biomarkers in AD are a model and reminder of the ways that PET imaging supports therapeutic trials by providing a window on primary pathophysiology. The recent, albeit controversial, FDA approval of the amyloid-targeting antibody aducanumab for AD serves as an example of the integration of PET biomarkers into clinical drug trials and hints to future clinical roles (20). Perhaps most relevant to the development of a radiotracer for the α-synucleinopathies is the recent history of the development of radiotracers for the tauopathies. The parallels between tau and a-syn are significant and could provide a road map of expectations for some potential paths toward the successful development of an a-syn PET agent.
Tau PET developed as follows. Briefly, phase 1 was the period of concept formation, articulation of need, and scientific and medical community buy-in as to the need or desire for targeted tau imaging biomarkers. Next was phase 2, a period of radiochemistry development called the wandering lost-in-the-desert-of-failed-compounds stage. Even so, while wandering, research teams were getting more sophisticated about binding affinities and selectivity of candidate structures. The move out of the desert was phase 3, when one or more promising structures were discovered and in vitro and nonclinical evaluation occurred. This led to phase 4, or the human proof-of-concept trials in AD, where there was characterization of the pharmacokinetics and validation of an outcome measure (21). Phase 5 was sharing of the pioneering compound among investigators and incorporation into clinical trials as an exploratory outcome. While this was happening, further development of second-generation tau tracers was initiated. Finally, phase 6 was the extensive incorporation of tau tracers as biomarkers in clinical trials and more nuanced understanding of differences in affinity to tau isoforms relative to the use of the radiotracer (22).
A-SYN PET: WHY HAS IT BEEN SO DIFFICULT?
Considering the tau PET experience just outlined, why has it been so difficult to develop an a-syn radiotracer for PD? Why not simply label the therapeutic? Although compelling, this strategy generally does not work out because the properties of a good therapeutic, such as high lipophilicity for brain penetrance, causes a higher background level and a lower PET signal-to-noise ratio. Further, radiotracers require nanomolar to subnanomolar affinity and high selectivity for a-syn, as well a preference for kinetics that offer fast washout of background signal and no confounding labeled metabolites. Fayyad et al. proposed several additional obstacles to developing an a-syn imaging biomarker: no good compound leads (e.g., dyes or tissue stains), lack of a library of compounds for selectivity screening against tau and β-amyloid, low target density, and poor PET resolution (23). Other issues include the potential confounders of isoform heterogeneity and cost. Recent work suggests that the field is addressing these concerns. For example, Ferrie et al. described an ultrahigh-throughput in silico screening strategy using idealized pseudo ligands (exemplars) to identify compounds, including confirmation of the binding site and evaluation of the structure–activity relationship of analogs for development of multiple molecules with nanomolar affinity for a-syn fibrils (24). More sophisticated understanding of the conformation-dependent binding sites on a-syn (25) can be used to inform radiotracer (and drug) development (26). Finally, there have been initial a-syn PET human studies with promising structures but mixed results to date (27,28).
CONCLUSION
Will we ever develop an a-syn PET tracer for clinical trials? Optimistically, there are several factors that suggest we will. There are increasingly pressing needs from the numbers of therapeutic trials with new a-syn–targeting treatments. The body of knowledge about a-syn function (binding sites, conformational states, and structure–activity relationship) and dysfunction (misfolding, aggregate formation, seeding, and spread) inform the medicinal and radiochemistry development as well as the in vivo validation of a-syn PET ligands. In addition, prior experience in AD suggests that imaging of proteinopathy in PD may be a useful clinical research tool and offers a road map for development of a-syn PET. Perhaps most encouraging is the recent presentation (March 2022) of the first apparently successful human a-syn PET agent, AC-12589, from AC-Immune and Oskar Hansson and colleagues at Lund University, Sweden. These very preliminary studies in multiple-system atrophy, PD, and controls demonstrated the expected increased uptake in cerebellar white matter in multiple-system atrophy, but not PD or healthy volunteers. The lack of specific uptake in PD could be related to a-syn conformational differences with multiple-system atrophy, relative target affinity, small sample size, or other factors.
In summary, we already know a great deal about pathologic a-syn formation and spread, as well as how to develop and validate imaging tools for clinical and research needs, and even a promising compound in initial human trials. We just need to keep going and make our way through the desert.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jul. 19, 2022.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 19, 2022.
- Accepted for publication April 7, 2022.





{kind=link}