


Abstract
Noninvasive methods to study glucocorticoid receptor (GR) signaling are urgently needed to elaborate the complexity of GR signaling in normal physiology and human disorders, as well as to identify selective GR modulators to treat diseases. Here, we report evidence supporting translational studies with (±)-11C-5-(4-fluorobenzyl)-10-methoxy-2,2,4-trimethyl-2,5-dihydro-1H-chromeno[3,4-f]-quinoline ((±)-11C-YJH08), a radioligand for PET that engages the ligand binding domain on GR. Methods: (±)-11C-YJH08 was synthesized by reacting the phenol precursor with 11C-methyl iodide. The biodistribution was studied in vivo. Specific binding was tested in vivo with adrenalectomy and ligand competition. A library of analogs was synthesized and studied in vitro and in vivo to understand the (±)-11C-YJH08 structure–activity relationship. Rodent dosimetry studies were performed to estimate the human-equivalent doses of (±)-11C-YJH08. Results: (±)-11C-YJH08 was synthesized by reaction of the phenolic precursor with 11C-methyl iodide, giving a radiochemical yield of 51.7% ± 4.7% (decay-corrected to starting 11C-methyl iodide). Specific binding was observed in many tissues, including the brain. An analysis of the (±)-YJH08 structure–activity relationship showed that (R)- and (S)-enantiomers are equally avid for GR by occupying discrete binding modes. A focused chemical screen revealed that the aryl fluoride motif on YJH08 is essential for high-affinity GR binding in vitro, high tissue uptake in vivo, and efficient passage across the blood–brain barrier. Lastly, we performed dosimetry studies on rodents, from which we estimated the human-equivalent doses of (±)-11C-YJH08 to be commensurate with the widely used 11C and 18F tracers. Conclusion: These studies reveal the molecular determinants of a high-affinity and high-selectivity ligand–receptor interaction and support the use of (±)-11C-YJH08 PET to make the first measurements of GR expression in human subjects.
The glucocorticoid receptor (GR) is a nuclear hormone receptor whose expression and activity (genomic and nongenomic) regulate a remarkable breadth of cellular processes (1). Moreover, dysregulation of GR signaling is known or suspected to drive the pathobiology of numerous human diseases, including endocrine disorders, pulmonary diseases, mood disorders, and even cancers (2,3). That said, fully articulating the role of GR in human biology has been challenging because the scientific community lacks safe and efficacious technologies to measure GR expression or activity in living human subjects. Moreover, whereas therapeutic modulators of GR are in routine clinical use (e.g., dexamethasone and mifepristone), their limitations are well characterized and due in part to on-target, off-tissue drug interactions (4). Thus, noninvasive biomarkers to study GR signaling could enable drug discovery efforts to identify more specific GR modulators.
Appreciating these unmet needs, the nuclear medicine community has attempted for over 3 decades to develop radiolabeled probes to measure GR expression in vivo with quantitative imaging modalities (5). Previous radioligands bore at least one of the following shortcomings: a low-yielding radiosynthesis, poor in vivo stability, or limited evidence of specific receptor binding in vivo. We recently developed (±)-18F-2,5-dihydro-10-methoxy-5-oxo-2,2,4-trimethyl-1H-[1]benzopyrano[3,4-f]quinoline ((±)-18F-YJH08), a promising nonsteroidal radioligand built on a benzopyrano[3,4-f]quinoline scaffold that appears to overcome these challenges (6). In vitro, the ligand is a potent and specific GR binder with milder agonism than that of dexamethasone. In vivo, the tracer specifically bound to GR in many normal mouse tissues of relevance to corticosteroid endocrinology (e.g., the adrenals) and achieved the highest level of specific brain uptake reported to date. Moreover, (±)-18F-YJH08 PET revealed an entirely unexpected tissue-selective pharmacologic interaction between GR and an inhibitor of the 70-kDa heat shock protein. These data inspired us to perform the current study, which was aimed at refining the structure and chemistry of the radioligand with an eye toward translation.
Herein, we provide evidence advocating for the clinical translation of (±)-11C-YJH08. For example, radiolabeling the phenol precursor of (±)-YJH08 using 11C-methyl iodide resulted in higher radiochemical yield and specific activity than were obtained with (±)-18F-YJH08. The in vivo stability of (±)-11C-YJH08 was also measured to be more than 90%. Studying the YJH08 structure–activity relationship showed that the (R) and (S) enantiomers had similar biodistribution profiles in vivo, as was consistent with in vitro affinity data and molecular dynamics simulations predicting approximately equal affinities for GR. A focused chemical screen revealed that the aryl fluoride group on the C5 position of the benzopyrano[3,4-f]quinoline scaffold was essential for high-affinity binding to GR. Moreover, a biodistribution study with 11C-YJH02 showed that the aryl fluoride motif is critical to tissue uptake and blood–brain barrier penetrance. Lastly, we performed a rodent dosimetry study with (±)-11C-YJH08 that showed the human-equivalent effective dose to be equal to or less than those of other 11C or 18F small-molecule radiotracers in widespread clinical use, such as 11C-Pittsburgh compound B and 18F-FDG (7–9). Collectively, these data support the translation of (±)-11C-YJH08 for human imaging trials to study GR expression in healthy subjects and patients and to parse the pharmacology of GR modulators.
MATERIALS AND METHODS
All synthetic details and additional experimental methods are provided in the supplemental materials (available at http://jnm.snmjournals.org). In vitro affinity assays were performed as previously reported (6).
Radiosynthesis of (±)-11C-YJH08, (R)-11C-YJH08, and (S)-11C-YJH08
11C-CO2 was produced in target by the 14N(p,a)11C nuclear reaction of 17-MeV protons on N2 at the University of California San Francisco radiopharmaceutical facility. 11C-CO2 was converted to 11C-MeI using the gas-phase method on a GE Healthcare FX/C Pro automated synthesis module. In a 3-mL V-type vial, the phenol 7 (1 mg, 2.5 μmol) was premixed with dimethylformamide (0.6 mL), a 1.1 equivalent of KOH (aqueous), and 20 equivalents of K2CO3 (aqueous) and then stirred at room temperature for 10 min. 11C-MeI (g) was transferred into the reaction vial at room temperature. The reaction mixture was stirred for another 10 min at 85°C and neutralized with HCl (aqueous). The reaction mixture was subjected to semipreparative high-performance liquid chromatography (HPLC) for purification, which took 10 min and provided the radioactive product in a decay-corrected radiochemical yield of 51.7% ± 4.7% (n = 8). The semipreparative HPLC conditions were as follows: a Waters C18 column (19 × 50 mm, 5 μm), a mobile phase consisting of acetonitrile/H2O (30%–95% CH3CN in water over 12 min and then 95% CH3CN for 8 min), and a flow rate of 10 mL/min.
To prepare the radiotracer for animal studies, (±)-11C-YJH08 was trapped on the cartridge and eluted with CH3CN (1 mL). After removal of the organic solvent, the pure product was dissolved in an injection formulation of 1/1/8 (v/v/v) dimethylsulfoxide/polysorbate 80/saline. The stability, ultraviolet purity, and radiochemical purity of (±)-11C-YJH08 in formulation were further confirmed by analytic HPLC equipped with a γ-ray radiodetector and a ultraviolet detector at 254 nm. The analytic HPLC conditions were as follows: a Waters C18 column (4.6 × 150 mm, 5 μm), a mobile phase consisting of acetonitrile/H2O (8:2 v/v), and a flow rate of 1 mL/min. Both the ultraviolet and the radioactive spectra comigrated with the analytic standard, and the purity of the tracer was more than 95%.
(R)-11C-YJH08 and (S)-11C-YJH08 were synthesized using the same procedure as above from corresponding enantiopure phenol precursor.
Animal Studies
All animal experiments were performed under the approval of the Institutional Animal Care and Use Committee at the University of California San Francisco. C57BL6/J mice (4−6 wk) were purchased from Charles River. All mice were housed in a dedicated vivarium with free access to food and water. For dexamethasone treatment studies, dexamethasone was administered via 2 routes involving oral gavage of 50 mg/kg (formulation, 0.5% hydroxy-propyl-methylcellulose and 0.2% polysorbate 80 in water) for 3 d before radiotracer injection or administration of a water-soluble dexamethasone–cyclodextrin complex via intraperitoneal injection at 10 mg/kg in phosphate-buffered saline 1 h before radiotracer injection. Adrenalectomized mice were purchased from Charles River and provided with drinking water supplemented with NaCl (aqueous) per instructions until the time of the tracer biodistribution study.
Serum Stability Studies
Mice received radiotracer (30 MBq in a 100- to 150-μL formulation) via tail injection, and were dissected at dedicated time points after injection. The blood was collected and an aliquot (50 μL) was mixed with CH3CN (50 μL). The serum proteins were precipitated out and followed by a centrifugation (10,000 rpm for 5 min). The cleared supernatant (50 μL) was collected and further analyzed by radioactive-instant thin-layer chromatography.
Plasma Protein Binding Studies
The inserts of the rapid-equilibrium-dialysis device, along with a Teflon (DuPont) base plate (Pierce), were used for the binding studies. The pH of the human or mouse plasma was adjusted to 7.4 before the experiment. Dimethylsulfoxide stocks (1 mM) were spiked into the plasma to make a final concentration of 2 μM. The spiked plasma solutions (300 μL) were placed into the sample chamber (indicated by the red ring), and 500 μL of phosphate-buffered saline, pH 7.4, were placed into the adjacent chamber. The plate was incubated at 37°C on an orbital shaker (250 rpm) for 4 h.
After 4 h, from the rapid-equilibrium-dialysis plate, aliquots (100 μL) were removed from each side of the insert (plasma and buffer) and dispensed into the 96-well plate. Subsequently, 100 μL of blank plasma were added to the buffer samples and 100 μL of blank buffer were added to all collected plasma samples. A 300-μL volume of quench solution (50% acetonitrile, 50% methanol, and 0.05% formic acid, warmed at 37°C) containing internal standards was added to each well. The plates were sealed, stirred in a vortex mixer, and centrifuged at 4°C for 15 min at 4,000 rpm. The supernatant was transferred to fresh plates for liquid chromatography with tandem mass spectrometry analysis. The samples were analyzed using liquid chromatography with tandem mass spectrometry on an AB Sciex API 4000 instrument, coupled to a Shimadzu LC-20AD LC pump system. Analytic samples were separated using a Waters Atlantis T3 dC18 reverse-phase HPLC column (20 × 2.1 mm) at a flow rate of 0.5 mL/min. The mobile phase consisted of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B).
The percentage of test compound bound to protein was calculated by the following equations:
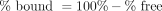
Small-Animal PET/CT Imaging
Data were acquired with a Siemens Inveon small-animal PET/CT scanner. For dynamic acquisitions, the mice were anesthetized beforehand with 1.5%–2% isoflurane and positioned on the scanner bed; then, approximately 30 MBq of (±)-11C-YJH08 were injected via the tail vein in a volume of 100−150 μL per mouse. All PET imaging data were decay-corrected, reconstructed with CT-based attenuation correction, and analyzed with AMIDE software. Regions of interest were manually placed to calculate SUVmean data from the dynamic acquisitions.
Biodistribution Studies
Mice were euthanized after radiotracer (7.4–18.5 MBq in 100–150 μL) injection with CO2 (gas) asphyxiation and dissected at dedicated time points after injection. The blood and tissues were collected, washed, dried, and weighed. The activity of each tissue was measured with a γ-counter. All data were decay-corrected. Prism software (GraphPad) was used to express percentage injected dose per gram of tissue.
Normal-Tissue Radiation Dose Estimation
Volumes of interest were drawn on coregistered CT images for brain, lungs, heart, liver, stomach, kidneys, and urinary bladder. All volumes of interest were either elliptic cylinders (5-mm long axis, 3-mm short axis, and 5-mm height, for the brain), cylinders (3-mm diameter and 3-mm height, for the kidneys), or spheres (3-mm diameter, for the lungs, heart, liver, stomach, and urinary bladder), and they were placed well within the anatomic boundaries to minimize spillover or spill-in of radioactivity. The mean values (in Bq/mL) in these volumes of interest were multiplied by standard mouse organ volumes (in mL) to estimate total activity (in Bq) within these organs. The total activity within the entire animal after subtraction of all organ activities was used as activity in the remainder of the body. The percentage injected activity within the defined organs was extrapolated to human-equivalent values using ratios of standard human organ weights to mouse organ weights.
These input percentage-injected-activity data for each organ and the remainder of the body were curve-fitted to derive time-integrated activity coefficients (also known as residence times) (in Bq-h/Bq), and organ and whole-body effective doses for human equivalents were estimated using each mouse’s data. The data from the 3 animals were averaged to derive organ-absorbed doses (in mGy/MBq) and whole-body effective dose (in mSv/MBq). Organ and effective doses were estimated using OLINDA, version 2.0, and International Commission on Radiological Protection Publication 103 tissue-weighting factors.
Autoradiography
After 20 min after injection of (±)-11C-YJH08 (∼30 MBq per mouse), mice were euthanized and perfused with cold phosphate-buffered saline via cardiac puncture. Tissues were immediately collected and flash-frozen in optimal-cutting-temperature compound on dry ice. Tissues were sectioned on a microtome at a thickness of 20 μm and immediately mounted on glass slides. The slides were then exposed on a GE Healthcare phosphor storage screen, and the screen was developed on an Amersham Typhoon 9400 phosphor imager. The autoradiography images were processed using ImageJ software.
Statistics
All statistical analysis was performed using Prism software, version 8.0. An unpaired, 2-tailed Student t test was used to determine statistically significant differences in the data. A P value of less than 0.05 was reported as statistically significant. For the determination of inhibition constant (K i ,) the data were fit with a 1-site nonlinear regression model.
Radiochemical Synthesis of 11C-YJH02
11C-MeI (g) was transferred from a Porapak N column into a glass reactor vial cooled in an ice-and-water bath containing 1 mg of precursor and 5 mg of Cs2CO3 dissolved in 600 μL of dimethylformamide. After transfer was complete, the vial was stirred at room temperature for 10 min and diluted with water. The solution underwent reverse-phase semipreparatory HPLC. The product-containing fraction was diluted with 30 mL of water and loaded onto a C18 Light Sep-Pak cartridge (Waters). The Sep-Pak was washed with water, and the product was eluted with 300 μL of ethanol. The solution was diluted with buffered saline and used in subsequent experiments. The tracer was obtained in 70% ± 8% (n = 4) decay-corrected yield with greater than 95% purity. The HPLC conditions were as follows: a Waters C18 column (19 × 50 mm, 5 μm), a mobile phase consisting of acetonitrile/H2O (6:4 v/v), and a flow rate of 10 mL/min. The stability, ultraviolet purity, and radiochemical purity of 11C-YJH02 in formulation were further confirmed by analytic HPLC equipped with a γ-ray radiodetector and a ultraviolet detector at 254 nm. The analytic HPLC conditions were as follows: a Waters C18 column (4.6 × 150 mm, 5 μm), a mobile phase consisting of acetonitrile/H2O (6:4 v/v), and a flow rate of 1 mL/min. Both the ultraviolet and the radioactive spectra comigrated with the analytic standard, and the purity of the tracer was more than 95%
RESULTS
Radiosynthesis of (±)-11C-YJH08 Was Achieved via Methylation of the Phenolic Precursor
The synthesis of the phenol 7 was achieved following the approach outlined in Supplemental Figure 1. The hydroxy group on the C10 position was chosen as the site for modification with 11C because of well-established chemical precedents for alkylating phenols with electrophiles such as 11C-methyl iodide or 11C-methyl triflate (10). To synthesize the precursor, the phenol on compound 2 was protected as a tert-butyldimethyl silyl ether. After addition of the 4-fluorobenzyl motif via Grignard chemistry, the silyl ether was removed to afford the phenol 7. Before the radiochemistry was conducted, a model cold alkylation was performed on 7 with methyl iodide. The reaction proceeded smoothly and conferred 8 in high yield.
The radiosynthesis of (±)-11C-YJH08 is shown in Figure 1A. After some optimization (Supplemental Table 1), the highest-yielding conditions were achieved by premixing 7 with dimethylformamide (0.6 mL), a 1.1 equivalent of of KOH (aqueous), and 20 equivalents of K2CO3 (aqueous) in the 3-mL vial, followed by stirring at room temperature for 10 min. 11C-methyl iodide (prepared from 11C-CO2; supplemental materials) was transferred into the reaction vial at room temperature. The reaction mixture was stirred for another 10 min at 85°C and neutralized with HCl (aqueous). The mixture was purified using semipreparative HPLC with a C-18 column (Fig. 1B). The final decay-corrected radiochemical yield from 14,800 MBq of 11C-MeI was approximately 2,590 ± 103.6 MBq, or 50% ± 4% (n = 6). The radiochemical purity, which was evaluated by reinjecting purified radiotracer over 2 HPLC conditions (Supplemental Fig. 2), was consistently greater than 95%, and the molar activity was 37 ± 7.4 GBq/μmol (Supplemental Fig. 3).

Summary of synthesis and in vivo assessment of (±)-11C-YJH08 biodistribution. (A) Summary of radiosynthesis of (±)-11C-YJH08. (B) Semipreparative ultraviolet HPLC traces showing retention of phenol precursor (7) and cold (±)-YJH08 (8). Below is shown ultraviolet trace of complex reaction mixture at 10 min and corresponding radioactive trace. Major peak at 10.12 min corresponds to product, (±)-11C-YJH08. (C) Time–activity curve from dynamic PET scan in wild-type male C57BL6/J mouse shows that (±)-11C-YJH08 rapidly cleared from serum, as expected, and primary mode of clearance appeared to be hepatobiliary. Data also show saturable radiotracer accumulation in brain and brown fat, 2 GR-rich tissues. At right are shown representative sagittal and coronal slices of mouse injected with (±)-11C-YJH08 PET/CT. DMF = dimethylformamide; ID = injected dose; RAD = radioactive; UV = ultraviolet.
In Vivo Imaging Studies with (±)-11C-YJH08 Show Specific Binding to GR in Numerous Mouse Tissues
We next conducted a dynamic PET acquisition on C57BL6/J mice to determine the radiotracer biodistribution in vivo. Region-of-interest analysis of the 60-min scan showed that (±)-11C-YJH08 rapidly cleared from the serum and that the primary mode of clearance appeared to be hepatobiliary (Fig. 1C). Moreover, tracer uptake in several organs of interest to GR biology (e.g., kidneys, brain, and adipose tissue) was above the reference blood and muscle levels at 90 min after injection and saturable over time (Fig. 1C). To confirm stability, we collected blood at 30 min after injection, a time point after which we previously showed specific binding in vivo with (±)-18F-YJH08. Resolution of the radiolabeled metabolites on instant thin-layer chromatography suggested that the radiotracer was more than 90% stable at this time point (Supplemental Fig. 4). We also conducted a plasma protein binding assay to determine the percentage of free compound versus protein-bound compound. (±)-YJH08 was almost completely bound to mouse or human serum protein (Supplemental Table 2), suggesting that the ligand, like endogenous corticosteroids, uses corticosteroid binding of globulin to traffic to tissues (11). Lastly, comparing the tissue uptake of 11C- versus 18F-labeled (±)-YJH08 in C57BL6/J mice at 20 min after injection showed no significant differences (Table 1).
Biodistribution Data for (±)-11C-YJH08 Vs. (±)-18F-YJH08
We then probed for evidence of specific binding by (±)-11C-YJH08 in vivo. We first treated wild-type C57BL6/J mice with dexamethasone for 3 d via oral gavage (50 mg/kg), an approach that we previously used to saturate GR in vivo (6,12). Dexamethasone treatment by this route significantly reduced (±)-11C-YJH08 uptake in numerous normal tissues (Fig. 2A; Supplemental Table 3). We also evaluated whether acute dosing with a more bioavailable formulation of dexamethasone–cyclodextrin suppresses (±)-11C-YJH08 binding in tissues. Significant reductions in (±)-11C-YJH08 tissue uptake were observed by treating mice with dexamethasone–cyclodextrin at 10 mg/kg via intraperitoneal injection 1 h before the radiotracer injection (Fig. 2A). Notably, both blocking approaches significantly reduced tracer binding in the brain (Fig. 2A). Comparing the relative suppression of radiotracer binding across tissues showed that chronic, high doses of dexamethasone generally resulted in more significant blockade of radiotracer binding per tissue, consistent with a GR-specific dose response (Fig. 2B). Visually inspecting the decay-corrected PET data showed (±)-11C-YJH08 suppression by dexamethasone in several tissue compartments, such as the brain, the supraspinal depot of brown fat, and the liver (Fig. 2C). We further studied specific binding in vivo by investigating relative tissue uptake in intact versus adrenalectomized mice (adrenalectomy depletes serum corticosteroids and elevates GR expression in tissues). As expected, tracer uptake was significantly higher in several tissues of adrenalectomized mice, including the brain (Supplemental Table 4). Moreover, the relative induction of radiotracer uptake per tissue in adrenalectomized versus intact mice was qualitatively very similar between (±)-11C- and 18F-YJH08 (Supplemental Fig. 5A). Lastly, we compared the radiotracer uptake within the brain using autoradiography and observed that the overall patterns of (±)-11C- and 18F-YJH08 binding were qualitatively similar between representative brain sections equidistant from bregma (Supplemental Fig. 5B).

In vivo blocking studies reveal that (±)-11C-YJH08 specifically binds GR in vivo. (A) Biodistribution data collected 20 min after injection showing suppression of (±)-11C-YJH08 uptake in several mouse tissues by dexamethasone (Dex., n = 5/arm). Dexamethasone was administered via 2 routes involving oral gavage of 50 mg/kg for 3 d before radiotracer injection or administration of water-soluble dexamethasone–cyclodextrin complex via intraperitoneal injection at 10 mg/kg 1 h before radiotracer injection. *P < 0.05. At right are shown blocking data observed in brain. *P < 0.01. (B) Percentage changes in radiotracer uptake per tissue among mice exposed to dexamethasone or dexamethasone–cyclodextrin versus vehicle. (C) Representative CT and PET/CT images showing suppression of (±)-11C-YJH08 binding in selected organs in vivo by dexamethasone treatment. Dex. = dexamethasone; Dex-CD = dexamethasone–cyclodextrin complex; ID = injected dose.
Enantiomers of YJH08 Are Equipotent for GR and Have Similar Biodistribution Profiles In Vivo
Since YJH08 is a racemic mixture, we next tested whether the (R) and (S) enantiomers behaved differently in vitro and in vivo—a finding that could provide a rationale for translating enantiomerically pure YJH08. The racemic mixture of the phenol 7 was resolved using a chiral Lux 5-μm cellulose-1 stationary phase with an isocratic mobile phase of 30% water, 70% acetonitrile, and 0.1% trifluoroacetic acid (v/v) (Fig. 3A). We isolated several milligrams of 1 peak and recrystallized the compound in hexanes. X-ray crystallography showed the peak eluting at 29.1 min to be the (R) enantiomer (Fig. 3B; Supplemental Table 5). We next tested the in vitro affinity of each enantiomer for GR on cells using a 3H-dexamethasone displacement assay. The (R) and (S) enantiomers had equivalent low-nanomolar affinity for GR, with K i values of 3.76 nM (95% CI, 2.93–4.8) and 2.03 nM (95% CI, 0.78–5.3), respectively (Fig. 3C). Thus, the enantiomers are equipotent with dexamethasone and have approximately 2-fold higher affinity than endogenous corticosteroids such as cortisol or corticosterone.

Resolution and analysis of (R)/(S) YJH08 enantiomers reveals similar affinity for GR in vitro and biodistribution patterns in vivo. (A) HPLC trace over chiral stationary phase shows separation of racemic YJH08 to provide (R) and (S) isomers. Starred peak at 29.1 min was isolated in and recrystallized in hexanes to give single crystal. (B) Oak Ridge thermal ellipsoid plot showing absolute conformation of crystal, which was (R) enantiomer of YJH08. Thermal ellipsoids are drawn at 40% probability. (C) 3H-dexamethasone displacement curves on DU145 cells showing relative affinities of (R)-YJH08, (S)-YJH08, and dexamethasone. Each ligand has affinity of less than 5 nM, and all are equipotent. R 2 values for curve fitting were 0.99 (R), 0.94 (S), and 0.97 (dexamethasone). (D) Mouse biodistribution data showing tissue uptake of (R)- and (S)-11C-YJH08. Data were acquired 20 min after injection in male C57BL6/J mice. (E) Structures of (S) and (R) enantiomers of YJH08. Bonds are colored-coded to enable distinction of small-molecule structure in ligand-binding domain of GR. Below each enantiomer are results of molecular dynamics simulation to identify lowest-energy interaction between ligand binding domain of GR and respective enantiomer. Receptor side chains within 5 Å of respective YJH08 enantiomer are shown. For clarity, 2 docked poses are shown for each ligand–receptor complex. At bottom is shown schematic representation of docked pose for each enantiomer within GR ligand binding domain, to highlight amino acids within 5 Å of respective enantiomer. ID = injected dose.
The (R) and (S) phenols were then radiolabeled with 11C-methyl iodide using the protocol outlined above. Each enantiomer labeled effectively to a 55% and 48% respective decay-corrected radiochemical yield from starting 11C-methyl iodide (Supplemental Fig. 6). A biodistribution study on C57BL6/J mice showed that tissue uptake of both enantiomers was very similar to each other (and to racemic 11C-YJH08) at 20 min after injection (Fig. 3D).
These somewhat surprising results prompted us to probe the structural basis of the YJH08/GR interaction with molecular dynamics simulations. For docking studies, we used as a scaffold a cocrystal structure of the GR ligand binding domain with the corticosteroid agonist budesonide (PDB: 5NFP). Both enantiomers of YJH08 (Fig. 3E) were prepared using the ligand preparation protocol in Maestro (Schrödinger, Inc.) and then docked into the binding site. We ran single-precision and extra-precision docking calculations with a flexible ligand and rigid receptor, within the Maestro docking suite. The resulting scores—single-precision, −7.3 (S) and −8.1 (R); extra-precision, −7.3 (S) and −8.1 (R)—were similar. On analysis of the binding pose within the site, we observed hydrophobic interactions (Fig. 3E). Collectively, these data show that the ligand binding domain on GR can accommodate both enantiomers through hydrophobic interactions.
Analysis of Structure–Activity Relationship for YJH08 Reveals Importance of Aryl Moiety on C5 Position to Affinity for GR and Uptake into Brain
To better understand the structure–activity relationship between YJH08 and GR, we performed a focused chemical screen of 5 YJH08 analogs bearing diverse moieties on the C5 position of the scaffold (supplemental materials). The in vitro affinities of YJH01-5 for GR were determined and show that an aromatic group extending from the C5 position is important for a high-affinity binding (Fig. 4; Supplemental Fig. 7).

Summary of Ki, 95% CIs, and coefficient of determination for YJH08. Data were calculated from 3H-dexamethasone displacement assay on cells. Ki was calculated using 1-site Ki fit nonlinear regression with Prism. Data are representative of 2 independent assays.
To understand the impact of the C5 moiety on in vivo biodistribution, we radiolabeled the phenolic precursor to YJH02 (which bears a carbonyl at the C5 position) via alkylation with 11C-methyl iodide (Fig. 5A; Supplemental Fig. 8). The final decay-corrected radiochemical yield from 18,500 MBq of 11C-MeI was approximately 4,670 ± 534 MBq, or 70% ± 8% (n = 4). The radiochemistry purity was greater than 95%. A biodistribution study on mice was conducted 20 min after injection to determine specific binding in tissues (Fig. 5B; Supplemental Table 6). Tracer uptake was suppressed in several tissues from mice treated intraperitoneally with dexamethasone–cyclodextrin compared with those treated with vehicle. Moreover, the overall pattern of tracer uptake was qualitatively similar to what was observed for (±)-11C-YJH08, with the highest uptake observed in the liver, large and small intestines, kidney, and heart. Directly comparing the uptake values in percentage injected dose per gram of tissue of 11C-YJH02 versus (±)-11C-YJH08 showed that 11C-YJH02 levels were generally lower (Fig. 5C). A stark difference in brain uptake was noted, and 11C-YJH02 uptake was approximately 3-fold lower than what was observed for (±)-11C-YJH08 (Fig. 5C). These data point to the importance of the benzyl fluoride group for high-affinity interactions with GR and access to GR within tissues of high importance to GR biology and pathobiology.

Biodistribution studies with analog 11C-YJH02 show importance of 4-fluoro-benzyl motif to specific GR binding in vivo. (A) Schematic of synthesis of 11C-YJH02. (B) Biodistribution data showing tissue uptake of 11C-YJH02 in mice. Data show tracer distribution in mice that received vehicle or dexamethasone-cyclodextrin at 10 mg/kg via intraperitoneal injection 1 h before radiotracer injection. Radiotracer uptake was evaluated 20 min after injection. *P < 0.01. (C) Comparison of 11C-YJH02 and (±)-11C-YJH08 uptake among tissues in normal mice. Uptake of 11C-YJH02 was generally lower than uptake of (±)-11C-YJH08. *P < 0.05. Dex-CD = dexamethasone–cyclodextrin complex; DMF = dimethylformamide; ID = injected dose; rt = room temperature.
Rodent Dosimetry Study Shows That Human Equivalent Organ Doses for (±)-11C-YJH08 Align with Other 11C and 18F Radiotracers Currently in Human Use
To explore the potential of (±)-11C-YJH08 for human translation, we conducted a rodent dosimetry study on male and female C57BL6/J mice (n = 4/sex). The human-equivalent doses for an average adult male (73 kg) or an average adult female (60 kg) were calculated using OLINDA, version 2.0, incorporating International Commission on Radiological Protection publication 103 tissue-weighting factors based on region-of-interest analysis from 90-min dynamic PET acquisitions (Table 2). The organs with the highest absorbed doses were the heart, stomach wall, liver, and thymus. All these tissues abundantly express GR, and in the case of the liver, the hepatobiliary mode of clearance (a slower process than renal clearance) for (±)-11C-YJH08 may contribute to the absorbed dose. The estimated whole-body effective doses were 0.0067 ± 0.002 mSv/MBq to an adult male and 0.0065 ± 0.004 mSv/MBq to an adult female. These values are comparable to those of other 11C radiotracers that have been studied in humans, for example 11C-Pittsburgh compound B, and smaller than a calculated effective dose for 18F-FDG (7–9).
Mouse Dosimetry Data for (±)-11C-YJH08
DISCUSSION
Here, we report data supporting the clinical translation of (±)-11C-YJH08 to study GR biology in humans. The radiosynthesis of (±)-11C-YJH08 had a higher yield than what we previously reported for (±)-18F-YJH08, and biodistribution studies showed equivalence to (±)-18F-YJH08. Importantly, the average final production yield of 2,590 ± 103.6 MBq is sufficient for subsequent clinical translation. Somewhat unexpectedly, both the (R) and the (S) enantiomers of YJH08 were predicted to have equal affinities for GR, and their in vivo patterns of biodistribution were mutually indistinguishable. Corticosteroid withholding and supplementation studies revealed evidence of specific binding for (±)-11C-YJH08 in vivo. Radioligand suppression was not achieved to equivalent extents in all tissues, although this finding could reflect our inability to safely saturate GR in vivo. Notably, we did previously observe dramatic suppression of (±)-18F-YJH08 uptake in the adipose tissue of an adipocyte-specific GR-knockout mouse, adding support to a model for GR-specific binding in vivo for this ligand structure.
Analysis of the YJH08 structure–activity relationship in vitro established the importance of the benzyl moiety on the C5 position to GR affinity. Moreover, a biodistribution study with 11C-YJH02 showed the importance of the benzyl fluoride group to tissue uptake in vivo, especially in compartments that can be challenging to access, such as the brain. The altered biodistribution of 11C-YJH02 may be attributable to somewhat lower affinity for GR (Fig. 4). In addition, lower blood-pool activity was observed for 11C-YJH02 at 20 min after injection, possibly indicating that lower tissue uptake is due to altered pharmacokinetics. Further studies are required to parse these mechanisms more systematically. Finally, a dosimetry study estimated the human-equivalent doses of (±)-11C-YJH08 to be within the range of those of 11C and 18F radiotracers that are in human use.
CONCLUSION
Collectively, these data support the feasibility of human imaging studies with (±)-11C-YJH08 to study GR biology. Moreover, with experimental GR modulators entering clinical trials (e.g., ORIC-101), (±)-11C-YJH08 could be a useful companion diagnostic to identify tissues with altered GR expression and to assess drug pharmacodynamics, as has been previously done with radioligands targeting the androgen receptor and the estrogen receptor (13–15).
DISCLOSURE
Michael J. Evans was supported by the National Institute of Mental Health (R01MH115043) and the Prostate Cancer Research Program of the Congressionally Directed Medical Research Programs (W81XWH-15-1-0552). No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Studying GR expression in normal human physiology and disease is an outstanding challenge for the field because of the lack of noninvasive biomarkers.
PERTINENT FINDINGS: We synthesized 11C-YJH08, a radioligand that potently and specifically binds GR. We further defined the structural determinants for the high-affinity GR–ligand interaction, as well as the molecular features that promote specific binding in vivo. Lastly, we calculated the human dosimetry estimates.
IMPLICATIONS FOR PATIENT CARE: These studies support the use of (±)-11C-YJH08 PET to make the first measurements of GR expression in human subjects. These measurements will further deepen our understanding of the role of GR in human physiology or disease and aid in the development of selective GR modulators.
Acknowledgments
We gratefully acknowledge M. James and A. Chaney of Stanford University for assistance with digital autoradiography and T. Huynh for technical assistance with small-animal PET/CT studies.
Footnotes
Published online Sep. 4, 2020.
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication May 15, 2020.
- Accepted for publication August 6, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.