


Abstract
99mTc is currently produced by an aging fleet of nuclear reactors, which require enriched uranium and generate nuclear waste. We report the development of a comprehensive solution to produce 99mTc in sufficient quantities to supply a large urban area using a single medical cyclotron. Methods: A new target system was designed for 99mTc production. Target plates made of tantalum were coated with a layer of 100Mo by electrophoretic deposition followed by high-temperature sintering. The targets were irradiated with 18-MeV protons for up to 6 h, using a medical cyclotron. The targets were automatically retrieved and dissolved in 30% H2O2. 99mTc was purified by solid-phase extraction or biphasic exchange chromatography. Results: Between 1.04 and 1.5 g of 100Mo were deposited on the tantalum plates. After high-temperature sintering, the 100Mo formed a hard, adherent layer that bonded well with the backing surface. The targets were irradiated for 1–6.9 h at 20–240 μA of proton beam current, producing up to 348 GBq (9.4 Ci) of 99mTc. The resulting pertechnetate passed all standard quality control procedures and could be used to reconstitute typical anionic, cationic, and neutral technetium radiopharmaceutical kits. Conclusion: The direct production of 99mTc via proton bombardment of 100Mo can be practically achieved in high yields using conventional medical cyclotrons. With some modifications of existing cyclotron infrastructure, this approach can be used to implement a decentralized medical isotope production model. This method eliminates the need for enriched uranium and the radioactive waste associated with the processing of uranium targets.
After 5 decades of use, the 99Mo/99mTc generator remains the principal source of radioisotopes for nuclear medicine. 99mTc is an ideal single-photon emitter (1), because of its favorable half-life, photon energy, and radiopharmaceutical chemistry (2). 99mTc is used in 30,000–40,000 procedures per day in the United States, with estimates of global consumption topping 40 million scans per year (3). The use of 99mTc continues to grow worldwide (4).
The fragility of 99Mo supply for 99Mo/99mTc generator production was highlighted by recent shutdowns at 2 of the leading production sites for 99Mo (4,5). The Canadian Chalk River nuclear reactor, which supplies 35%–40% of the global demand of 99Mo, will terminate its isotope production service in 2016 (6). Other reactors supplying 99Mo are typically more than 40 y old and are at risk of prolonged or permanent shutdown within a few years, creating a risk for loss of a long-term, stable supply of 99Mo for medical purposes.
The fission of enriched 235U was a cost-effective approach to produce large quantities of high-specific-activity 99Mo using legacy research reactors. However, the cost of building new nuclear reactors for isotope production is extremely high. The international community has recommended, through the Nuclear Energy Agency of the Organization for Economic Co-operation and Development, to implement full cost recovery on radioisotope production, which will lead to higher 99Mo pricing in upcoming years (7). This price increase will be compounded by the effects of a shift from highly enriched uranium to low enriched uranium targets for 99Mo production (8,9).
Alternative methods for 99Mo and 99mTc production have been known for years (10). These include nuclear reactors for neutron enrichment of 98Mo (11), accelerators to produce spallation neutrons for enrichment of 98Mo, linear accelerators for the fission of 238U or the 100Mo(γ,n)99Mo reaction (12), and the direct production of 99mTc through the 100Mo(p,2n) 99mTc reaction (13–15). This latter method was pioneered by Beaver and Hupf in 1971 (13), and experiments were later conducted by Lagunas-Solar (16), Takacs et al. (17), and others (18) to measure the cross-sections for the 100Mo(p,2n)99mTc reactions (19).
Despite some successes with low-current irradiations, the implementation of a robust production system for routine, large-scale production of 99mTc has remained elusive to date. We report the results of our successful experiments in designing a fully engineered solution for high-yield production of 99mTc by conventional medical cyclotrons.
MATERIALS AND METHODS
Cyclotron System
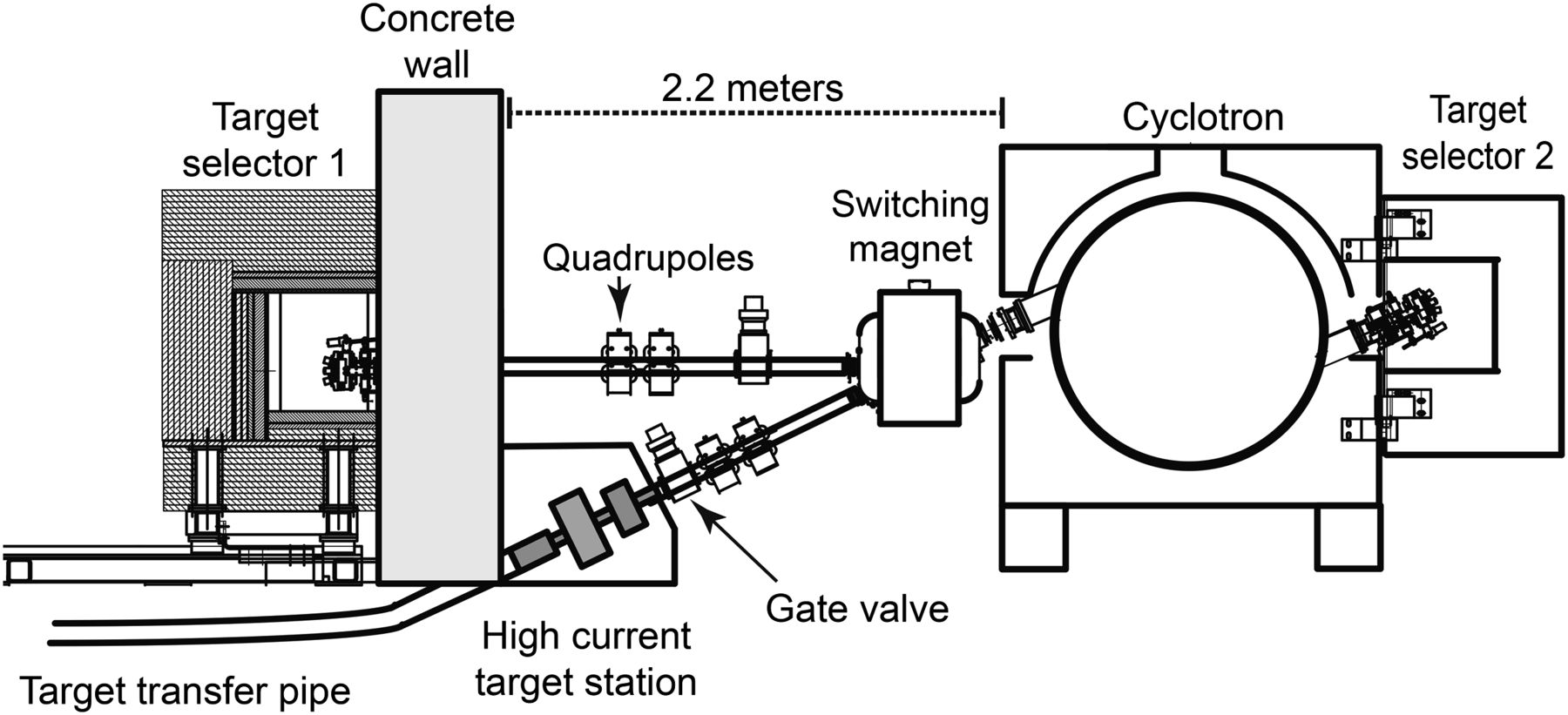
The experiments were conducted on a 19-MeV cyclotron (300 μA, TR PET cyclotron; ACSI) at the BC Cancer Agency. This cyclotron had 2 beam ports, one connected to a target selector and the other to a switching magnet to direct the beam to either a solid target station or a target selector positioned horizontally for conventional targets. The cyclotron configuration is illustrated in Figure 1. In addition to the concrete shielding of the vault, both target selectors and the solid target station were surrounded by mobile local shields.
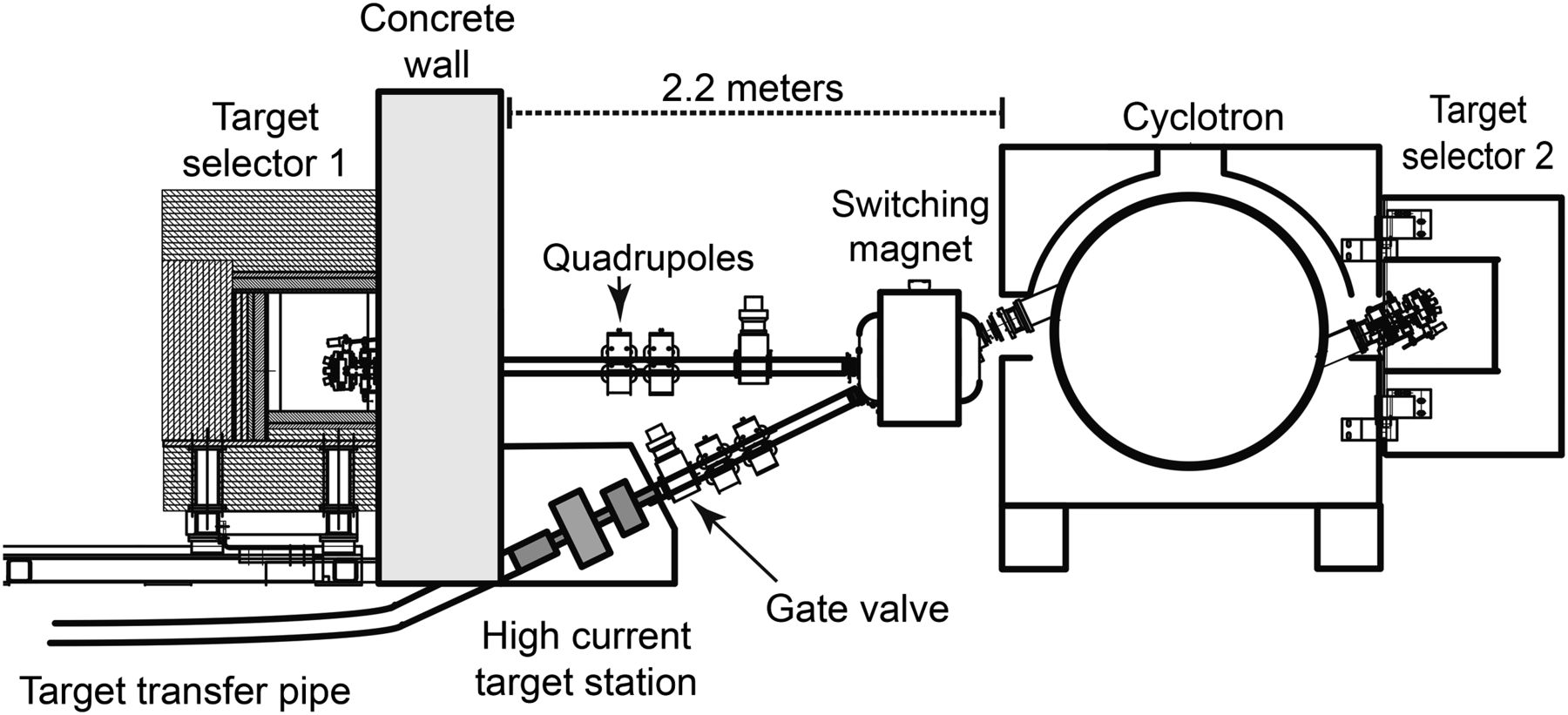
Cross-sectional view of BC Cancer Agency TR-PET cyclotron showing beamline facilitating both standard 4-port target selector and solid target station for high-current 100Mo irradiations.
Target System Design
Two target systems were designed for 99mTc production. The initial design consisted of an aluminum target holder accommodating a 10 × 20 mm plate with an aluminum cold finger at the back to dissipate heat from the target plate (cold finger target, further described in the supplemental data [supplemental materials are available at http://jnm.snmjournals.org]). The target plate was automatically dropped into a shielded container after irradiation.
The second target holder was designed for high-current irradiations over a larger surface (referred to as the large target) (Fig. 2). This target station accommodated a capsule containing the target plate. This capsule could be automatically connected to, and retrieved from, the target station. The assembly is described in the supplemental data section. The target station alignment and the beam collimator adjustments were performed by irradiating blank aluminum or tantalum target plates, exposed to radiographic film (Gafchromic EBT2; International Specialty Products) to measure the beam distribution on the target plate.

Photograph of high-current target station and target capsule (bottom right).
Target Plate Preparation
Tantalum target-backing plates were mechanically etched at the target surface by introducing shallow grooves to maximize adhesion of the target material. The targets were prepared by electrophoretic deposition (EPD) of 100Mo on tantalum plates as described by Gutierrez et al. (20). The deposition of 100Mo was achieved within minutes. Two lots of 100Mo were tested (99.01% and 97.4% 100Mo content) (Table 1). The third batch (99.8%) listed in Table 1 was not irradiated but is included to highlight the specifications of enriched material that is now available. After EPD, the targets were placed in a sintering oven (CTF 18/300; Carbolite) and heated to 1,700°C for 5 h. Electron microscopy images of the target were obtained on a Hitachi S-2300 instrument.
Composition of Various Batches of 100Mo
Target Irradiations
The targets were irradiated at progressively higher currents to test their ability to withstand the proton beam. The energy was 18 MeV for all irradiations. The cold finger target was irradiated at 20, 30, and 50 μA. For the large target, experiments were performed at beam currents of 100, 200, and 240 μA. The current was progressively ramped up over a period of approximately 15 min, followed by constant current. The total irradiation time ranged from 1.3 to 6.9 h.
Target Dissolution Process and Apparatus
The small target plates were dissolved into a beaker containing 30 mL of 30% H2O2 preheated to 50°C. In early experiments, the dissolved target solution was mixed with 2 M NaOH and evaporated to dryness. The dry powder was then reconstituted in 4–6 mL of water. For later experiments, the dissolution mixture was first evaporated to dryness while heating the beaker at 80°C, and 7 mL of 2 M NaOH were added as a final step to dissolve the residue.
The target capsules were directly used as dissolution vessels to extract 99mTc from the target plates (supplemental data). The target dissolution containing the dissolved 99mTc and approximately 1 g of 100Mo was reconstituted in 4 M NaOH.
Pertechnetate Purification
The 99mTc pertechnetate purification followed our published solid-phase extraction procedures (21). Several resins were compared for the separation of pertechnetate from molybdate, including the ABEC-2000 resin (Eichrom Technology) and the AnaLig Tc02 resin (IBC Advanced Technologies).
The ABEC-2000 resin was used as previously described, with the exception that potassium hydroxide was replaced by NaOH (21).
Initial experiments with the AnaLig TcO2 resin were performed manually to optimize separation conditions at low activity levels using a solution of 1.8 g of molybdenum metal powder dissolved in 15 mL of 30% H2O2. The solution was evaporated to dryness and dissolved in 7 mL of 2 M NaOH. A small amount of 99mTc was added to this solution to simulate a target dissolution mixture. We experimented with various resin contents, ranging from 100 to 400 mg, various flow rates (0.2–0.6 mL/min), and water elution flow rates (0.5–5 mL/min). Further experiments were done using a purification system as described in Morley et al. (21) and implemented under real irradiation conditions of 100Mo targets, dissolved as described above. One key modification was the addition of a syringe pump to control the loading rate of the target dissolution mixture on the AnaLig Tc02 resin.
To summarize the AnaLig purification process, the 2 M NaOH solution containing 99mTc and 100Mo was passed through a solid-phase extraction cartridge containing 100 mg of AnaLig resin. The resin was loaded at a flow rate of 0.2 mL/min. At high pH, 100Mo, along with small quantities of 99Mo and radioactive niobium impurities, was recovered in the first waste vessel. The resin was rinsed with 2–3 mL of 2 M NaOH and then eluted with water.
The 99mTc was recovered in 10 mL of water. The water/99mTc solution was neutralized over a cation exchange resin (On-Guard II H; Dionex) and trapped on a small Alumina A cartridge (Waters). The alumina cartridge was eluted with 0.9% saline solution, passed through a 0.22-μm filter and recovered in the product vial.
Measurement of Radionuclidic Purity
Samples for γ-ray spectroscopy were taken from the dissolved target solution before purification. Samples were counted 3 h after the end of beam and at 24 h, 3 d, 7 d, and 21–30 d after end of beam. For late samples, longer acquisitions were used (up to 24 h for the 21- to 30-d samples). All samples were measured on a high-resolution γ-ray spectrometer (Canberra) equipped with a high-purity germanium detector. The HyperLabs 2009 software (Hyperlabs Software Inc.) was used for data analysis. The γ-ray spectrometer was energy- and efficiency-calibrated using National Institute of Standards and Technology–traceable radioactive sources (Eckerd & Ziegler).
Quality Control and Kit Radiolabeling
The 99mTc-pertechnetate solution was tested for alumina with a colorimetric test kit (Biodex), compared with a test solution of 10 μg of aluminum per milliliter. The presence of residual 100Mo was tested with a colorimetric assay, using a molybdenum assay kit (Quantofix Molybdenum; Macherey-Nagel).
The presence of hydrolyzed-reduced 99mTc was tested by thin-layer paper chromatography using Whatman paper developed in acetone. Kits for neutral (99mTc-exemetazime; GE Healthcare), cationic (99mTc-sestamibi; Covidien), and anionic (99mTc-medronate; Edmonton Radiopharmacy Centre) radiopharmaceuticals were reconstituted and tested according to the manufacturer’s instructions.
Recycling of 100Mo
The first waste solution containing 100Mo in NaOH and radioactive impurities was decayed for 6 mo before reprocessing. The solution was purified by ion exchange chromatography using a Dowex 50W-X8 cation exchange column (50 × 2 cm diameter). The eluate containing molybdic acids (MoO3·n H2O) was evaporated to dryness and reduced to molybdenum metal in a tube furnace at 1,100°C under a hydrogen atmosphere (22,23).
RESULTS
Target Plate Preparation and Electron Microscopy
With EPD, fine molybdenum powder was rapidly deposited on the tantalum backing, forming a soft layer on top of the plate. Up to 1.5 g of molybdenum metal powder were deposited over a surface ranging from 15 to 18 cm2 for the large-area targets. After high-temperature sintering, the deposited material formed a hard, adherent layer, which bonded with the tantalum backing (Fig. 3). The texture and bonding strength was dependent on the particle size of the 100Mo metal powder—empirically we observed better results with finer (<10 μM) powder. The deposited layer could not be removed from the target backing by dropping or bending the tantalum plate. Scanning electron microscope images showed that the powder grains were adherent to each other, with residual gaps and voids (Fig. 4).

Picture of target plate for large target station, with 100Mo layer prepared by EPD and high-temperature sintering (bottom). Corresponding beam shape is shown on film exposed to target plate (top).

Scanning electron micrographs of target section. There is good bonding of molybdenum layer to tantalum target-backing plate (white arrow). Deposited material is porous as noted by voids in deposited target material.
Target Irradiations
Several cold finger targets were irradiated to test the feasibility of the EPD/sintered targets. Two batches of 100Mo were irradiated, containing 97.4% (Trace Sciences) and 99.01% (Isoflex) 100Mo content, respectively. Irradiation conditions ranged from 20 to 50 μA peak proton current, for durations ranging from 22 to 377 min (total integrated current, 700–7,200 μA·min). The results are reported in Table 2. The total collected activity reflects measured 99mTc activity before purification, whereas the final product assay is the measured 99mTc pertechnetate after purification. Both values are decay-corrected to end-of-beam time. In some cases, we noticed that the target plate could be introduced in the holder without reaching optimal contact between the aluminum cooling finger and the target plate, leading to poor target performance due to inadequate thermal conductivity.
Production Yields with Small-Area Cold Finger Target
The large targets were irradiated at proton beam currents ranging from 100 to 240 μA, for durations ranging from 85 min to 6.9 h. These results are reported in Table 3. For these experiments, the total activity was assayed after target dissolution and before pertechnetate purification. As measured by the dose calibrator, the total 99mTc activity was likely overestimated by the presence of a small but significant quantity of higher energy isotopes present in the irradiated 99.01% 100Mo targets.
Large Target Production Runs
Target Dissolution
For cold finger targets, the dissolution of the molybdenum target with H2O2 was an exothermic process, which required careful handling. The addition of NaOH to the molybdic solution was also exothermic. The target dissolution and evaporation process was achieved in 40 min.
The large targets were readily and completely dissolved within the capsule, with no trace of residual 100Mo on the target at the end of the process. No burn marks were visible on the large-area targets after high-current irradiation.
Pertechnetate Purification
For the ABEC-2000 resin, the mean purification efficiency of 99mTc was 78.7%, with an SD of 9.5%. The content of 99mTc not trapped by the ABEC-2000 resin was 18.2% ± 9.3% (mean ± SD). Remaining losses were minimal (0.7% ± 0.9%, 2.1% ± 1.2%, and 0.3% ± 0.2% for the ABEC-2000 resin, alumina cartridge, and cation exchange resin, respectively).
With manual and eventually automated purification using the AnaLig Tc02 resin, optimal results were obtained using 100 mg of resin preconditioned with 5 mL of deionized water, loaded at a flow rate of 0.2 mL/min. The AnaLig Tc02 resin was readily eluted with water (2 mL/min).
For the cyclotron runs performed using the AnaLig Tc02 resin, the purification efficiency was 83.9% ± 1.7%, with 5.2% ± 1.5% losses in waste, some activity remaining trapped on the AnaLig resin (7.5% ± 1.8%), and minimal activity remaining on the alumina or cation exchange cartridges (3.0% ± 0.6% and 0.4% ± 0.1%).
Radionuclidic Purity Results
Radionuclide purity analyses were performed on batches of 99.01% and 97% 100Mo irradiated at high currents, and the results are shown in the supplemental data. The technetium radionuclide contents showed that 99mTc constituted over 99.5% of all technetium radioisotopes produced. The content of 96gTc, 95gTc, 94mTc, and 94gTc was lower using the 97.4% 100Mo target material, which had a lower content of 92–97Mo. The results agreed with extrapolations from experimental data (24) and showed lower amounts of long-lived impurities than predicted based on Empire III calculations (19). The quantity of 94mTc was higher than predicted based on theoretic models. The proportion of 96mTc could not be quantified because of its short half-life, low γ-emission rate, and overlap with other radioisotopes peaks. Because of chemical identity with 99mTc, other technetium radioisotopes could not be separated from the final 99mTc-pertechnetate solution. Radioisotopes other than technetium that were recovered in waste are reported in Supplemental Table 2.
Quality Control of Pertechnetate and Kit Radiolabeling
Three batches of cyclotron-produced 99mTc-pertechnetate produced using the cold finger targets were tested according to the quality control procedures used for generator-produced 99mTc-pertechnetate. The radiochemical purity of 99mTc-pertechnetate was 99.7% ± 0.5% (United States Pharmacopeia [USP] criteria > 95%). In all cases, the aluminum ion content was lower than 10 μg/mL. The radiochemical purity of 99mTc-sestamibi was 99.6% ± 0.7% (USP criteria > 90%). The radiochemical purity of 99mTc-medronate was 98.1% ± 3.2% (USP criteria > 90%). The radiochemical purity of 99mTc-exemetazime was 91.3% (USP criteria > 90%; n = 2). No traces of molybdenum were found in the pertechnetate solution.
Recycling of 100Mo
The radioactive impurities in the waste included 99Mo, 99mTc, and 95Nb, with 95Nb having the longest half-life (35 d). After 6 mo of decay, the waste solution contained significantly less than the exemption quantity of all radioisotopes. 100Mo from the waste solution was recycled with an average yield of 85% (range, 80%–92%). High-temperature reduction under a hydrogen atmosphere yielded a light gray metallic powder that could be reused for deposition on tantalum target backings.
DISCUSSION
Our research effort was prompted by the need to find alternative solutions for medical isotope production after the Canadian government’s decision to shut down isotope production at the Chalk River reactor in 2016 (6). The current supply is vulnerable and relies on a limited number of aging nuclear reactors.
The well-established use of 99mTc in nuclear medicine practice makes it unlikely to be replaced by another radioisotope over a short period of time (25). Given the kit chemistry of 99mTc (26), any replacement technology must be transparent to end users.
With an increasing number of medical cyclotrons installed for 18F production for PET imaging, it was time to explore the practical feasibility of the direct production method using the 100Mo(p,2n)99mTc reaction. Conveniently, the peak cross section for the 100Mo(p,2n) 99mTc occurs at approximately 15 MeV (17–19), which is within the range of many existing medical cyclotrons used for PET radioisotope production.
Our team tackled several challenges to implement a simple, reliable method for routine production of 99mTc. This was made possible by prior work of Beaver and Hupf (13), Lagunas-Solar (14,16), and others (15,17), who had studied the feasibility of this approach.
The first challenge was the development of a target system that could be prepared using accessible laboratory equipment and small quantities of enriched target material. Molybdenum metal has a high melting point (>2,600°C) and is difficult to work with given its propensity for oxidation at lower temperatures (∼440°C). As such, melting or sputtering the metal potentially leads to high losses.
We explored various methods for target production including electroplating (27) and sugar codeposition of molybdenum (28). These methods were not useful to produce high quantities of 99mTc. We developed an EPD method that makes thick, robust 100Mo coatings for routine use. These targets were successfully used for high-current irradiations at up to 240 μA. The target plates faced the beam at either a 20° (cold finger target) or a 10° angle (large target) relative to the proton beam. These angles increased the effective thickness of the material by a factor of 2.9–5.7. Thus, the deposited 100Mo layer was sufficiently thin to enable adequate thermal heat transfer from the cooled backing during irradiation. The porosity of the molybdenum layer facilitated the recovery of 99mTc.
Another challenge was the design of a target station that allowed the routine production of 99mTc. Our solution was to use a mechanical push-and-retrieve system that directly retrieved the target capsule to a hot cell. The target dissolution occurred directly inside the target capsule, allowing the safe handling of the targets.
The purification of 99mTc-pertechnetate from molybdenum at various specific activities was reviewed in a recent publication (29). Several methods are not applicable to cyclotron-produced 99mTc because of the quantity of nonradioactive molybdenum present in the target dissolution mixture. We tested several approaches, including the ABEC-2000 resin and the AnaLig resin, and found that these methods were suitable for the purification of 99mTc-pertechnetate from the irradiated 100Mo target solution.
To reduce the cost of 100Mo, we developed a recycling method for 100Mo and achieved a recovery efficiency of up to 92%. This process can be optimized further to reduce 100Mo losses.
The radionuclidic purity of 99mTc was analyzed using a high-resolution γ-ray spectrometer. The results highlighted the fact that the isotopic composition of the target material was more important than the absolute enrichment level. In particular, 94Mo, 95Mo, 96Mo, and 97Mo are significant contributors to the production of radionuclide impurities. We observed lower levels of long-lived impurities than theoretically predicted (19,24).
It is now possible to obtain batches of high-quality 100Mo achieving 99.8% isotopic purity (Table 1), with 94–97Mo content below the detection limit, in sufficient quantities to meet clinical and commercial needs. The radionuclidic purity of cyclotron-produced 99mTc will change depending on the target material, irradiation conditions, and the time after production at which the 99mTc is used. No USP or European Pharmacopeia standards exist for cyclotron-produced 99mTc. We previously showed that the impact of these impurities on patient radiation dose was minimal when using batches of 100Mo with low 94–97Mo content (30).
The specific activity of 99mTc was not measured in the current study but was previously reported (18). Both experimental and theoretical results suggest that similar ratios of 99mTc to 99gTc can be expected when comparing typical cyclotron irradiation parameters (e.g., 3–6 h) with a generator eluted at a 24-h frequency (18,19). We radiolabeled various radiopharmaceutical kits using cyclotron-produced 99mTc. All radiopharmaceuticals successfully passed routine quality control tests.
Our production system was able to reliably produce 99mTc in saturated yields exceeding 3 GBq/μA at a 18-MeV proton beam. In a single irradiation, we achieved the production of 347 GBq (9.4 Ci) of 99mTc in just over 6 h. The target dissolution and 99mTc purification were completed in approximately 75 min. We achieved average purification yields of 84%. Further optimization is possible to reduce the processing time and improve yields.
The precise production costs of cyclotron 99mTc were not compared with those of generator-produced 99mTc in this study. The cost estimates need to take into account overhead costs, equipment amortization, and implementation of good manufacturing practice–compliant production and various distribution models in addition to direct expenses. These factors will be important considerations for eventual market acceptance of this production method.
CONCLUSION
The production of large quantities of 99mTc is possible using a medical cyclotron. These results demonstrate that it is feasible to supply a large population base from a single cyclotron. The 99mTc produced by cyclotrons at 18 MeV can be obtained with high radionuclidic purity and is a potential alternative for routine clinical use. With some modifications of existing cyclotron infrastructure, this approach could be used to implement a highly decentralized medical isotope production model, eliminating the requirement for enriched uranium and radioactive waste associated with nuclear fission.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the Natural Sciences and Engineering Research Council, the Canadian Institutes for Health Research and Natural Resources Canada. Salary support was provided by the Leading Edge Endowment Fund and the Michael Smith Foundation for Health Research. Jean-Pierre Appiah is now an employee of ACSI. A provisional application for patent was filed for some of the material presented in this article. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Dr. Sarah Mullaly for editing the manuscript.
Footnotes
Published online Apr. 10, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication October 7, 2013.
- Accepted for publication January 21, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Clinical Trial with Sodium 99mTc-Pertechnetate Produced by a Medium-Energy Cyclotron: Biodistribution and Safety Assessment in Patients with Abnormal Thyroid Function
- Radioisotopic Purity of Sodium Pertechnetate 99mTc Produced with a Medium-Energy Cyclotron: Implications for Internal Radiation Dose, Image Quality, and Release Specifications
- Diversification of 99Mo/99mTc Separation: Non-Fission Reactor Production of 99Mo as a Strategy for Enhancing 99mTc Availability
- Cross-Linked Polyethylene Glycol Beads to Separate 99mTc-Pertechnetate from Low-Specific-Activity Molybdenum
- Diversification of 99Mo/99mTc Supply