


Abstract
Post-processing by means of a cation-exchanger–based protocol is an efficient strategy for purification and concentration of generator-derived 68Ga. It ensures the removal of 68Ge before 68Ga-radiopharmaceutical preparation and high labeling yields of 68Ga-labeled radiopharmaceuticals for routine medical application. Methods: In an effort to overcome the problem associated with acetone in the currently applied method, we have investigated the feasibility of replacing it with ethanol. The purification of 68Ga from coeluted metallic impurities (68Ge4+, Fe3+, Zn2+, and Ti4+) on various cation-exchange columns has been investigated with a variety of post-processing solutions. As a proof of principle, the post-processed 68Ga was used to radiolabel DOTATOC in combination with high-purity water and various buffer solutions. Results: An effective protocol for the processing of generator-produced 68Ga on the basis of cation-exchange chromatography using EtOH/HCl medium has been developed. Up to 95% of the initially eluted 68Ga activity can be collected in a 1-mL fraction of 90% EtOH/0.9N HCl after removal of 68Ge-breakthrough in a washing step. The post-processed eluate has been used to radiolabel DOTATOC in yields of approximately 97% ± 0.25% at 80°C in 5 min. Conclusion: The described novel protocol improves the radiolabeling efficiency and efficacy of DOTATOC, providing yields of greater than 99% (decay-corrected). As a result, further purification to separate the desired product from uncomplexed 68Ga is not necessary. The developed post-processing and labeling protocols permit reliable and high-yield preparation of injectable 68Ga-DOTATOC (or other 68Ga-labeled radiopharmaceuticals) that are suitable for routine application. It is possible to incorporate this protocol into existing automated modules.
An enticing characteristic of the positron-emitting radionuclide 68Ga is its cyclotron-independent availability via 68Ge/68Ga generators. Also attractive is the high positron yield (89% positron branching) and accompanying low photon emission (1.077 keV, 3.22%) from 68Ga. As a result, 68Ga is considered to be one of the more promising radionuclides to carry PET into wider clinical application, with 68Ge/68Ga generators already being used extensively in research and to a lesser extent for clinical applications.
68Ga-radiopharmaceuticals are often used in clinical applications. Recently, 68Ga-DOTATOC has been officially designated an orphan drug by the U.S. Food and Drug Administration. Widespread application of the 68Ge/68Ga generator requires the development of protocols to make 68Ga available for radiolabeling, because the eluate obtained is not necessarily suitable for direct medical use. First, the 68Ga eluate contains measurable activities of the long-lived 68Ge, which is a critical parameter in the context of the routine clinical application of 68Ga-radiopharmaceuticals (1,2). Second, the relatively large volume and high acidity of the eluate hinder efficient radiolabeling and, thus, application in vivo. Third, the presence of metal ion contaminants can result in reduced labeling yields and specific activities.
Several methods have been developed to reduce the metallic impurities and concentrate the eluate, of which cation-exchange (CEX) resin–based post-processing has been particularly successful (3). The method pioneered by Zhernosekov et al. provides high recovery of 68Ga and complete removal of 68Ge, as well as a decrease in acidity, volume, and metallic impurities (3). After post-processing, the 68Ga is available as the 68Ga3+ species in 400 μL of a 97.56% acetone/0.05N HCl solution and is suitable for direct radiolabeling of DOTA-functionalized vectors (3,4).
The procedure permits efficient production of 68Ga-labeled radiopharmaceuticals and is successful, having been incorporated into various commercial module systems for generator elution, post-processing, and synthesis.
The acetone content of the final injectable 68Ga-radiopharmaceutical prepared using this method is estimated to be less than 0.5 μg (<8.61 nmol) (3). Although acetone is a class 3 solvent (low risk to human health) (5), it is evident from conversations with colleagues in the field that certain sectors believe an injected radiopharmaceutical should not contain acetone. The aim of this work was to investigate the use of other, more accepted solvents to replace acetone for the post-processing of the generator eluate.
Another class 3 solvent commonly applied as an additive and solvent in medical applications is ethanol, which is used to increase the solubility of pharmaceuticals and act as a preservative. In radiopharmaceutical chemistry, ethanol is known to inhibit ionization-induced radiolysis. Specifically, this work focuses on substituting ethanol for acetone within the successful CEX post-processing method (3,4). The objective is to provide a more widely accepted constitution for the final injectable solution. A further potential benefit of this approach is that the presence of ethanol enhances the rate of radiolabeling with certain metallic radionuclides (6). Indeed, the use of ethanol-based CEX post-processing may represent an important step toward clinical application of the 68Ge/68Ga generators and even kit-type formulations of 68Ga-radiopharmaceuticals.
MATERIALS AND METHODS
Metals Used for Ion-Exchange Distribution Measurements
A commercial 1,100-MBq generator with 68Ge4+ adsorbed on a TiO2 support was obtained from Cyclotron Co. Ltd.
An 85-MBq quantity of 68Ga in 5 mL of 0.1N HCl was obtained from an 18-month-old 1,100-MBq generator, which had been eluted more than 200 times previously. The activity of 68Ge breakthrough in the eluted 68Ga fraction was about 7 kBq.
59Fe was produced by a neutron-capture nuclear reaction of naturally occurring metallic iron. Iron (200 mg) was irradiated for 21 d at the Hahn-Meitner-Institute Berlin neutron source at 1.6 × 1014 n cm−2 s−1, yielding 456 MBq of 59Fe. The iron was dissolved in concentrated HCl solution and then diluted to give a solution of 0.1N HCl. TiCl4 was obtained from Sigma-Aldrich as 0.09 M titanium solution in 20% HCl. It was diluted to give a solution of 0.1N HCl before use.
Chemicals and Equipment
Only the highest-grade chemical reagents and TraceSelect water were used. These were purchased from Sigma-Aldrich and used without further purification, unless otherwise stated. AG 50W-X8 (<400 mesh) and AG 50W-X4 (200–400 mesh) (BioRad), STRATA-X-C (Phenomenex), Bond Elut SCX (Varian), and KGG LiChrolut SCX CEX (Merck) resins were used to prepare microchromatography columns (50 mg of resin, 2-mm inner diameter, 5-mm length). Labeling reactions were performed in 11-mL glass vials (Mallinckrodt) using a block thermostat (TK13; Ditabis) for temperature control and agitation. The 68Ga-DOTATOC was purified with 30-mg C-18 cartridges (Strata-X Tubes; Phenomenex), with subsequent sterile filtration using a 0.22-μm membrane filter (Millex GV; Millipore). The activity of 68Ga3+ and 59Fe3+ was measured using a curie meter (Isomed 2010; Nuklear-Medizintechnik Dresden GmbH). 68Ge decays exclusively by electron capture and therefore can be radiochemically detected only indirectly through the photon emission of its daughter nuclide, 68Ga. For the γ-spectroscopic determination, the samples were stored for 48 h until the secular equilibrium with 68Ga had been reached. 68Ge activity was determined by γ spectrometry using a high-purity germanium detector (68Ge-detection limit, 12 Bq). Ti4+ content was determined using inductively coupled plasma mass spectrometry (7500ce; Agilent). pH was measured using a calibrated pH meter (SevenEasy pH; Mettler Toledo). Thin-layer chromatography plates (aluminum-backed silica gel 60; Merck) were analyzed using a flat-bed scanner (Instant Imager; Canberra Packard).
Separation of 68Ga3+ from 68Ge4+, 59Fe3+, and Ti4+
The microchromatography columns were prepared using 50 mg of each CEX resin. 68Ga was eluted from the generator with 0.1N HCl (5 mL) and transferred online to the microchromatography CEX column. The column was then washed with 1 mL of a washing solution comprising ethanol (70%–90%) and HCl (0.1, 0.15, or 0.2 M), and the washing fraction was collected. Remaining traces of the washing solution were removed by passing air through the CEX resin using a syringe. This washing step is designed to eliminate the unwanted chemical and radiochemical impurities without loss of 68Ga from the CEX. In the final step, the column was filled (∼200 μL) with a second solution of varied EtOH/HCl composition and allowed to stand for 2 min. Subsequently, the column was eluted with varying amounts (200–800 μL) of the same solution and collected in an Eppendorf vial (2 mL). The protocol is designed such that this eluent should contain the purified and concentrated 68Ga3+. The column was reconditioned with 4N HCl (1 mL) and then H2O (1 mL). In each experimental setup, 5 fractions were analyzed for their 68Ga and 68Ge contents: (i) 0.1N HCl, (ii) washing solution (80% EtOH/0.1–0.2N HCl), (iii) elution mixture (EtOH/HCl), (iv) 4N HCl, and (v) H2O. The distributions of 68Ga3+ and 68Ge4+ in the different fractions were investigated to determine the most suitable setup. To analyze the behavior of relevant impurities, the distributions of 59Fe3+ and Ti4+ in 0.1N HCl were determined using the described protocol.
68Ga Radiolabeling
The 68Ga eluate obtained after elution with 1 mL of 90% EtOH/0.9N HCl (9 × 10−4 mol HCl) was used for radiolabeling experiments. Labeling was performed in water (4 mL) or suitably prepared buffer solutions (NaOAc, NH4OAc, or N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid [HEPES]) in open standard reagent vials containing DOTATOC (21 nmol). The volume and concentrations of the buffer were varied to establish the optimal conditions. Solutions were preheated to the desired temperature (80°C or 95°C depending on the setup used) and reacted for either 10 or 15 min. For comparison, the labeling kinetics of DOTATOC were determined using 68Ga purified by the standard acetone post-processing method. In this case, labeling was performed in water (4.6 mL) at 80°C and 95°C for 15 min.
Purification of 68Ga-Labeled Peptide and Quality Control
68Ga-DOTATOC was separated from unreacted 68Ga species by reverse-phase chromatography. The reaction mixture was cooled to about 50°C and passed through a C-18 cartridge. The peptide was quantitatively retained on the reverse phase. After the cartridge had been washed with 1 mL of H2O, the 68Ga-DOTATOC was eluted using 0.4 mL of ethanol.
Thin-layer chromatography was used to analyze the reaction yields. The reaction solution (2 μL) was spotted on thin-layer chromatography plates and developed with 0.1 M Na3-citrate water solution (pH 4), and the activity population distribution was measured using a flat-bed scanner.
Each experiment was performed in triplicate to accurately determine the SD. All reported yields are decay-corrected.
RESULTS
Distribution of 68Ga
The initial 68Ga adsorption from the generator eluate onto the CEX resins was about 99%. Subsequent washing using an 80% EtOH/0.15N HCl solution caused less than 3% of the 68Ga to be removed from the CEX and lost to the waste fraction in each case. In fact, this percentage loss remained constant for varying constitutions (70%–90% ethanol and 0.2–0.1N HCl) of the washing solution. Subsequent desorption of the 68Ga from the resin was achieved using a 90% EtOH/0.9N HCl CEX eluting solution with desorption yields that varied depending on the CEX used. Desorption efficiencies were greater than 75% (mean) for the AG 50W-X8/AG 50W-X4 columns, approximately 23% (maximum) for the Bond Elut Strata-X-C columns, and less than 1% (maximum) for the LiChrolut Strata-X-C columns. The distribution of 68Ga between the different fractions (i–v), using 80% EtOH/0.15N HCl as the washing solution (ii) and 90% EtOH/0.9N HCl as the CEX eluting solution (iii), is shown in Table 1 for each CEX investigated.
Relative Distribution (%) of 68Ga3+ on All Investigated CEX Microchromatographic Columns (n = 3)
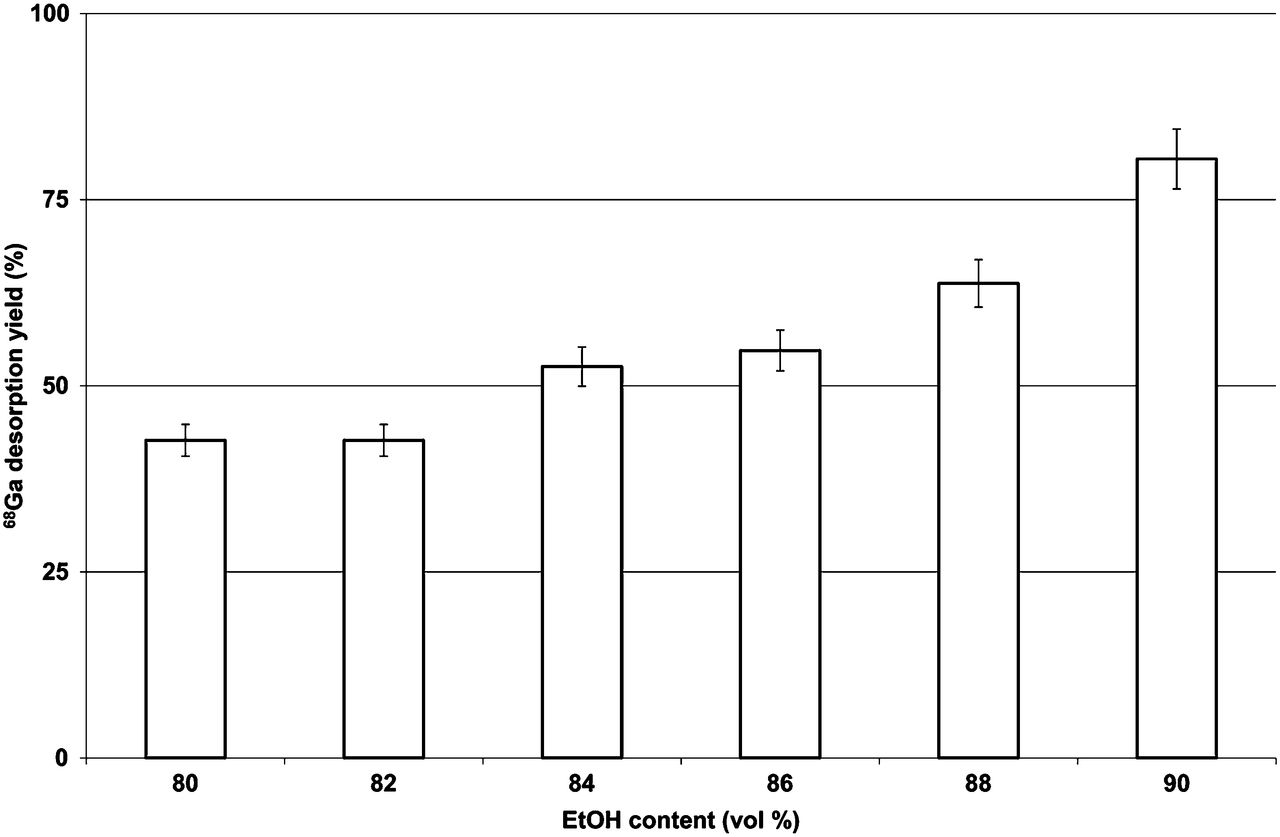
Desorption was optimal using the AG 50W-X8 and AG 50W-X4 resins, and these became the focus of subsequent investigations. Desorption of 68Ga from the resin was also found to be influenced by the acidity, ethanol content, and volume of solution iii used. Figures 1, 2, and 3 show the dependency of the desorption yield on the acidity, ethanol content, and eluting volume, respectively (with all other parameters kept constant). For a constant ethanol content and volume, the desorption yield increases with increasing acidity up to 0.90N HCl (Fig. 1). Maximum desorption was obtained when solution iii consisted of 90% ethanol (Fig. 2). It is clear that the AG 50W-X8 and AG 50W-X4 CEX resins are suitable for the adsorption and desorption of 68Ga. A further important consideration is the efficacy of the washing step at separating 68Ga from metallic impurities. This efficacy was evaluated in terms of the distribution of 68Ge4+, 59Fe3+, and Ti4+ after post-processing.

Dependence of 68Ga-desorption yield on HCl concentration for AG 50W-X8 resin eluted with 0.4 mL of 90% EtOH/0.05–0.95N HCl solutions (n = 3).
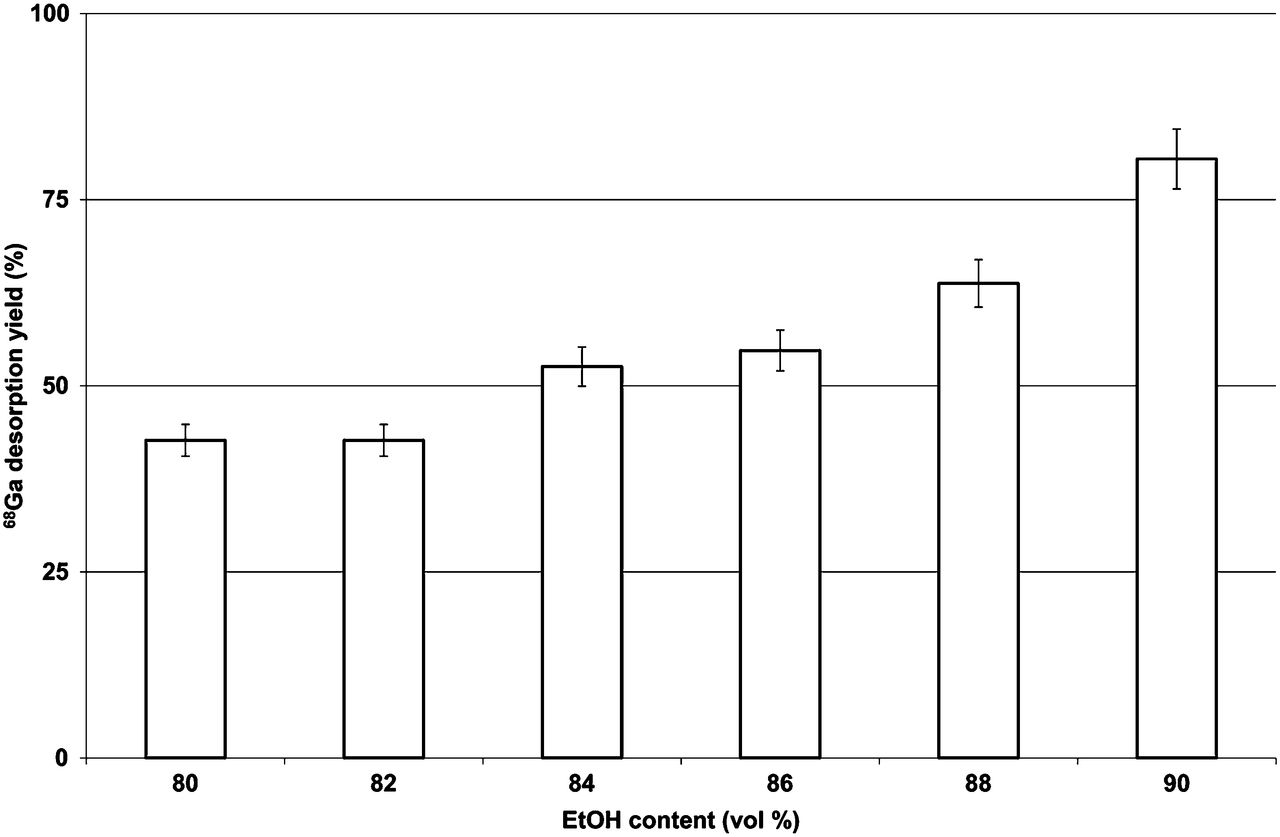
Dependence of 68Ga-desorption yield on ethanol content for AG 50W-X8 resin eluted with 0.4 mL of 80%–90% EtOH/0.9N HCl solution (n = 3).
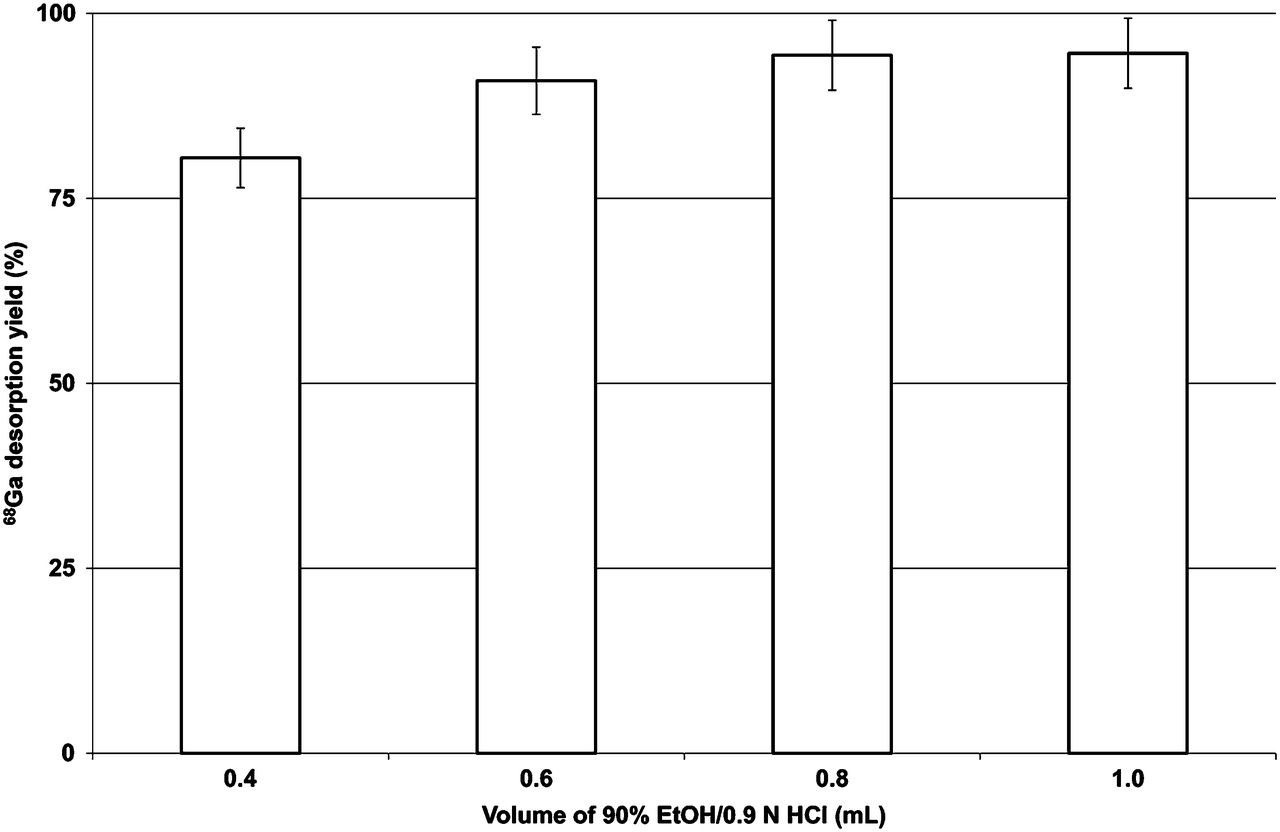
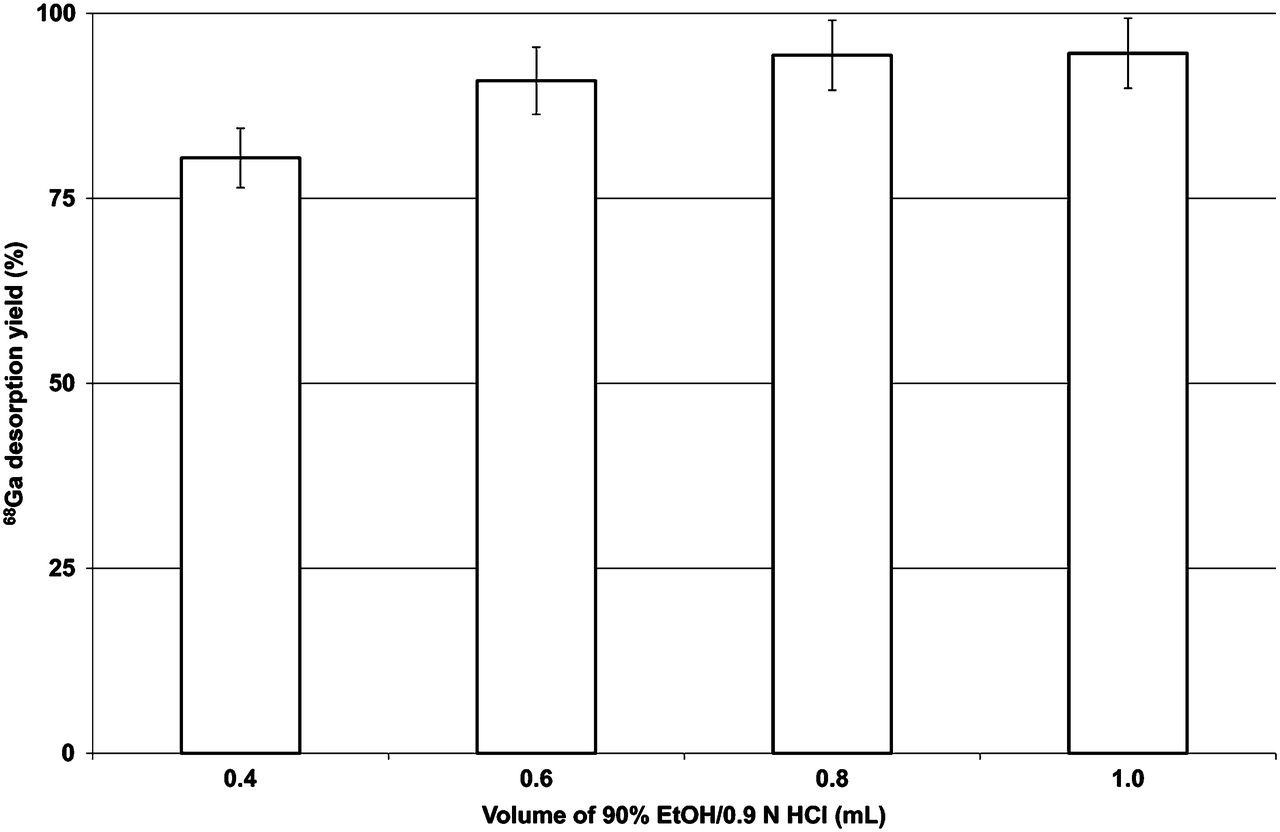
Dependence of 68Ga-desorption yield on total volume of elution mixture for AG 50W-X8 resin eluted with 90% EtOH/0.9N HCl (n = 3).
Distribution of 68Ga for 1-mL Mixtures of 90% EtOH/0.9N HCl
A profound effect was observed when the volume of the solution was increased (Fig. 3), with the yield reaching a maximum (∼95%) at 1 mL.
The relative distributions of 68Ga3+, 68Ge4+, Fe3+, and Ti4+ on CEX columns AG 50W-X8 (−400 mesh) and AG 50W-X4 (200–400 mesh) (50 mg), using 1 mL of the 90% EtOH/0.9N HCl as the CEX eluting solution, are summarized in Table 2. In a setup that does not include the washing step, such as those used within automated modular systems, the elution with the 90% EtOH/0.9N HCl solution would produce 68Ga desorption yields of about 97%.
Relative Distribution (%) of 68Ga3+, 68Ge4+, 59Fe3+ and Ti4+ on AG 50W-X8 (−400 mesh, 50 mg) and AG 50W-X4 (200–400 mesh, 50 mg) (n = 3)
Distribution of 68Ge4+
For both CEX resins, approximately 97% of 68Ge4+ passes through the column with the initial 0.1N HCl solution (i). The remaining traces are further reduced (∼3%) by the washing step with solution ii. After solution v, less than 0.25% of the initially eluted 68Ge remains trapped on the resin. There was no detectable 68Ge in fractions iii, iv, or v within 8 h of measurement. Because the 68Ge detection limit for the γ spectrometer was 12 Bq, this equates to a purification factor of more than 400.
Distribution of 59Fe3+
The relative distribution of Fe3+ varied considerably between the AG 50W-X8 and AG 50W-X4 CEX columns. The amount of Fe3+ in fraction iii could be reduced to approximately 14% and 33% of the initial content with AG 50W-X8 and AG 50W-X4, respectively. Purification factors for the respective CEX columns are therefore about 7 and 3.
Distribution of Ti4+
Ti4+ is almost entirely adsorbed on the CEX columns from solution i. Up to 11% of the initial Ti4+ content is removed with solution iii, with the AG 50W-X8 CEX performing slightly better in this regard (∼7% in purified 68Ga fraction). The largest titanium content is observed for solutions iv and v, indicating that the column regeneration is effective.
Labeling
Before labeling, the generator eluate was post-processed using the protocol outlined by Table 2 with AG 50W-X4, to give the purified 68Ga eluate in 1 mL of solution iii. The acidic content of the solution (HCl ≡ 9·10−4 mol H+) produced a pH of 1.6 ± 0.05 after dilution to 5 mL using H2O. When this solution was used directly, a labeling yield of 10% was achieved at 95°C after 15 min. The poor yield is not surprising and can be attributed to the strongly acidic environment protonating some of the ligand donor atoms, which hinders approach of the metal ion and creates an energy barrier to complexation of the metal ion.
To improve yields, labeling was repeated with different buffer solutions of varying molarity, pH, and volume (Table 3). NH4OAc, NaOAc, and HEPES were evaluated in all combinations of the following parameters: pH 4 and 5, total labeling volume of 1 and 4 mL, and buffer concentrations of 1.0, 0.5, 0.25, and 0.1 M. The labeling yields and corresponding specific activities are shown in Table 3.
68Ga-DOTATOC Radiolabeling Yield for Various Buffer Solutions at 95°C for 10 Minutes (n = 3)
As expected, the smaller labeling volumes produced higher specific activities, provided that the yields did not differ significantly. However, only at certain pH and buffer concentrations did labeling in 1 mL of buffer result in a reliable yield of greater than 89% (specific activity of 22–23 MBq/mol). Specifically, these conditions were NaOAc, pH 5, 0.5–1.0 M; NH4OAc, pH 5, 0.1–1.0 M; and HEPES, pH 5, 0.1–1.0 M. The equivalent higher-volume (4 mL) analogs of these experiments generally, but not always, gave rise to slightly lower yields. This result is not surprising given that the reactants are more dilute and thus have a detrimental effect on the rate of the reaction. It was possible to achieve yields of greater than 89% at a pH of 4 with NaOAc (0.5 M and 0.1 M), NH4OAc (0.1–1.0 M), and HEPES (0.25–1.0 M) with 4 mL of the buffer, albeit with low specific activities (9 MBq/nmol). There was only a single example of a similar yield at the same pH when the buffer volume was reduced to 1 mL (0.25 M NH4OAc). Optimal buffers are NH4OAc (1 M, pH 5) and HEPES (1 M, pH 5).
The labeling experiments were also repeated at 80°C for pH 5 buffers with a concentration of 0.5 M. All other parameters were varied as before. For comparison, the data are illustrated graphically in Figure 4 along with the analogous radiolabeling experiments performed at 95°C. Labeling yields of more than 90% at 80°C were obtained in all cases except for NaOAc (4 mL) and NH4OAc (4 mL). More remarkable was that the only small difference in yield between 80°C and 95°C labeling occurred when 1 mL of the buffer was used. In contrast, this yield was as much as 20% higher at 95°C when 4 mL of the buffer were used.
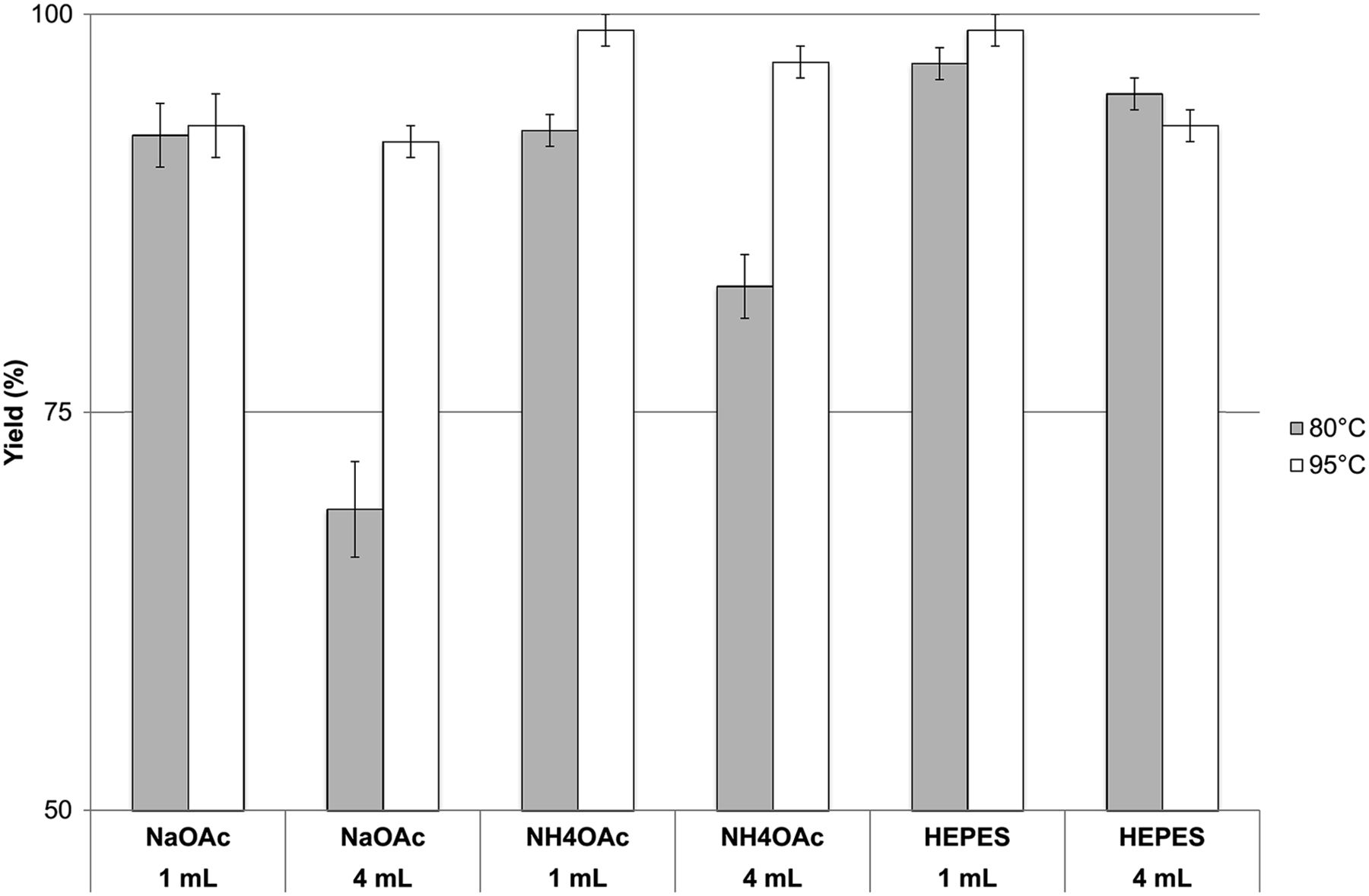
Comparison of 68Ga-DOTATOC radiolabeling yield obtained at 80°C and 95°C for 3 different buffer solutions (NaOAc, NH4OAc, and HEPES, all at 0.5 M) at pH 5, using 1 and 4 mL of buffer solution (n = 3).
Labeling with the HEPES buffer proved to be the most reliable, effective, and versatile and was used to directly compare the 2 post-processing methods. In this evaluation, DOTATOC was labeled at 80°C and 95°C using post-processed 68Ga obtained from the acetone- and ethanol-based methods. Acetone-post-processed 68Ga was used to label DOTATOC using the standard procedure, whereas ethanol-post-processed 68Ga was used with HEPES buffer (1.0 M, 1 mL). The labeling kinetics are shown in Figure 5; 95°C labeling using the ethanol-based post-processed 68Ga has been excluded for clarity. With ethanol-processed 68Ga, labeling yields of 97% ± 0.25% (specific activity, 23 ± 0.06 MBq/nmol) and 98% ± 0.2% (specific activity, 23 ± 0.05 MBq/nmol) were obtained at 5 and 15 min, respectively, at 80°C. Compared with the labeling kinetics using acetone-processed 68Ga, it is evident that there is a significant increase in the rate of labeling using the ethanol-based method (Fig. 5). This finding is highlighted by the difference between the ethanol method at 80°C and the acetone method at 95°C of 4% (5 min) and 1% (15 min) in favor of the ethanol method.
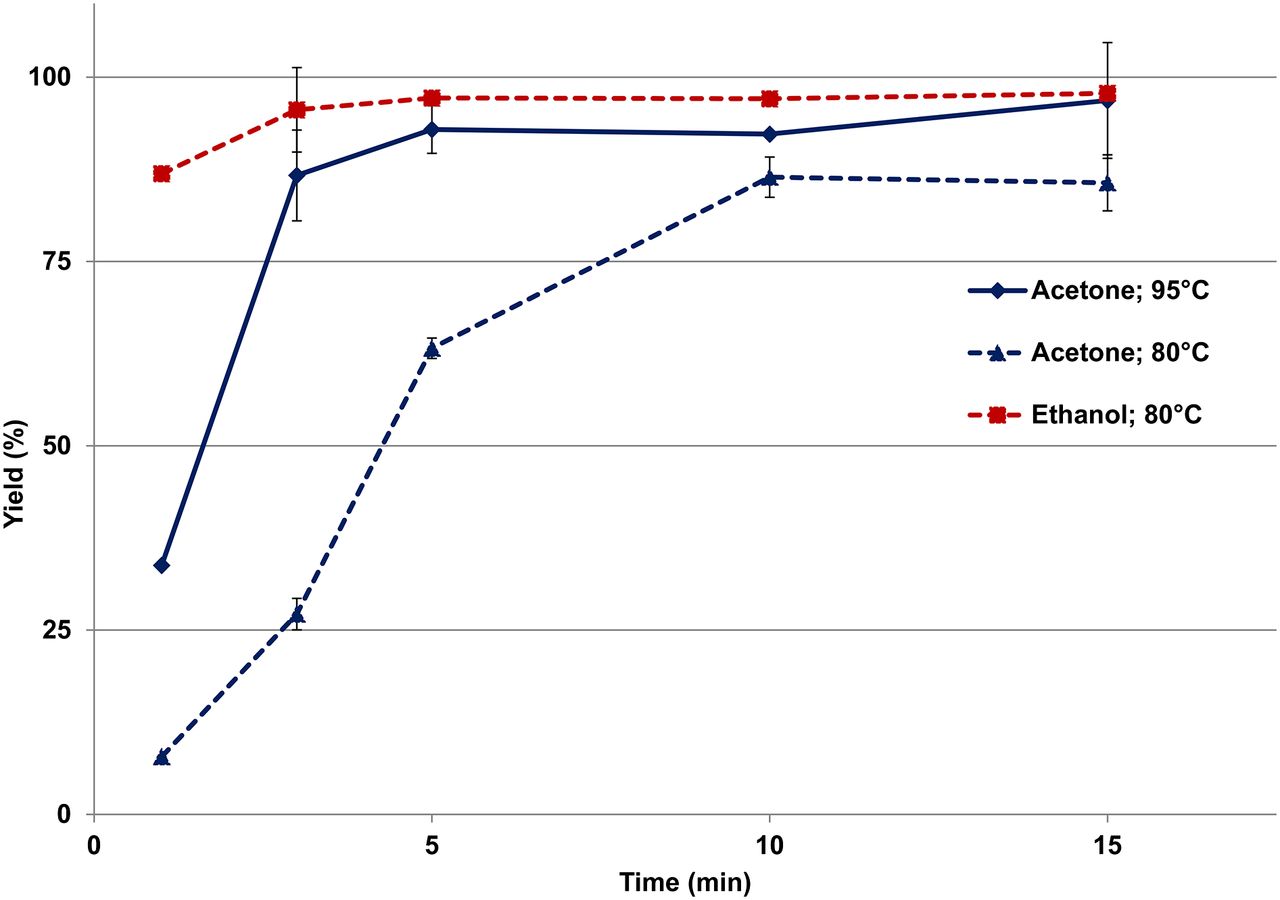
Radiolabeling kinetics (0–15 min) of 68Ga-DOTATOC obtained in 1 M HEPES buffer (80°C, 2 mL) after ethanol-based post-processing and in pure water (5 mL, 80°C and 95°C) after acetone-based post-processing (80°C and 95°C) (n = 3).
Purification of 68Ga-DOTATOC and Quality Control
Purification on Strata-X (C18 cartridge) did not significantly improve the radiochemical purity when the initial labeling yield was greater than 99%. Quality control was performed by thin-layer chromatography or high-performance liquid chromatography.
DISCUSSION
Five CEX resins were evaluated in terms of their ability to adsorb 68Ga from the initial generator eluate and to subsequently release the purified 68Ga when solutions in which the acetone is substituted for ethanol were used. Each of the resins was effective for the initial trapping of 68Ga from solution I; however, only AG 50W-X4 and AG 50W-X8 resins permitted desorption with solution iii in an acceptable yield. In comparison to AG 50W-X4, AG 50W-X8 was more effective for the initial 68Ga adsorption but produced a slightly smaller desorption yield with eluting solution iii.
Further improvements in the processing method were sought through changes to the constitution and volume of the eluting solution (iii). On the basis of these changes, the optimal combination for the post-processing was found to be resin AG 50W-X4, 1 mL of 80% EtOH/0.15 M HCl as the washing solution (ii), and 1 mL of 90% EtOH/0.9 M HCl as the eluting solution (iii). The steps involved are similar to those of the acetone-based method, which has been incorporated into the successful EZAG modules (Eckert and Ziegler Radiopharma Inc.). Therefore, it is feasible that this new ethanol-based protocol could be easily implemented within the same module and others.
On the basis of these findings, the 2 AG 50W resins were evaluated further for ability to separate 68Ga from unwanted metal ion impurities. Given that both resins provide an acceptable desorption yield of 68Ga, the next important consideration is the purity of the fraction used for labeling. Both resins were effective for the removal of 68Ge4+ and lowered the Ti4+ and Fe3+ content to an acceptable level. The 68Ge4+ content of the purified 68Ga-eluate is below the detection limit of γ spectroscopy, equating to a purification factor of less than 400. Significantly, the reduction of the initial 68Ge breakthrough on the cation-exchangers using the ethanol-based protocol fulfills legal requirements for the routine use of the 68Ge/68Ga generator in nuclear medicine. It is also possible to envision further applications such as the use of 2 or more 68Ge/68Ga generators connected in 1 line eluting onto a single CEX resin. Resin AG 50W-X8 provided a more than 2-fold greater reduction in 59Fe3+ removal than AG 50W-X4. Iron content is dictated by the HCl used, and therefore the resin with the largest 68Ga desorption yield is preferred. Thus, AG 50W-X4 is the preferred resin for post-processing of 68Ga by the ethanol-based method. Significantly, the reduction in Fe3+ and 68Ge content was more than 3-fold greater for the ethanol-based method than for the acetone-based method (3).
The processed 68Ga eluate facilitates high labeling yields and specific activities of 68Ga-DOTATOC because of the increased chemical and radiochemical purity. In contrast to the acetone-based protocol, the purified eluate resulting from the ethanol method is more acidic and of little practical use for radiolabeling after simple dilution. However, through the inclusion of a simple buffer formulation it is possible to produce nearly quantitative yields of 68Ga-DOTATOC within 10 min. Significantly, the labeling efficiency using ethanol-processed 68Ga was better than that using acetone-processed 68Ga at a lower temperature. The rate of radiolabeling was such that it is possible to obtain the radiolabeled product ready for injection 5 min after initial generator elution. Recently, significant increases in radiochemical yields of a trivalent radiometal–ligand complex formation were reported in the presence of ethanol in the aqueous labeling solutions (6). It is likely that the presence of ethanol in the labeling solution is in part responsible, but the differences in labeling pH and volume between the 2 methods should not be ignored.
Certain commercial synthesis modules routinely added 1 vol% ethanol to act as a radiolytic stabilizing agent. With the ethanol-based procedure, the addition of ethanol is already incorporated, thus reducing the number of overall steps. Depending on the volume of the labeling solution used, ethanol makes up between 18 and 45 vol% of the final preparation.
CONCLUSION
An appropriate and efficient protocol for the post-processing of generator-produced 68Ga, based on cation-exchange chromatography with ethanol/hydrochloric acid medium, has been described. Like the acetone-based protocol, the ethanol-based protocol allows for concentration of 68Ga generator eluate with only small losses of eluted 68Ga. Quantitative removal of 68Ge-breakthrough is routinely possible and ensures that the final injectable radiopharmaceutical fulfills legal requirements relating to the 68Ge content of the injectable solution. A significant and improved reduction in the Ti4+ and Fe3+ content was also demonstrated, which promotes the synthesis of radiopharmaceuticals with higher specific activities. Furthermore, the use of ethanol facilitates more efficient radiolabeling and enhances the radiolytic stability of the radiolabeled compound. It has been shown that it is possible to prepare 68Ga-DOTATOC suitable for medical application with a radiochemical yield of more than 99% at a lower temperature than that of the standard acetone-based procedure. The entire post-processing protocol, including generator elution, can be completed within 5 min and may easily be incorporated into commercially available automated modules. The absence of acetone, 68Ge, and unchelated 68Ga in the final formulation using ethanol-based post-processing is an important step toward the kit-type synthesis of 68Ga radiopharmaceuticals.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. No potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Nils Stöbener and Tobias Reich (Institute of Nuclear Chemistry, Johannes Gutenberg University, Mainz, Germany) for liquid chromatography–mass spectrometry measurements.
Footnotes
Published online Apr. 21, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication October 7, 2013.
- Accepted for publication February 18, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}