


Abstract
The gastrin-releasing peptide receptor (GRPr) is overexpressed in prostate cancer and is an attractive target for radionuclide therapy. In addition, inhibition of the protein kinase mammalian target of rapamycin (mTOR) has been shown to sensitize various cancer cells to the effects of radiotherapy. Methods: To determine the effect of treatment with rapamycin and radiotherapy with a novel 177Lu-labeled GRPr antagonist (177Lu-RM2, BAY 1017858) alone and in combination, in vitro and in vivo studies were performed using the human PC-3 prostate cancer cell line. PC-3 cell proliferation and 177Lu-RM2 uptake after treatment with rapamycin were assessed in vitro. To determine the influence of rapamycin on 177Lu-RM2 tumor uptake, in vivo small-animal PET studies with 68Ga-RM2 were performed after treatment with rapamycin. To study the efficacy of 177Lu-RM2 in vivo, mice with subcutaneous PC-3 tumors were treated with 177Lu-RM2 alone or after pretreatment with rapamycin. Results: Stable expression of GRPr was maintained after rapamycin treatment with doses up to 4 mg/kg in vivo. Monotherapy with 177Lu-RM2 at higher doses (72 and 144 MBq) was effective in inducing complete tumor remission in 60% of treated mice. Treatment with 37 MBq of 177Lu-RM2 and rapamycin in combination led to significantly longer survival than with either agent alone. No treatment-related toxicity was observed. Conclusion: Radiotherapy using a 177Lu-labeled GRPr antagonist alone or in combination with rapamycin was efficacious in inhibiting in vivo tumor growth and may be a promising strategy for treatment of prostate cancer.
Prostate cancer continues to be the second leading cause of cancer-related deaths for men in developed countries. Although the 5-y disease-specific survival rate of localized disease treated by surgery or radiotherapy is more than 90%, the effectiveness of systemic therapy for advanced prostate cancer is limited. Currently, depleting or blocking the action of androgens is the standard of care for men with advanced prostate cancer. Androgen deprivation results in a decrease in the concentration of prostate-specific antigen, in tumor regression, and in relief of symptoms in most patients, but the response to treatment is not durable and with time prostate-specific antigen concentrations increase, indicating reactivated androgen-receptor signaling and a transition to a castration-resistant state that is invariably fatal (1).
Docetaxel prolongs survival in the treatment of castration-resistant prostate cancer (2) and is currently the first-line agent for this stage of disease. There is a wide range of toxicities associated with docetaxel given its nonspecific inhibition of microtubule assembly (3,4); therefore, targeted therapies such as protein kinase inhibitors are being evaluated in conjunction with radiotherapy for treatment of advanced prostate cancer (5,6).
When deprived of androgen stimulation, prostate cancer cells develop the ability to survive and thus become androgen-insensitive by upregulating oncogenic pathways in which protein kinases play a crucial role. Phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling is upregulated in 30%–50% of prostate cancers, often through loss of the tumor suppressor gene phosphatase and tensin homolog, and alterations of this pathway are associated with increasing tumor stage, grade, and risk of biochemical recurrence (7,8). Rapamycin, a potent and specific inhibitor of mTOR, inhibits tumor growth, angiogenesis, and metastasis as well as induces apoptosis in cancer cell lines and mouse models of cancer (9–11). mTOR inhibition is also known to confer radiosensitivity on some types of human cancers, such as breast, head and neck, and colon cancer, and induce the nonapoptotic cell death pathway of autophagy for improved destruction of prostate cancer cells (12).
The gastrin-releasing peptide receptor (GRPr) is overexpressed in a variety of human tumors and is present in a high percentage of prostate cancers; thus, radiolabeled peptides specific for this receptor are of potential use for imaging and targeted radionuclide therapy (13–23). Compared with other targeted therapies, targeted radiotherapy has the advantage of cross-fire irradiation, meaning that an individual tumor cell lacking receptor expression may still receive an adequate dose of radiation from neighboring cells with receptor expression. This concept has become especially important as our understanding of human tumor heterogeneity has evolved.
For imaging and targeted radiotherapy of prostate cancer, several GRPr agonists have been developed (14). The 177Lu-labeled GRPr agonist 177Lu-DO3A-CH2CO-G-(4-aminobenzoyl)-QWAVGHLM-NH2 (177Lu-AMBA) (24) underwent phase I clinical trials in patients with metastatic prostate cancer (25). More recently, we developed the DOTA-conjugated GRPr antagonist 4-amino-1-carboxymethyl-piperidine-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 (RM2, BAY 1017858), which can be easily labeled with various radionuclides (26). RM2 and other GRPr antagonists have shown a more favorable biodistribution than GRPr agonists (such as 177Lu-AMBA) for imaging of prostate cancer in preclinical models. Specifically, pancreatic and intestinal retention of the antagonists has been found to be markedly lower than that for GRPr agonists (26,27). On the basis of these imaging and biodistribution data, we hypothesized that RM2 labeled with the β-emitter 177Lu may be an effective tool for therapy of prostate cancer.
Agents that sensitize malignant cells to radiation would amplify tumor response while minimizing toxicity to surrounding organs by lowering effective therapeutic doses, a strategy that has been successfully used in other malignant diseases to improve local control and survival outcomes (28–30). In this study, we investigated the novel GRPr antagonist 177Lu-RM2 (BAY 1017858) alone and in combination with the mTOR inhibitor rapamycin using in vitro and in vivo models of PC-3, an androgen-independent human prostate cancer cell line with known upregulation of the PI3K/Akt/mTOR pathway, with the rationale of sensitizing prostate cancer cells to the effects of radiation by mTOR kinase inhibition.
MATERIALS AND METHODS
Cell Culture and Reagents
The human prostate cancer cell line PC-3 was obtained from American Type Culture Collection and cultured in Dulbecco modified Eagle medium supplemented with vitamins, amino acids, penicillin and streptomycin, and 10% fetal bovine serum in a humidified 5% CO2 atmosphere at 37°C. Rapamycin (Sigma-Aldrich) was obtained in a stock solution of 2.74 mM and diluted in dimethyl sulfoxide (DMSO) for in vitro experiments. RM2 was synthesized and characterized as previously described (26). A 68Ge/68Ga generator was used to produce 68Ga (Eckert & Ziegler). 177LuCl3 was purchased from Isotope Technologies Garching GmbH.
Radiolabeling
68Ga-RM2 was prepared as described by Zhernosekov et al. (31). Briefly, purified 68Ga (250–300 MBq) was used directly for labeling of RM2 (20 μg) in 0.2 M NH4-acetate buffer (2 mL) at pH 4, followed by incubation for 10 min at 95°C. 177Lu-RM2 was prepared by dissolving 10 μg of peptide in 250 μL of sodium acetate buffer (0.4 M, pH 5.0) and by incubating with 177LuCl3 (110–220 MBq; 30–60 μL) for 30 min at 95°C. Quality controls were performed by analytic reversed-phase high-performance liquid chromatography (Knauer) with a photo diode array detector (1200 series; Agilent Technologies) and a flow-through RamonaStar (Raytest GmBH) γ-detector using a Macherey-Nagel Nucleosil 120 C18 column or a Chromolith Merck RP-18e column (eluents: A, 0.1% trifluoroacetic acid in water, and B, acetonitrile; gradient 1: 0–30 min, 95%–30% A, and flow, 0.750 mL/min; gradient 2: 0–3 min, 95%–50% A, and flow, 2.5 mL/min). For injection, the radioligand was diluted in 0.9% NaCl with 0.1% bovine serum albumin.
Proliferation Assay
PC-3 cells were plated at a density of 100,000 per well in 12-well plates and incubated overnight. The following day, rapamycin was added to the wells at concentrations ranging between 0.1 and 10 nM. After 24, 48, and 72 h, viable cells were counted using a trypan blue exclusion assay. Experiments were performed in triplicate.
GRPr Expression After Rapamycin Treatment
PC-3 cells were seeded at a density of 50,000–100,000 per well to account for rapamycin-induced growth inhibition in 12-well plates and incubated overnight. Experiments were performed in triplicate. On the day of the experiment, cells were washed twice with phosphate-buffered saline (PBS), and rapamycin-containing medium (0.1–10 nM) or control medium (medium with DMSO) was added to each well. The plates were incubated at 37°C for 24 and 48 h. At the selected time point, the medium was removed, cells were washed with PBS, and fresh medium containing 177Lu-RM2 (0.8 pmol/well) was added. After incubation at 37°C for 2 h, a modified uptake assay based on previously described methods was performed (16). Briefly, 177Lu-RM2–containing medium was removed, and cells were washed twice with ice-cold PBS. Cells were trypsinized for 5 min at 37°C, after which serum containing medium was added to each well. Half of the contents from each well was used for cell counting, and the other half was used for γ-counting. γ-counting was done on a Cobra 5003 γ-system well counter (Packard Instruments).
In Vitro Therapy
PC-3 cells were seeded at a density of 25,000–100,000 per well depending on the time point in 12-well plates and incubated overnight. Cells were treated with 10 nM rapamycin for 6 h at 37°C, after which the drug-containing medium was removed and was replaced with fresh medium containing 1,850 kBq (50 μCi) of 177Lu-RM2. The cells were returned to the 37°C incubator, and cell viability was assessed by a trypan blue exclusion assay after 24, 48, 72, and 96 h.
In Vivo 68Ga-RM2 Uptake After Rapamycin Treatment
Animals were maintained and treated in compliance with the guidelines of the German Law for the use of living animals in scientific studies. Ten female athymic nude mice (age, 6–7 wk; weight, 16–18 g) (Harlan) with subcutaneously implanted PC-3 tumors (106 cells/mouse) were randomly divided into 2 groups 13 d after implantation and given intraperitoneal injections of rapamycin (4 mg/kg) in DMSO daily for 3 d or PBS (untreated). Tumor size at the start of treatment averaged 7 × 7 mm. After treatment, 0.5 MBq (0.1 nmol) of 68Ga-RM2 was injected via lateral tail vein cannulation. Animals were anesthetized with 1.5% isoflurane, and 10-min static scans were acquired at 1 h after injection using a microPET focus 120 scanner (Siemens Preclinical Solutions). PET images were reconstructed with an ordered-subset expectation maximization algorithm provided by the manufacturer. Image counts per pixel per second were calibrated to activity concentrations (Bq/mL) by measuring a 3.5-cm cylinder phantom filled with a known concentration of radioactivity. Images were generated using AMIDE software. The color scale was set from 0% to 5% to allow for qualitative comparison among the images. Biodistribution studies were performed after completion of small-animal PET scanning. Samples from the tumor, muscle, and liver were collected for each mouse, rinsed of excess blood, blotted dry, weighed, and measured in a γ-counter (Cobra 5003; Packard Instruments). Results were expressed as percentage of injected activity per gram of tissue (%IA/g). A separate group of 6 mice were treated as described above, and GRPr expression was analyzed via autoradiography with [125I-Tyr4]-bombesin (18) on sections of snap-frozen tumor tissue. Results were reported in disintegrations per minute.
177Lu-RM2 Biodistribution and Tumor Radiation Dose
To study the biodistribution of 177Lu-RM2, groups of 5 athymic nude mice bearing PC-3 tumors were sacrificed at 4 and 72 h after injection of 0.185, 0.925, 1.85, and 4.63 MBq of 177Lu-RM2 (10, 50, 100, and 250 pmol). Organs were collected as described above, and results were expressed as %IA/g. To estimate the tumor radiation dose, groups of 3–7 animals were sacrificed at 1, 4, 24, 48, and 72 h after injection of 0.185 MBq of 177Lu-RM2 (10 pmol). The radioactivity concentration of RM2 was measured as %IA/g of tumor tissue (without correction for radioactive decay). The area under the tumor time–activity curve was calculated by the trapezoid rule. After the last measured time point, the curve was extrapolated to infinity by assuming that the activity concentration decreases by physical decay only. The SD of the time–activity curve was calculated as described by Yuan (32). Because of the short range of the β-particles emitted by 177Lu, the tumor dose was estimated by multiplying the number of decay events within the tumor tissue with the mean β-energy of 177Lu and dividing by the tumor mass (i.e., it was assumed that all β-particles are absorbed within the tumor tissue).
177Lu-RM2 Therapy In Vivo
To assess the therapeutic effect and associated toxicities of 177Lu-RM2, a fractionated dosing scheme was performed in 25 female athymic nude mice subcutaneously implanted with PC-3 tumors (106 cells/mouse). Thirteen days after implantation, mice were randomly divided into 5 groups. Tumor size at the start of treatment averaged 7 × 7 mm. Three of the groups (groups 1–3) received 3 injections of 177Lu-RM2 per week (days 0, 2, and 4), and the procedure was repeated (days 14, 16, and 18) after a drug-free week. The total activity injected corresponded to 36, 72, and 144 MBq given in doses of 6, 12, and 24 MBq (100, 200, and 400 pmol) for groups 1, 2, and 3, respectively. The fourth group received cold peptide (200 pmol) labeled with the nonradioactive isotope 175Lu (175Lu-RM2), and the fifth group received PBS (untreated). The mice were monitored 3 times per week by measuring tumor size, appearance, and body mass for up to 150 d. Animals with loss of more than 15% of their original mass or with a maximum tumor diameter of greater than 20 mm were sacrificed. Tumor size was measured with calipers in 2 dimensions, and tumor volume was calculated assuming an elliptical shape. Tumor, kidneys, and pancreas were prepared for histologic evaluation.
In Vivo Combination Therapy with 177Lu-RM2 and Rapamycin
The effect of combination therapy on tumor growth was evaluated in vivo using female athymic nude mice with subcutaneous PC-3 xenografts treated with 37 MBq (300 pmol) of 177Lu-RM2 alone or rapamycin daily (4 mg/kg) for 72 h, followed by 37 MBq of 177Lu-RM2. Control groups consisted of animals receiving PBS (untreated) or rapamycin (4 mg/kg) daily for 72 h. Tumor size at the start of treatment averaged 7 × 7 mm. Animals were monitored daily throughout the course of therapy as described above for up to 160 d. Organs (tumor, kidney, pancreas, and liver) were collected for histology. The experiment was performed twice, first with 5–6 animals per group then repeated with 7 animals per group.
Histology
Selected mouse organs were dissected from the sacrificed animals and immediately fixed in 10% neutral buffered formalin at 4°C. After fixation and routine dehydration, all tissue samples were embedded in paraffin, and 4- to 6-μm-thick sections were stained with hematoxylin and eosin for microscopic examination using standard protocols. Macroscopic and microscopic findings were assessed in terms of affected number of organs and severity.
Statistical Analysis
All in vitro studies were performed in triplicate. Quantitative data were compared by an unpaired 2-tailed t test or 1-way ANOVA. A log-rank test was used to test differences in survival for different groups of animals. All statistical tests were performed with GraphPad Prism (GraphPad Software Inc.).
RESULTS
177Lu-RM2: Favorable In Vivo Biodistribution Profile
RM2 was synthesized using solid-phase peptide synthesis (Fmoc chemistry). The 177Lu-RM2 conjugate was obtained with greater than 95% radiolabeling yield at a maximum specific activity of 37 GBq/μmol. 68Ga-RM2 was obtained with a specific activity of 25 GBq/μmol and a greater than 95% radiolabeling yield.
Biodistribution data demonstrate favorable tumor–to–normal tissue ratios at 4 and 24 h. Data from mice injected with 4.63 MBq of 177Lu-RM2 (250 pmol) are shown in Table 1. No significant difference in radiopeptide biodistribution was seen among mice injected with 10, 50, 100, or 250 pmol of 177Lu-RM2 (other datasets not shown). Initial uptake of bone marrow and liver was relatively low at 4 h and further decreased by 24 h, whereas robust tumor uptake of 177Lu-RM2 was seen at both times. Renal uptake decreased by approximately 44% over a 24-h period. Supplemental Figure 1 (supplemental materials are available online only at http://jnm.snmjournals.org) shows the tumor time–activity curves for 177Lu-RM2 in tumor-bearing mice. Clearance of activity from the tumor approximates physical decay at 72 h after injection. The total (measured plus extrapolated) area under the curve was 1.42 × 103 %IA/g per h (SD, 5.00 × 103 %IA/g per h). The estimated tumor dose was 0.84 Gy/MBq.
Biodistribution Results and Tumor–to–Normal Tissue Ratios of 177Lu-RM2 in Nude Mice Bearing PC-3 Tumors
177Lu-RM2 Therapy as Effective Single Agent in Treating PC-3 Tumors In Vivo
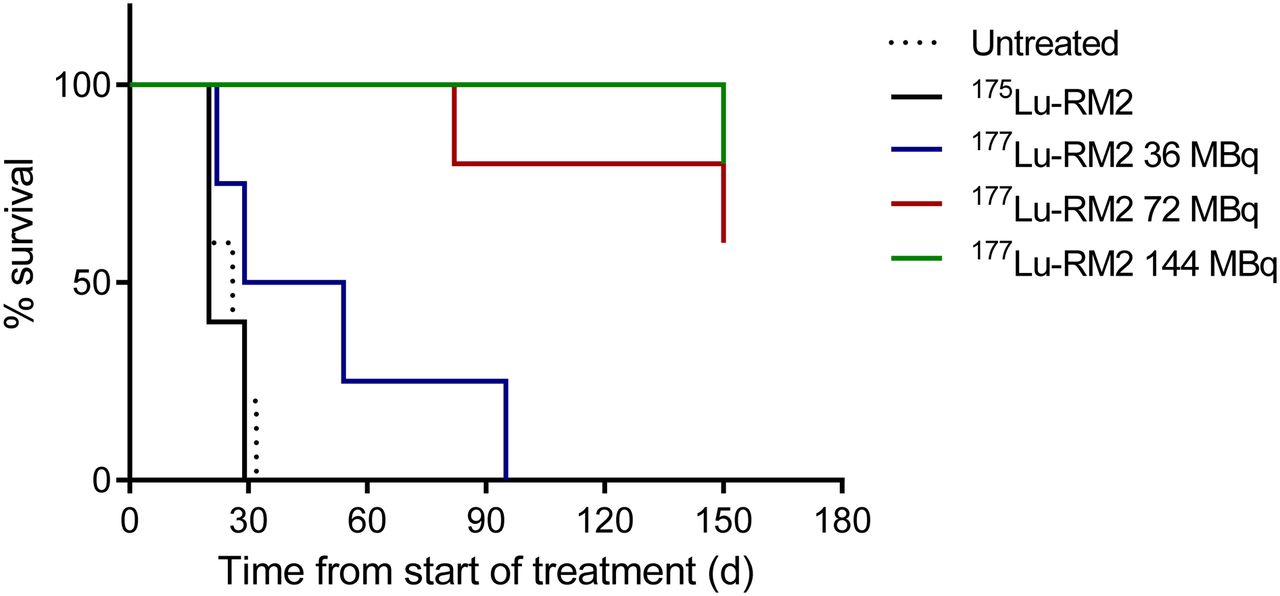
PC-3 tumor growth was rapid in control groups (untreated animals and animals receiving the cold peptide 175Lu-RM2), and all mice were sacrificed within 30 d after the start of therapy (Fig. 1). All animals treated with higher doses (a total of 72 or 144 MBq of 177Lu-RM2) initially demonstrated partial reduction in tumor size during the first 50 d. Roughly 2 mo after treatment, 4 of 10 animals (40%) experienced rapid recurrence of the tumor and were sacrificed. The remaining 6 mice (60%) showed complete tumor remission and were sacrificed at the end of the study after 150 d. Mice treated with a total of 36 MBq showed an intermediate response, with a median survival of 42 d before sacrifice because of the large tumor size.
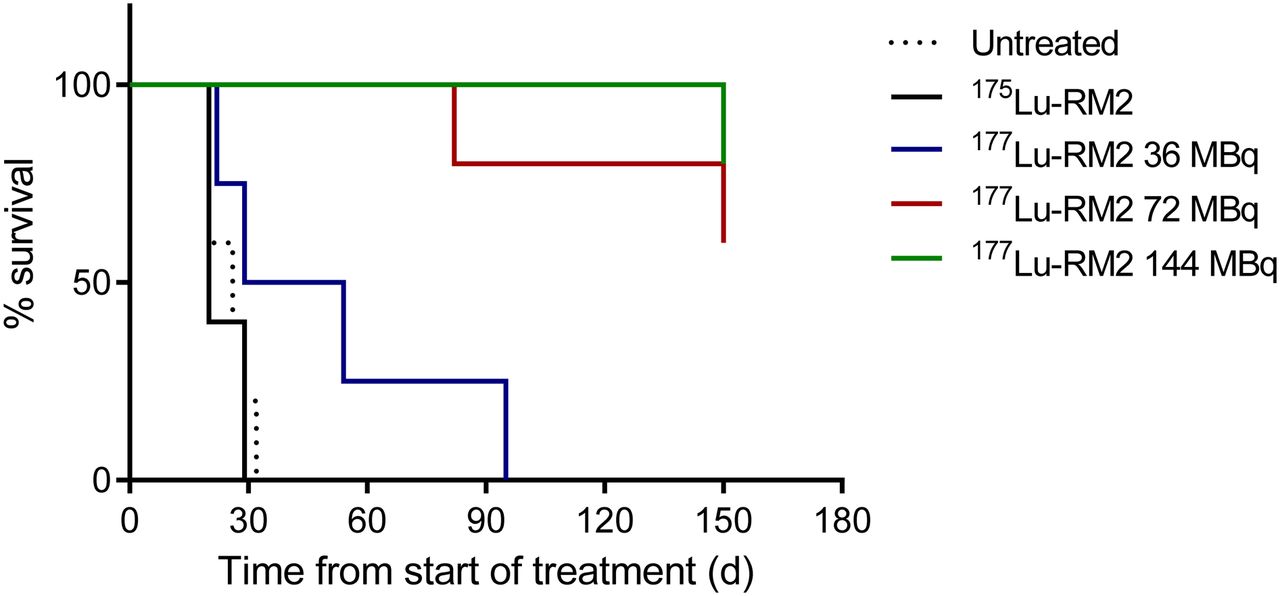
Survival curves of untreated and treated mice.
Effect of Rapamycin Treatment on GRPr Expression In Vitro and In Vivo
177Lu-RM2 uptake was preserved after treatment with rapamycin (cellular uptake 48 h after start of treatment: 104%, 95%, and 120% of untreated cells with 0.1 nM rapamycin, 1 nM rapamycin, and 10 nM rapamycin, respectively) (Fig. 2 A). After rapamycin treatment, a dose-dependent reduction in PC-3 proliferation was seen (viable cells 72 h after start of treatment: 10.00 × 105, 8.58 × 105, 6.75 × 105, and 4.84 × 105 in untreated, 0.1 nM rapamycin, 1 nM rapamycin, and 10 nM rapamycin, respectively) (Supplemental Fig. 2). Treatment with rapamycin did not significantly affect the in vivo biodistribution of 68Ga-RM2 as shown by PET imaging and ex vivo tissue sampling (Figs. 2B and 2C). In fact, there was a trend for a higher tumor uptake of 68Ga-RM2 in rapamycin-treated animals (6.21 ± 0.74 %IA/g vs. 4.87 ± 0.69 %IA/g, P = 0.04) (Fig. 2C). Ex vivo autoradiography confirmed that expression of GRPr does not decrease after treatment with rapamycin (Fig. 2D).
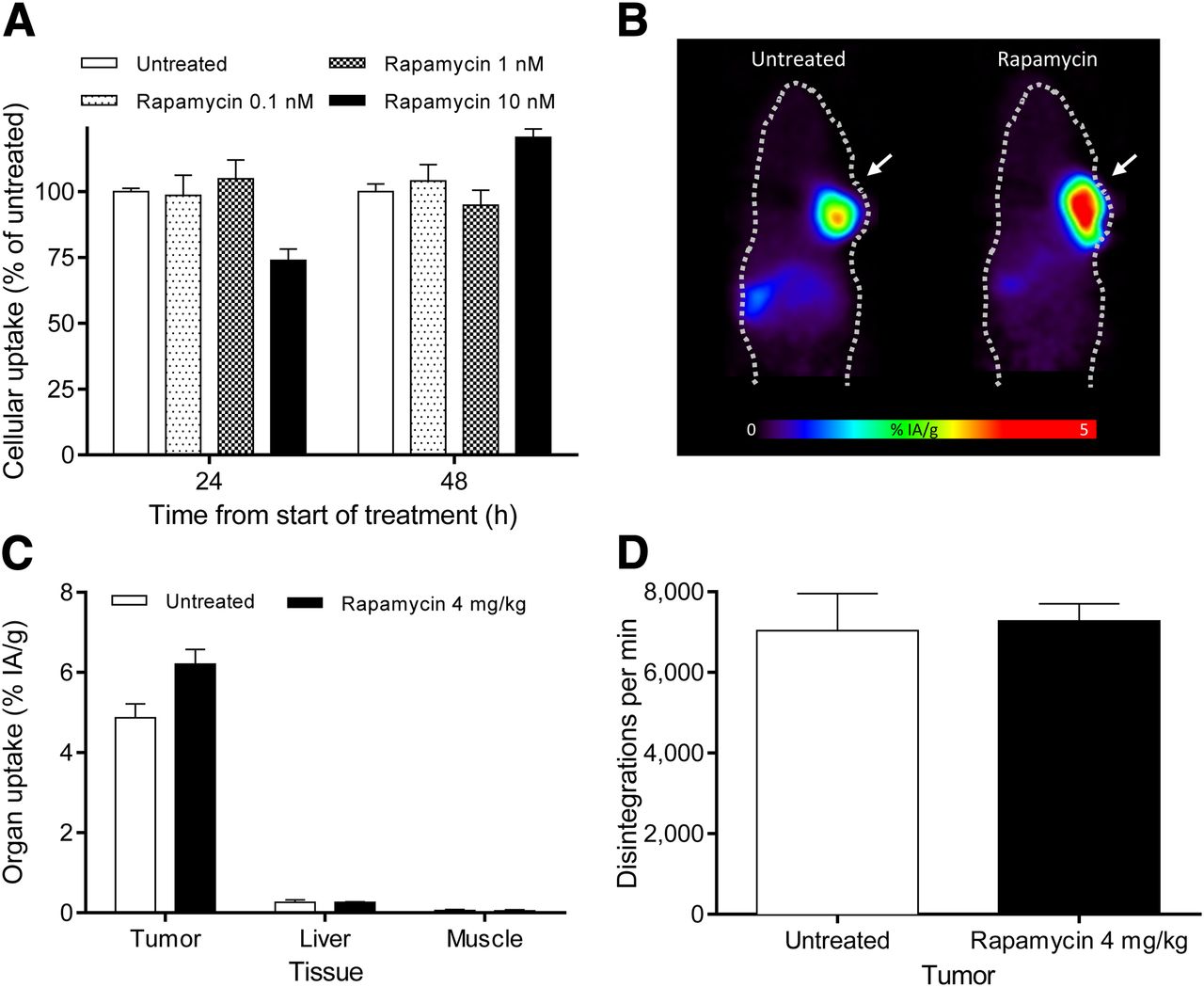
(A) In vitro 177Lu-RM2 uptake assay after rapamycin treatment. (B) 68Ga-RM2 small-animal PET scans of PC-3 tumor–bearing mice (arrows) with and without 72 h of treatment with rapamycin. Biodistribution of tumor, muscle, and liver (C) and autoradiography of tumor samples with 125I-bombesin (D) from untreated and rapamycin-treated animals.
Improved Efficacy of Combination Therapy with 177Lu-RM2 and Rapamycin In Vitro and In Vivo
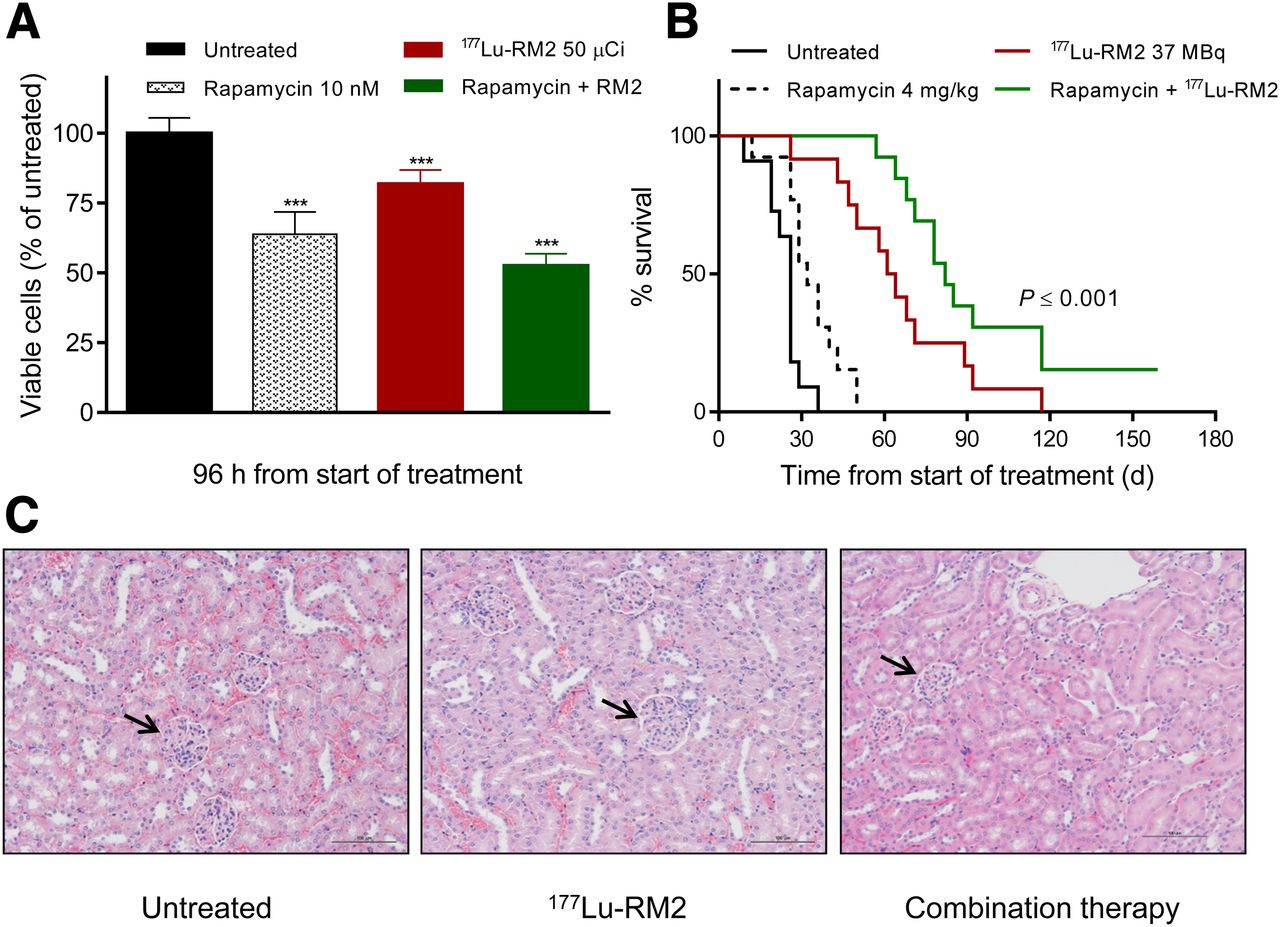
Rapamycin at 10 nM had the greatest cytostatic effect in PC-3 cells without causing impairment of 177Lu-RM2 uptake (Supplemental Fig. 2); thus, this dose was chosen for in vitro therapy studies. Combination treatment was more effective than either agent alone (cell viability after 96 h: 64%, 82%, and 53% of untreated cells with 10 nM rapamycin, 1,850 kBq [50 μCi] of 177Lu-RM2, and combination therapy; P < 0.0001) (Fig. 3A).

(A) In vitro therapy study evaluating effect of combination therapy in PC-3 cells. (B) Survival curves of untreated mice and mice treated with rapamycin alone, 177Lu-RM2 alone, or combination therapy. (C) Representative hematoxylin and eosin sections from kidneys of untreated mouse, mouse treated with 144 MBq of 177Lu-RM2, and mouse from combination-therapy group. Arrows demonstrate intact nephrons. Samples from both treated mice were obtained at conclusion of respective studies. Control sample was obtained 3 wk after start of treatment. ***P < 0.0001.
Combined treatment with rapamycin and 177Lu-RM2 resulted in the most pronounced inhibition of tumor growth and the longest survival (Fig. 3B; Supplemental Fig. 3). Median survival was 26 d for untreated mice and 32, 62, and 82 d for mice treated with rapamycin alone, with 177Lu-RM2 alone, and with combination therapy, respectively (P < 0.0001, log-rank test). Tumor regression was observed for all animals receiving combination therapy, and complete remission was observed in 5 of 13 animals from this group during the 160-d follow-up period. In contrast, tumors progressed rapidly in the untreated and rapamycin-alone groups, and all animals were sacrificed by day 50 of the study (Supplemental Fig. 3). Tumors in animals treated with 37 MBq of 177Lu-RM2 monotherapy grew more slowly than those treated with rapamycin alone; however, tumors treated with monotherapy progressed more rapidly than tumors in the combination therapy group.
There was no evidence of treatment-related toxicity as evaluated by animal appearance, by body weight loss, or on histologic examination of harvested organs for any of the 177Lu-RM2 doses used in this study. Animals also tolerated the 4-d course of rapamycin without side effects as evaluated by animal appearance and body weight. Two animals in the combination-therapy group were sacrificed because of intestinal problems, one at day 57 and one at day 82. Given the relatively long delay between drug administration and radiotherapy and onset of symptoms, it is unclear if this represented true treatment-related toxicity. No abnormalities were found on histologic examination of organs collected from animals in the monotherapy and combination-therapy studies (Fig. 3C).
DISCUSSION
Radiopeptide therapy with the somatostatin analogs 90Y-DOTATOC and 177Lu-DOTATATE is frequently used in Europe for the treatment of neuroendocrine tumors. Developed and introduced into clinical practice in 1997, the clinical benefit of this therapy has been documented in several studies. However, despite their successes, the applicability of somatostatin-based radiopeptides remains limited to malignancies expressing the somatostatin receptor, which accounts for a relatively small percentage of all human tumors. In creating the GRP analog RM2, we aimed to develop a peptide-based platform by which to deliver targeted radionuclide therapy to GRPr-expressing tumors, among which is prostate cancer, one of the most common malignancies in men.
Here, we show preclinical evidence that the GRPr antagonist 177Lu-RM2 can induce remission in tumor-bearing mice in a safe and effective manner. The targeted and specific nature of radiopeptide therapy lends itself to a favorable side-effect profile. In our study, mice tolerated doses of 177Lu-RM2 up to a total of 144 MBq without evidence of adverse effects. The GRPr is expressed in normal pancreatic tissue with resultant uptake of bombesin-based radiotracers; however, washout of 177Lu-RM2 from the pancreas occurs after injection and by 24 h approximates uptake seen in tissues such as the blood pool and muscle. This decrease in tracer retention likely explains the absence of pancreatic damage in the present study.
The most common and concerning side effect seen with radiopeptide therapy is renal toxicity; however, there was no observed damage to the nephrons or renal parenchyma on histologic evaluation of treated mice. Previously, we have reported the favorable biodistribution of 111In- and 68Ga-labeled RM2 conjugates, whereby renal excretion led to moderate renal uptake initially with substantial washout by 2 h. Favorable tumor–to–background tissue (blood, kidney, liver, and muscle) ratios were observed at all times ranging from 20 min to 72 h, and bone marrow uptake was low across all evaluated times (26). We saw similar biodistribution results with the 177Lu-labeled RM2 conjugate, which showed favorable tumor–to–background tissue ratios at both 4 and 24 h after injection. Uptake of 177Lu-RM2 is maintained in prostate cancer xenografts at 24 h with an estimated tumor dose of 0.84 Gy/MBq. Fractionated dosing of 72 or 144 MBq of 177Lu-RM2 resulted in complete tumor remission in 60% of animals and prolonged survival in the remaining 40%. Minimal differences were observed in response and survival between animals treated with a total of 72 or 144 MBq, which may be due to a nonstochastic effect of the radiation doses on cell death.
Previous work on radiopeptide therapy of prostate cancer has been based on GRPr agonists. Lantry et al. described the efficacy of bombesin agonist 177Lu-AMBA in slowing tumor progression in PC-3 tumor–bearing mice with mild kidney toxicity, work that led to the initiation of a phase I clinical trial of 177Lu-AMBA in patients with prostate cancer (24). The bombesin agonist 177Lu-DOTA-8-AOC-BBN(7-14)NH2 was evaluated in combination with the chemotherapeutic agents docetaxel and estramustine by Johnson et al. (33). The animals treated with combination therapy survived significantly longer than those treated with either radiopeptide or chemotherapeutic agent alone. Although these results are encouraging, the long uptake and retention of GRPr agonists by the pancreas and intestines is a significant concern for the use of this form of radiopeptide therapy in humans. In addition, therapeutic doses of radiolabeled GRPr agonists have been found to cause significant acute side effects such as nausea and vomiting, most likely due to stimulation of GRPrs in the gastrointestinal tract and subsequent release of intestinal hormones (25).
Previous work has shown that GRPr expression is highest in well-differentiated prostate carcinoma, with one series demonstrating GRPr expression in 30 of 30 locally invasive human prostate cancers. Poorly differentiated and metastatic prostate cancers can show reduced GRPr expression or lose expression altogether (18). Before therapy with 177Lu-RM2 in humans, a pretherapeutic scan with 68Ga-RM2 would be required to demonstrate adequate receptor expression. This method is currently used in patients receiving other forms of radiopeptide therapy (e.g., DOTATOC or DOTATATE).
In the present study, the effect of 177Lu-RM2 on inhibiting or preventing tumor progression was enhanced when combined with the mTOR inhibitor rapamycin. After mice were pretreated with rapamycin, we saw improved efficacy with 37 MBq of 177Lu-RM2, a dose that was modestly effective alone. Because of the inhibition of tumor growth, mice lived significantly longer in the combination-therapy group than in the radiopeptide-alone group. Overall, rapamycin was well tolerated in the study alone and in combination with 177Lu-RM2 at a dose of 4 mg/kg. These findings suggest that the amount of radioactivity needed for effective radiopeptide therapy, and the risk for radiation-induced damage to normal organs, can be decreased by combining radiopeptide therapy with mTOR inhibitors.
mTOR inhibitors are routinely used in the clinic to prevent transplant rejection, and several agents are currently approved for the treatment of various malignancies. Everolimus was approved by the Food and Drug Administration in 2009 for treatment of refractory renal cell carcinoma and received approval earlier this year for use in patients with progressive pancreatic tumors of neuroendocrine origin. Generally, these medications are considered safe, with side effects that are limited and clinically manageable. There is a strong rationale for mTOR inhibition in advanced prostate cancer given the high prevalence of activation of the PI3K/AKT pathway due largely to the loss of expression–function of the tumor suppressor phosphatase and tensin homolog and the association of this pathway with adverse pathologic features, recurrence after radical prostatectomy, and systemic treatment resistance (7,8,34).
Although the exact mechanism underlying radiosensitization of rapamycin-treated tumors is unknown, mTOR inhibition is associated with induction of autophagy in irradiated cells (12). Furthermore, inhibition of the PI3K/AKT/mTOR pathway has been found to arrest cancer cells in the G2 M phase of the cell cycle (35,36). Given that cells are often most radiosensitive in the G2 M phase, the cell cycle regulatory effects may contribute to the enhanced antitumor effects that are seen when mTOR inhibitors are combined with ionizing radiation by synchronizing cancer cells to the stage of growth in which they are most likely to experience irreparable damage.
Further work to elucidate the synergistic interaction between 177Lu-RM2 and mTOR inhibition in the treatment of prostate cancer is necessary, as are experiments to determine ideal dosing schemes and therapy regimens. On the basis of evidence suggesting mTOR inhibition sensitizes cells to ionizing radiation, we chose to pursue a pretreatment strategy with rapamycin because we felt that this was the most straightforward method to evaluate whether rapamycin before 177Lu-RM2 treatment would have an additive effect. However, alternative methods such as concurrent mTOR inhibitor or radiopeptide therapy or mTOR inhibitor treatment with fractionated radiopeptide dosing should be evaluated. Furthermore, a plethora of preclinical data have emerged indicating that treatment with various protein kinase inhibitors confers radiosensitivity on prostate cancer models, including those that target Akt, sphingosine kinase, aurora kinase A and B, and Wee1 (37–41). Most of these studies used external-beam radiation; however, 1 group reported additive effects when a 177Lu-labeled anti-Lewis Y monoclonal antibody was combined with an epidermal growth factor receptor kinase inhibitor in an in vivo prostate cancer model (42).
CONCLUSION
We demonstrated that the GRPr antagonist 177Lu-RM2 is suitable for targeted radiotherapy of prostate cancer. Because of its high tumor uptake and rapid clearance from normal organs, 177Lu-RM2 was effective in treating PC-3 xenografts without significant side effects. The efficacy of 177Lu-RM2 therapy was further increased by pretreatment with rapamycin. These data warrant further research investigating radiolabeled GRPr antagonists alone and in combination with other molecularly targeted therapies in the treatment of prostate cancer.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported in part by Bayer Pharma AG, European Community for the TARCC (no. HEALTH-F2-2007-201962) Project within the 7th Framework project, the Swiss National Science Foundation (320000-11833), the Deutsche Forschungsgemeinschaft (DFG) SFB 850, and COST BM0607. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Dr. Melpomeni Fani, Yvonne Kiefer, and Roswitha Tönnesmann for assistance with animal experiments and radiopeptide labeling. We thank Joerdis Luebke and Dr. Michael Mix for support with the small-animal PET.
Footnotes
Published online Mar. 14, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 1, 2012.
- Accepted for publication November 26, 2012.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- SMER28 attenuates PI3K/mTOR signaling by direct inhibition of PI3K p110 delta
- Mechanisms of Resistance to Prostate-Specific Membrane Antigen-Targeted Radioligand Therapy in a Mouse Model of Prostate Cancer
- New Developments in Peptide Receptor Radionuclide Therapy
- Expression of Gastrin-Releasing Peptide Receptor in Breast Cancer and Its Association with Pathologic, Biologic, and Clinical Parameters: A Study of 1,432 Primary Tumors
- Molecular Imaging and Therapy with a Purpose: A Renaissance of Nuclear Medicine
- Radiopeptides for Imaging and Therapy: A Radiant Future
- In Vitro and In Vivo Application of Radiolabeled Gastrin-Releasing Peptide Receptor Ligands in Breast Cancer
- Cerenkov Luminescence Imaging for Radiation Dose Calculation of a 90Y-Labeled Gastrin-Releasing Peptide Receptor Antagonist