


Visual Abstract

Abstract
Prostate-specific membrane antigen (PSMA)–targeted radioligand therapy (RLT) is effective against prostate cancer (PCa), but all patients relapse eventually. Poor understanding of the underlying resistance mechanisms represents a key barrier to development of more effective RLT. We investigate the proteome and phosphoproteome in a mouse model of PCa to identify signaling adaptations triggered by PSMA RLT. Methods: Therapeutic efficacy of PSMA RLT was assessed by tumor volume measurements, time to progression, and survival in C4-2 or C4-2 TP53−/− tumor-bearing nonobese diabetic scid γ-mice. Two days after RLT, the proteome and phosphoproteome were analyzed by mass spectrometry. Results: PSMA RLT significantly improved disease control in a dose-dependent manner. Proteome and phosphoproteome datasets revealed activation of genotoxic stress response pathways, including deregulation of DNA damage/replication stress response, TP53, androgen receptor, phosphatidylinositol-3-kinase/AKT, and MYC signaling. C4-2 TP53−/− tumors were less sensitive to PSMA RLT than were parental counterparts, supporting a role for TP53 in mediating RLT responsiveness. Conclusion: We identified signaling alterations that may mediate resistance to PSMA RLT in a PCa mouse model. Our data enable the development of rational synergistic RLT-combination therapies to improve outcomes for PCa patients.
Prostate-specific membrane antigen (PSMA)–targeted radioligand therapy (RLT) with the β-particle emitter 177Lu (e.g., [177Lu]Lu-PSMA-617) yields responses (>50% prostate specific antigen decline) in 57%–66% of prostate cancer (PCa) patients; however, remissions are short-lived (1). α-particle emitters (e.g., 225Ac) may be superior to [177Lu]Lu-PSMA RLT because of their higher-energy radiation delivery per injected activity and increased density of ionizations responsible for DNA damage (2). Responses to [225Ac]Ac-PSMA RLT have been reported in 63%–76% of PCa patients (2). However, neither [225Ac]Ac- nor [177Lu]Lu-PSMA RLT is curative, and relapse occurs invariably.
Ionizing radiation, as delivered by RLT, induces DNA damage. This, in turn, engages the DNA damage response/replication stress response (DDR/RSR) pathway, which is a critical compensatory mechanism to cytotoxic stress in tumor cells and initiates either DNA repair or triggers cell death (3). This pathway consists of an extensive signaling cascade coordinated by the serine threonine kinases ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia-and-rad3-related protein (ATR) and their downstream effectors checkpoint kinases CHEK2, CHEK1, and WEE1. A similarly important mediator of DDR/RSR-induced cell cycle arrest is TP53, which induces the cyclin-dependent kinase inhibitor p21 after cytotoxic stress.
Defects in DDR/RSR genes are common in cancer and increase the reliance on parallel DNA repair pathways (such as ATR, ATM, and TP53 signaling) for survival after genotoxic stress (4). In PCa, TP53 (43% of metastatic PCa patients) together with androgen receptor (AR; 57%), phosphatase and tensin homolog (PTEN; 35%), ETS (e.g., TMPRSS2:ERG fusion, ∼30%), and MYC (21%) are among the most frequently mutated genes (5). TP53 loss of function has been linked to radioresistance and tumor cell survival (6). Inactivating mutations in ATM, ATR, and BRCA1/2 (which are known ATR substrates) have been linked to PCa. The mutation rates of BRCA1/2 (15% combined), ATR (9%), and ATM (7%) in PCa patients are significantly higher in lethal than in localized disease and are associated with earlier age at death and shorter survival (5,7). Although recent clinical data have associated mutations in DDR genes with response (8) or resistance (9) to α-particle therapy, the roles of DDR/RSR and TP53 pathways in modulating responses to RLT in PCa have not been investigated systematically.
Here, we investigate the biologic responses of a PCa mouse model to PSMA RLT with the goal of identifying actionable mechanisms potentially underlying resistance to PSMA RLT in PCa. We profiled RLT-induced proteomic and phosphoproteomic changes in tumors to elucidate compensatory cellular stress responses and identify potential liabilities that could be exploited by novel RLT-based combination therapies. Lastly, we demonstrated that TP53 loss of function decreases sensitivity to PSMA RLT in our PCa model.
MATERIALS AND METHODS
Cell Culture
C4-2 cells were provided by Dr. Georg N. Thalmann (Department of Urology, Inselspital Bern). C4-2 TP53−/− cells were generated by Crispr/Cas9 knockout of tp53 using a tp53 guide RNA (GTGTAATAGCTCCTGCATGG (10)) in the lentiCRISPRv2 backbone (plasmid 52961, addgene; the full targeting vector was a gift of Dr. David Nathanson, UCLA) and validated as shown in Supplemental Figure 1 (supplemental materials are available at http://jnm.snmjournals.org). Cells were maintained in Rosewell Park Memorial Institute 1640 medium/10% fetal bovine serum at 37°C and 5% CO2, monitored for Mycoplasma contamination using the Venor GeM Mycoplasma detection kit (Sigma Aldrich), and authenticated by short tandem repeat sequencing (August 2019; Laragen).
Therapy Studies
The UCLA Animal Research Committee approved all animal studies (approval 2005-090). Male 6- to 8-wk-old nonobese diabetic scid γ-mice (UCLA Radiation Oncology Animal Core) were housed under pathogen-free conditions with food and water ad libitum and a 12-h/12-h light/dark cycle. The mice were observed daily to ensure animal welfare and determine whether humane endpoints were reached (e.g., a decreasing body condition score, including severe weight loss, a hunched and ruffled appearance, apathy, ulceration, tumor burden impeding with normal movement, or tumor volume ≥ 3 cm3).
In vivo PSMA expression was verified by [68Ga]Ga-11 PET/CT (Supplemental Figs. 1C and 2) (11).
To optimize therapeutic activity of [225Ac]Ac- and [177Lu]Lu-PSMA RLT in mice with subcutaneous C4-2 tumors (5 × 106 cells, 100% Matrigel; Corning), the mice were randomized on the basis of tumor volume into 2 studies. Mice in study 1 either were untreated or received 30 or 120 MBq of [177Lu]Lu-PSMA-617 intravenously (11) (6 mice per group), whereas mice in study 2 either were untreated or received 20, 40, or 100 kBq of [225Ac]Ac-PSMA-617 intravenously (12) (8 mice per group). Therapeutic efficacy was assessed by caliper (volume = ½[length × width2]; study 1) or CT (study 2), time to progression to half-maximal tumor volume (TTP), and survival.
To investigate the impact of TP53 status on RLT efficacy, mice bearing subcutaneous C4-2 or C4-2 TP53−/− tumors were randomized into vehicle (0.9% saline) or [177Lu]Lu-PSMA-617 (15 MBq, intravenously) groups (8–10 mice per group). Therapeutic efficacy was assessed by CT, TTP, and survival.
Radiochemical Synthesis
PSMA-617 precursor (ABX GmbH) was stored in aliquots (1 mg/mL) in 0.1% aqueous trifluoroacetic acid until use. No-carrier-added 177LuCl3 was obtained from Spectron MRC. 225Ac was supplied by the Isotope Program within the Office of Nuclear Physics in the Department of Energy’s Office of Science. Radiolabeling was performed at the UCLA Biomedical Cyclotron Facility as previously described, resulting in molar activities of 84 GBq/μmol and 130 MBq/μmol for [177Lu]Lu-PSMA-617 and [225Ac]Ac-PSMA-617, respectively (13).
Immunoblot
TP53 (catalog no. 2527; all antibodies were obtained from Cell Signaling), phospho-Chk2 (Thr68; catalog no. 2197), p21 (catalog no. 2947), phospho-Histone H2A.X (Ser139; catalog no. 9718), and β-actin (catalog no. 3700) were detected as previously published (14).
PSMA Expression and Radiosensitivity
PSMA expression was flow-cytometrically quantified using an anti–human PSMA-APC antibody; the in vitro radiosensitivity of tumor cells was assessed by propidium iodide staining (flow cytometry) and as days to confluence after irradiation (13).
Mass Spectrometry (MS)
Tandem mass tagging and normal-phase liquid chromatography–MS/MS were used to quantify the total proteome and phosphoproteome of C4-2 tumors 48 h after treatment with 40 kBq of [225Ac]Ac-PSMA-617 or 120 MBq of [177Lu]Lu-PSMA-617 (3–5 tumors per group). MS data were processed using Elucidata’s Polly software packages. The complete proteomic and phosphoproteomic datasets can be accessed on MassIVE (University of California, San Diego [https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp]; identifier MSV000086408). A detailed description is provided in the supplemental materials.
Statistics
Statistical analysis of data was performed using GraphPad Prism, version 8. Statistical significance was set to a P value of 0.05 or less. The log-rank (Mantel–Cox) test was used for survival and TTP analyses. Therapeutic efficacy data were analyzed with 1-way ANOVA with Bonferroni adjustment. For analysis of MS data, differential expression events were defined by identifying proteins or phosphopeptides with between-treatment variance significantly larger than within-replicate variance using 1-way ANOVA. Significantly altered proteins or phosphopeptides were filtered using the Benjamini–Hochberg procedure at a 5% false-discovery rate. All statistical analyses of proteomic or phosphoproteomic MS data were performed using Python. Nucleotide pool measurements in RLT versus control tumors were compared with 2-tailed unpaired t tests.
RESULTS
Optimizing PSMA RLT in the C4-2 PCa Model
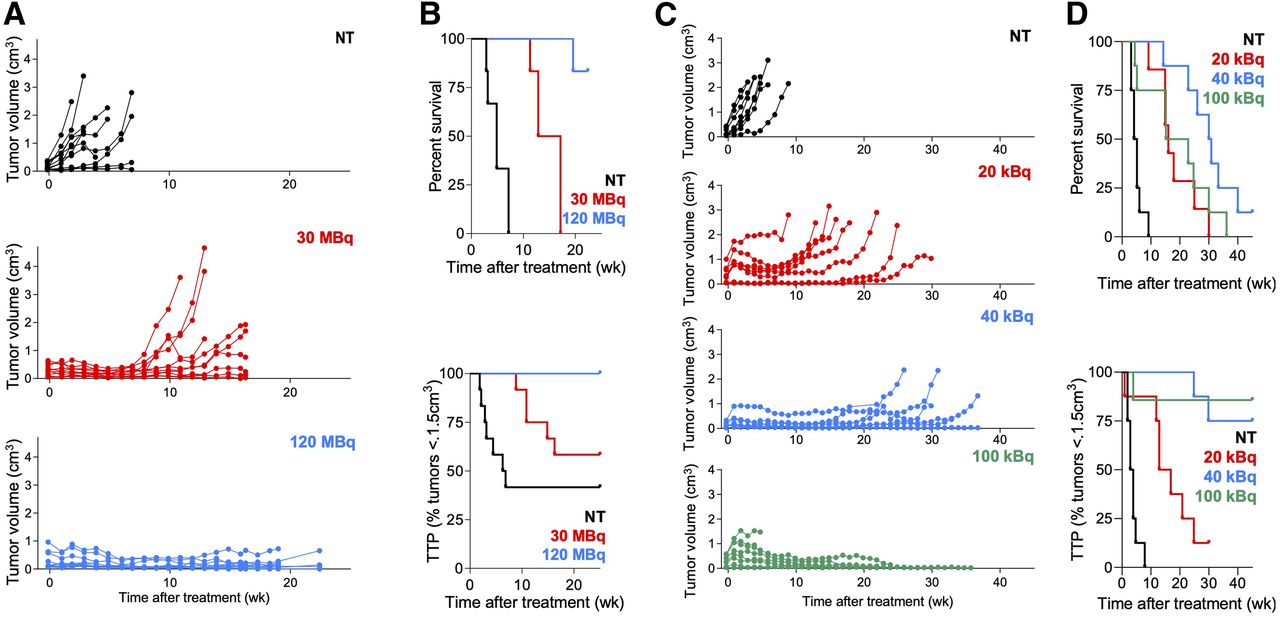
To identify the treatment activity resulting in the best antitumor effects without toxicity, mice were treated with varying activities of [225Ac]Ac- or [177Lu]Lu-PSMA RLT. PSMA RLT induced significant, dose-dependent tumor shrinkage and increased TTP and survival (Fig. 1). [225Ac]Ac-PSMA RLT with 100 kBq achieved the best tumor control, but the mice experienced toxicity as evidenced by a deteriorating condition leading to a humane endpoint, explaining their shorter survival. No severe weight loss was observed after [177Lu]Lu-PSMA RLT (Supplemental Fig. 3). Comparing the efficacy of 40 kBq of [225Ac]Ac-PSMA RLT versus 30 MBq of [177Lu]Lu-PSMA RLT, we found that [225Ac]Ac-PSMA RLT resulted in longer survival (P = 0.0019) but not TTP (P = 0.147) (treatment activities of 30 MBq and 40 kBq were chosen for comparison because we have observed them to be most comparable in terms of energy deposition and tumor dose (Catherine Meyer, unpublished data, March 2019]).

Optimizing treatment activities for [225Ac]Ac- and [177Lu]Lu-PSMA RLT. (A) Individual tumor growth curves after [177Lu]Lu-PSMA (12 tumors, 6 mice per group; NT vs. RLT, P ≤ 0.0018 [7 wk]; 30 vs. 120 MBq, P > 0.99 [7 wk] and P = 0.032 [16 wk]). (B, top) Survival: 4.8 wk (NT), 15 wk (30 MBq), not reached (120 MBq) (6 mice per group: all P ≤ 0.001). (B, bottom) TTP: 6.6 wk (NT), not reached (30 and 120 MBq) (6 mice per group; NT vs. 30 MBq, P = 0.153; all other P ≤ 0.014). (C) Individual tumor growth curves after [225Ac]Ac-PSMA RLT (8 mice per group; NT vs. RLT, P ≤ 0.027 [6 wk]; 20 vs. 40 or 100 kBq, P < 0.023 [15 wk], 40 vs. 100 kBq, P > 0.99). (D, top) Survival: 4.5 wk (NT), 16 wk (20 kBq), 30 wk (40 kBq), 19 wk (100 kBq) (mice per group; P ≤ 0.0137, except 40 vs. 100 kBq [P = 0.0783]; 20 vs. 100 kBq [P = 0.6203]). (D, bottom) TTP: 3.5 wk (NT), 15 wk (20 kBq), not reached (40 kBq, 100 kBq) (8 mice per group; P ≤ 0.0012, except 40 vs. 100 kBq [P = 0.679]).
PSMA RLT Induction of DDR/RSR, Cell Cycle Arrest, and TP53 Signaling
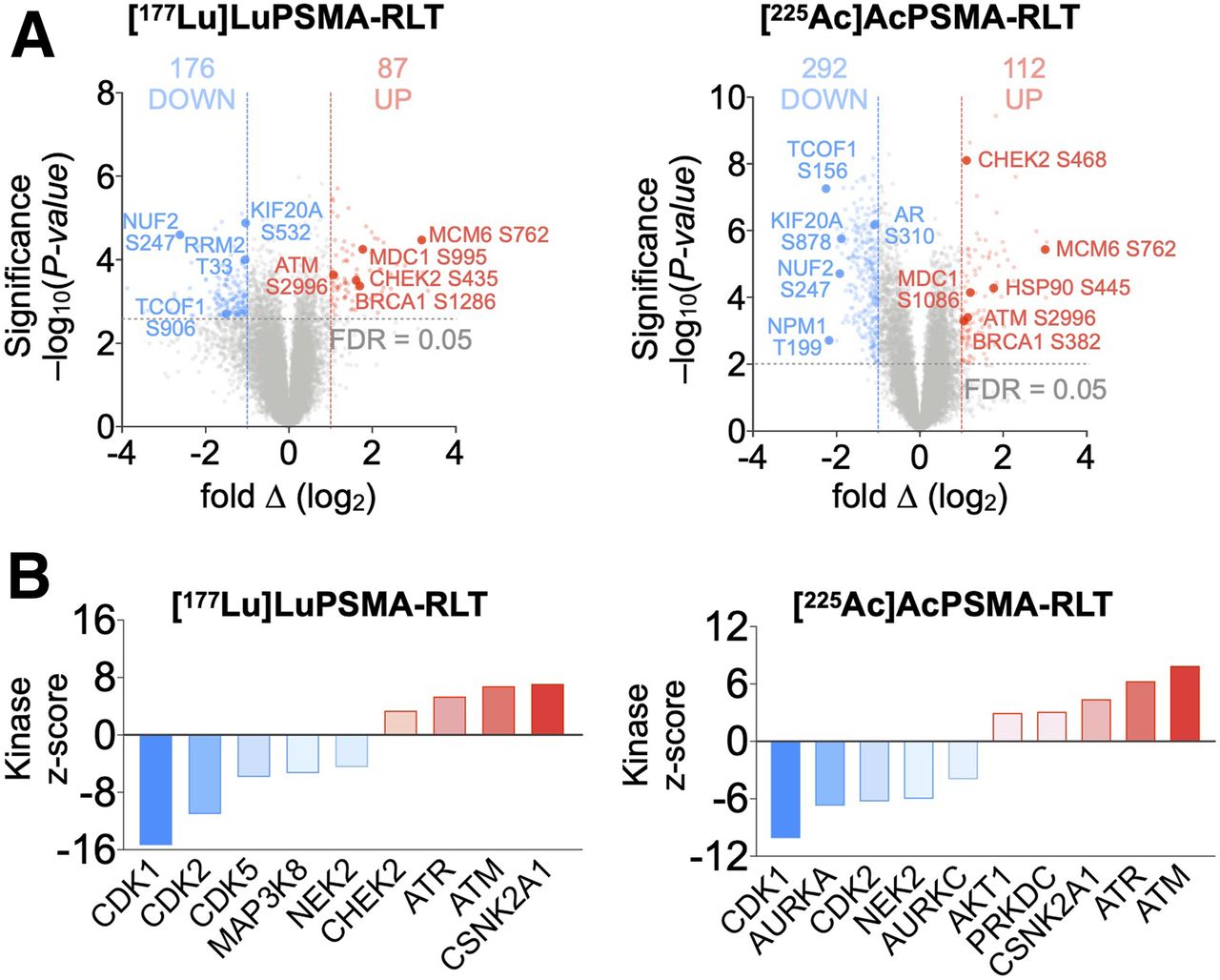
To investigate the molecular alterations in tumors induced by PSMA RLT, we used global proteomics and phosphoproteomics. [225Ac]Ac-PSMA RLT altered 3.3% of the proteome and 2.8% of the phosphoproteome; in [177Lu]Lu-PSMA RLT–treated tumors, 2.5% of the total identified proteome and phosphoproteome exhibited significant differences between control and RLT groups (Fig. 2A). The difference in the number of significant alterations in [225Ac]Ac- versus [177Lu]Lu-PSMA RLT samples most likely reflects technical improvements in MS methodology during the time between the 177Lu and the 225Ac studies—for example, a transfer from manual to semiautomatic tissue homogenization that improved sample quality. Because of this technical development, we focused on the alterations common to [225Ac]Ac- and [177Lu]Lu-PSMA RLT. However, it cannot be excluded that [225Ac]Ac- versus [177Lu]Lu-PSMA RLT can induce different alterations in the proteome or phosphoproteome due to differences in energy deposition to the surrounding tissue and the resultant differences in biological effect.
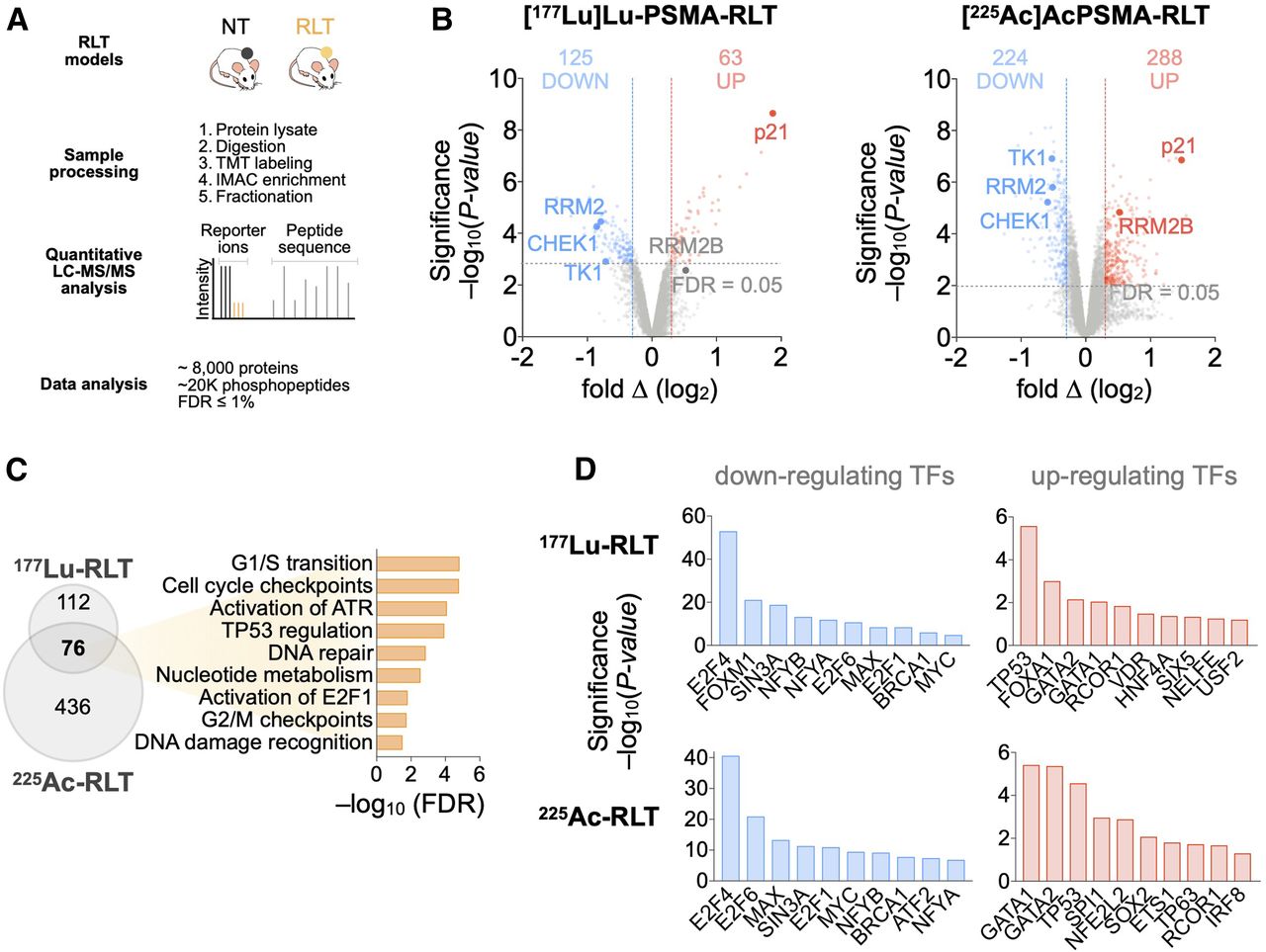
Proteomic analyses of PCa tumors reveal PSMA RLT–induced alterations. (A) Experimental workflow (also applies to Fig. 3). (B) Volcano plots highlighting changes in protein levels relative to untreated groups (188 significant proteins for [177Lu]Lu-PSMA RLT and 241 for [225Ac]Ac-PSMA RLT). (C) Gene ontology analysis of 2 datasets revealing commonly activated pathways. (D) Transcription factor enrichment analysis on differentially expressed proteins. Identified transcription factors have at least 4 associated targets. Graphs represent data from 3 tumors per group for [177Lu]Lu-PSMA RLT and 5 tumors per group for [225Ac]Ac-PSMA RLT. FDR = false-discovery rate; LC = liquid chromatography.
Differential regulation of DDR/RSR signaling, accompanied by cell cycle arrest, was one of the most significant alterations in PCa after PSMA RLT (Fig. 2C). RLT enhanced ATM, ATR, and casein kinase 2A.1 (CSNK2A1) and suppressed cyclin-dependent kinase activity by increasing phosphorylation of ATM at Ser2996 and of known ATM, ATR, and CSNK2A1 substrates (e.g., BRCA1) while decreasing the phosphorylation of cyclin-dependent kinase substrates (e.g., ribonucleotide reductase regulatory subunit M2; RRM2) (Fig. 3). Upregulation of p21 and the TP53-inducible subunit RRM2B indicated enhanced TP53 activity. RLT modulated the activity of additional transcription factors involved in responses to genotoxic stress and cell cycle progression: GATA1 (15) and REST corepressor 1 (16) were upregulated, whereas BRCA1, MYC, and E2F family members were downregulated (Fig. 2D).

Phosphoproteomic analyses of PCa tumors reveal PSMA RLT–induced alterations. (A) Volcano plots of identified phosphopeptides (512 significant phosphopeptides for [177Lu]Lu-PSMA RLT and 405 for [225Ac]Ac-PSMA RLT). (B) Kinase-substrate enrichment analysis identifying kinases with at least 5 substrates. Graphs represent data from 3 tumors per group for [177Lu]Lu-PSMA RLT and 5 tumors per group for [225Ac]Ac-PSMA RLT. FDR = false-discovery rate.
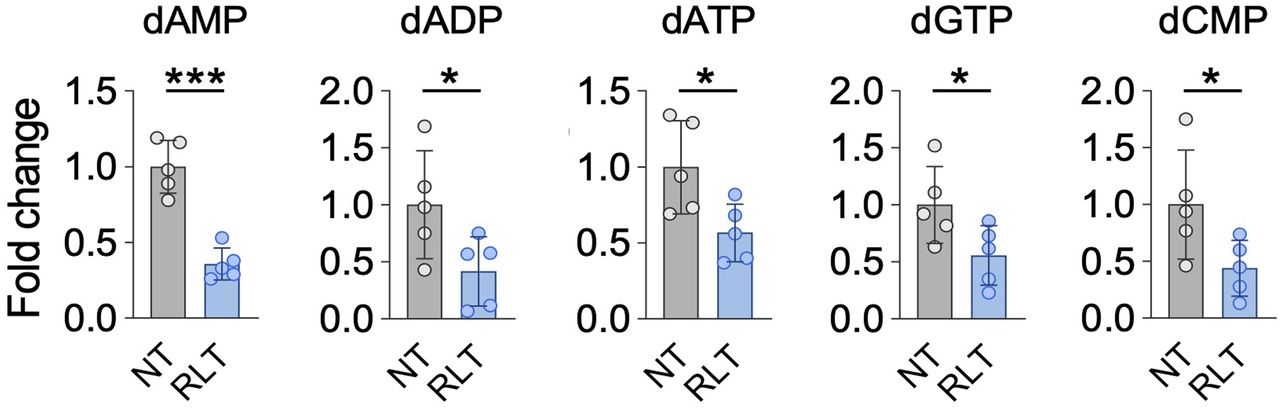
Supporting the notion of RLT-induced replication stress, and consistent with alterations in nucleotide metabolism pathways in response to RLT (Fig. 2C), nucleotide levels and expression of RRM2 and thymidine kinase 1 (TK1) were decreased by PSMA RLT (Fig. 4).
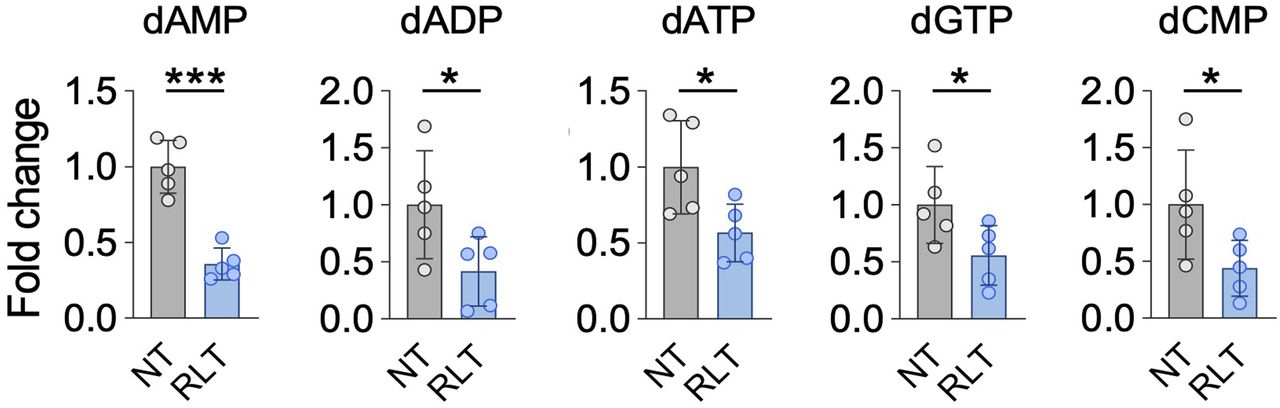
Nucleotide levels are significantly altered by [225Ac]Ac-PSMA RLT compared with untreated controls (P ≤ 0.0499). Mean NT fold-change ± SD is shown (5 tumors per group). *Significant difference. dADP = deoxyadenosine diphosphate; dAMP = deoxyadenosine monophosphate; dATP = deoxyadenosine triphosphate; dCMP = deoxycytidine monophosphate; dGTP = deoxyguanosine triphosphate.
RLT deregulated AR (diminished phosphorylation of AR Ser310) and proteins regulated by or regulating AR, such as CSNK2A1, heat shock protein 90 AB (HSP90AB), and RAC-α-serine/threonine-protein kinase (AKT1) (Fig. 3) (17–19).
TP53-Loss–Induced Reduction in Tumor Responsiveness to PSMA RLT
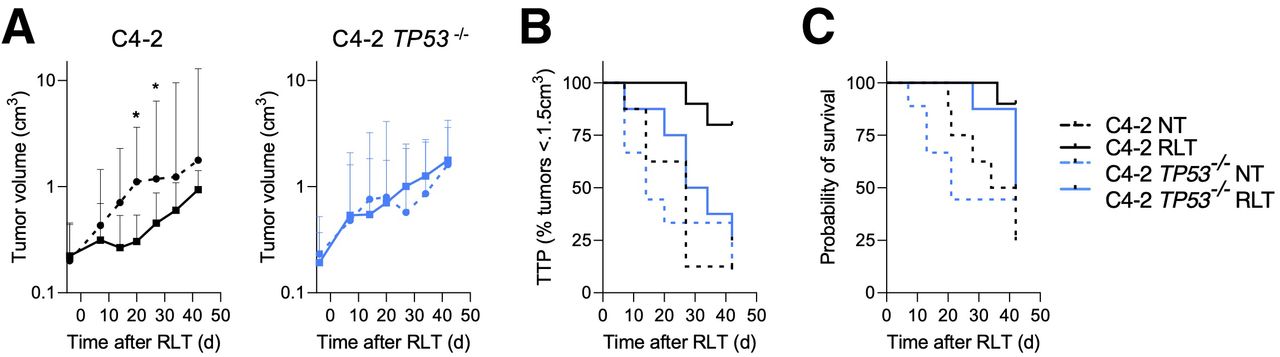
On the basis of the proteomic and phosphoproteomic analyses (Fig. 2C) and the observation that TP53 alterations are among the most common mutations in metastatic PCa, we tested whether TP53 status impacts RLT responsiveness in PCa. RLT resulted in excellent disease control in parental C4-2 tumors, with a significantly reduced tumor burden (P ≤ 0.0376) and an increased TTP (normalized to nontreated [NT], 27 d; RLT, not reached; P = 0.016) and survival (NT, 38 d; RLT, not reached; P = 0.044) (Fig. 5B). Mice with C4-2 TP53−/− tumors were less responsive to RLT than those with C4-2 tumors; neither tumor growth nor TTP (NT, 14 d; RLT, 30.5 d; P = 0.3250) or survival (NT, 21 d; RLT, 42 d; P = 0.3939) was significantly reduced (Fig. 5C).

TP53 loss renders PCa resistant to PSMA RLT. (A) Tumor growth of C4-2 (left) and C4-2 TP53−/− (right) tumors treated with 15 MBq of [177Lu]Lu-PSMA RLT. Geometric mean and 95% CI (n = 8–10 mice per group) are shown. (B) TTP: C4-2 NT, 27 d; C4-2 RLT, undefined (P = 0.0016); C4-2 TP53−/− NT, 14 d; C4-2 TP53−/− RLT, 30.5 d (P = 0.3250). (C) Survival: C4-2 NT, 38 d; C4-2 RLT, undefined (P = 0.0044); C4-2 TP53−/− NT, 21 d; C4-2 TP53−/− RLT, 42 d (P = 0.33939). *Significant difference. C4-2p53−/− NT = C4-2 TP53−/− NT.
DISCUSSION
Using proteomic and phosphoproteomic analysis, we identified compensatory tumor cell mechanisms that might confer treatment resistance by mitigating the cytotoxic effects of RLT and be exploited for synergistic RLT-combination therapies.
Unbiased investigation of adaptive tumor cell mechanisms in response to PSMA RLT revealed that PSMA RLT was associated with activation of genotoxic stress response pathways and TP53-dependent cell cycle checkpoints. To our knowledge, the current study is the first systematic evaluation of the effect of RLT on DDR/RSR pathways. The relevance of DDR/RSR signaling for RLT efficacy is supported by first clinical data showing aberrations in the DDR/RSR system in 6 of 7 patients resistant to [225Ac]Ac-PSMA RLT (9) and in 28 (of 93) PCa patients responding to 223Ra treatment (8). The exact consequence of defects in or deregulation of the DDR/RSR are likely context-dependent. DDR/RSR deregulation can drive tumor evolution and progression because of increased genomic instability and might contribute to mitigating the cytotoxicity from RLT to facilitate tumor cell survival. At the same time, these defects create vulnerabilities, such as by enhancing the reliance on parallel DNA repair pathways for survival after genotoxic stress (4); these pathways could be targeted to trigger excessive DNA damage and, thus, cell death. In addition, high DNA damage levels and disturbance of the DDR/RSR pathway might lead to a more error-prone DNA repair, which might support tumor immunogenicity by increasing tumor mutational burden.
Clinical stage inhibitors of ATM, ATR, and poly(adenosine diphosphate–ribose)polymerase (PARP) are synthetically lethal in DNA repair-compromising settings (20,21) and have radiosensitizing properties (3). Two studies demonstrated synergistic efficacy of 227Th radioimmunotherapy and inhibition of ATR or PARP (22,23), and the combination of [177Lu]Lu-PSMA RLT with the PARP inhibitor olaparib is being investigated for the treatment of genetically unselected metastatic PCa (NCT03874884). [177Lu]Lu-DOTA-octreotate combined with talazoparib improved disease control and survival compared with RLT alone in murine neuroendocrine tumors (24). In this context, the deregulation of ATM, ATR, CHEK2, and BRCA1 after PSMA RLT observed in the current study supports integration of ATR (e.g., AZD6738 and BAY1895344), ATM (e.g., AZD1390), and PARP (e.g., olaparib and talazoparib) inhibitors as radiosensitizers into RLT regimens.
TP53 is another key effector of the DDR/RSR and mediates cell cycle arrest to allow for repair of damaged DNA. Conversely, TP53 aberrations have been linked to radioresistance and proliferation despite severe DNA damage (6,25), and TP53 knockout rendered PCa less responsive to PSMA RLT than their TP53 wild-type counterparts (Fig. 5). This finding validates our proteomic and phosphoproteomic approach to elucidating potential RLT resistance mechanisms and suggests that TP53 status is one factor impacting the outcome of PSMA RLT. Drugging mutant TP53 to restore wild-type TP53 function has proven difficult because of the multitude of TP53 mutations with different properties (26); only one compound, APR-246, is currently being tested in clinical trials and has achieved Food and Drug Administration breakthrough therapy designation. However, mutant TP53 PCa exposed to RLT may particularly depend on ATM and ATR for survival (27). Future studies will investigate whether inhibition of these kinases sensitizes mutant TP53 (and wild-type TP53) PCa to RLT.
In agreement with the activation of DDR/RSR signaling, RLT impacted nucleotide metabolism in our PCa model. Synthesis of nucleotides is essential for DNA repair, as cellular nucleotide pools are limited and produced on demand (28). The 2 major nucleotide biosynthesis pathways are the de novo pathway, which relies on glucose and amino acids, and the salvage pathway, in which preformed nucleosides are recycled (29). RLT downregulated key effectors in de novo (RRM2, carbamoyl-phosphate synthetase 2) and salvage nucleotide biosynthesis (TK1) and decreased nucleotide levels. However, expression of deoxycytidine kinase, the second key effector in the nucleotide salvage pathway next to TK1, was not impacted by RLT. This finding suggests that deoxycytidine kinase–mediated nucleotide salvage might contribute to RLT resistance in PCa by enabling cells to sustain nucleoside salvage biosynthesis after RLT, thereby avoiding severe nucleotide depletion, which may impair DNA repair capacity.
Further analysis of the proteome and phosphoproteome revealed targetable signaling alterations beyond canonic DDR/RSR signaling, including deregulation of AR, AKT1, MYC, and HSP90. AR transcriptional activity was enhanced after RLT, possibly because of reduced phosphorylation of AR Ser310 (30). RLT-induced AR activation may have important consequences, as AR has been associated with regulation of the DDR and suppression of PSMA expression (31). Thus, AR might confer resistance to PSMA RLT by facilitating DNA repair and reducing target expression. However, inhibition of AR signaling, which is widely used in PCa, could be exploited to upregulate PSMA expression before [68Ga]Ga-PSMA PET/CT and PSMA RLT with the aim of improving tumor detection, targeting, and radiation dose delivery (NCT04419402, NCT04279561, and NCT03977610).
The phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway is a complex pathway of special interest in PCa because of its extensive crosstalk with AR signaling, and PTEN aberrations in about 35% of metastatic PCa render AKT constitutively active (5). These characteristics have been associated with radioresistance, regulation of TP53 signaling, aggressive PCa phenotypes, and resistance to AR-targeted therapies (32). Although PI3K/AKT/mTOR inhibitors have shown promising in vitro and in vivo anti-PCa efficacy (33,34), only the mTOR inhibitor everolimus has undergone clinical testing in combination with radiotherapy (NCT01548807) (35). However, combined inhibition of AR and PI3K/AKT signaling is being explored (e.g., NCT01485861 and NCT02525068), as AR inhibition can upregulate PI3K/AKT signaling (and vice versa) to maintain tumor cell survival and promote resistance. Given the radiosensitizing properties of both AR and PI3K/AKT inhibition (36), this treatment combination might sensitize PCa to RLT while preventing the development of resistance associated with either inhibitor alone.
MYC is an important PCa driver that has been associated with numerous protumorigenic signaling and metabolic alterations resulting in, for example, androgen-independent proliferation, metastasis, genomic instability, and, importantly, transition to neuroendocrine PCa (37,38). Compounds reducing MYC expression or activity have shown promise for the treatment of PCa (39) and may radiosensitize PCa (36), which supports their exploration in RLT combination therapies. Among those compounds are inhibitors of bromodomain proteins, cyclin-dependent kinases, and HSP90. Interestingly, RLT increased the activity of HSP90AB1, a molecular chaperone for many proteins implicated in DNA repair signaling cascades, including AR (40). HSP90 is an emerging target in PCa, and its inhibition can upregulate PSMA expression on PCa cells (Supplemental Fig. 1G) and synergize with [177Lu]Lu-DOTATATE in murine neuroendocrine tumors (41,42).
CONCLUSION
Causes of RLT resistance are not well understood, representing a major barrier to urgently needed more effective RLT approaches. Although the generalizability of our findings is limited by use of a single immunocompromised mouse model, our study identifies tumor cell intrinsic mechanisms that might allow PCa cells to survive RLT, as well as key effectors of these mechanisms that represent attractive targets for synergistic RLT-combination therapies (e.g., DDR/RSR, AR, MYC, and PI3K/AKT/mTOR pathways). In ongoing studies, we are exploring these combination therapies with the aim of providing rationales for clinical studies. Association of PSMA RLT (43) and identified key effectors with tumor immunogenicity, including the upregulation of PD-L1 on tumor cells by radiation-induced ATR activation (44), supports the combination of RLT with DDR/RSR inhibitors and immunotherapies. Lastly, our data advocate for the systematic molecular profiling of PCa patients eligible for RLT with the goal of identifying determinants and predictors of RLT responsiveness that will guide patient stratification and the selection of RLT combination therapies.
DISCLOSURE
Funding was provided by the Prostate Cancer Foundation (17CHAL02 and 19CHAL09), UCLA SPORE in Prostate Cancer (P50 CA092131), the Broad Stem Cell Research Center (BSCRC) Innovation Award, a Swedish Research Council International postdoctoral grant (2015-00452, to Susan Evans-Axelsson), and grants from the German Research Foundation (807454 and 807122 to Katharina Lückerath and Wolfgang P. Fendler). Johannes Czernin and Caius Radu are cofounders of and hold equity in Sofie Biosciences and Trethera Therapeutics. Intellectual property has been patented by UCLA and licensed to Sofie Biosciences and Trethera Therapeutics but was not used in the current study. Jeremie Calais performs consulting activities outside the submitted work for Advanced Accelerator Applications, Blue Earth Diagnostics, Janssen, Curium Pharma, GE Healthcare, Progenics, Radiomedix, and Telix. Wolfgang P. Fendler is a consultant for Endocyte and BTG and received fees from RadioMedix and Bayer outside the submitted work. No other potential conflict of interest relevant to this article was reported.
ACKNOWLEDGMENTS
We thank Liu Wei, Larry Pang, Joel Almajano, Chloe Cheng, and Mark Girgis for their excellent assistance.
KEY POINTS
QUESTION: Which signaling mechanisms mediate resistance to PSMA RLT and might be targeted to increase RLT efficacy?
PERTINENT FINDINGS: PSMA RLT induced alterations in the PCa proteome and phosphoproteome, including deregulation of compensatory DDR and RSR pathways. TP53 status impacted the response to PSMA RLT in vivo.
IMPLICATIONS FOR PATIENT CARE: Our results elucidate mechanisms underlying RLT resistance and facilitate development of rational synergistic combination therapies.
Footnotes
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication September 2, 2020.
- Revision received November 11, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}