


Abstract
The growing worldwide health challenge of atherosclerosis, together with advances in imaging technologies, have stimulated considerable interest in novel approaches to gauging this disease. The last several decades have witnessed a burgeoning in understanding of the molecular pathways involved in atherogenesis, lesion progression, and the mechanisms underlying the complications of human atherosclerotic plaques. The imaging of atherosclerosis is reaching beyond anatomy to encompass assessment of aspects of plaque biology related to the pathogenesis and complication of the disease. The harnessing of these biologic insights promises to provide a plethora of new targets for molecular imaging of atherosclerosis. The goals for the years to come must include translation of the experimental work to visualization of these appealing biologic targets in humans.
The growing worldwide health challenge of atherosclerosis, together with advances in imaging technologies, have stimulated considerable interest in novel approaches to gauging this disease. The last several decades have witnessed a burgeoning in understanding of the molecular pathways involved in atherogenesis, lesion progression, and the mechanisms underlying the complications of human atherosclerotic plaques (1–6). This progress in our fundamental understanding of the pathogenesis of atherosclerosis has opened up new horizons for imaging. The processes identified by basic science studies offer novel targets for visualizing aspects of atherosclerosis that were unimagined just a decade ago. Appropriate deployment of imaging strategies requires acquaintance with some current concepts of the basic and clinical biology of this disease.
With respect to current understanding of clinical aspects of atherosclerosis, a new focus on molecular aspects of lesion biology, rather than on the degree of luminal compromise, has emerged. Traditional vascular imaging of atherosclerosis centered on anatomic issues and primarily assessed the degree of segmental arterial stenosis and vascular calcification. Many treatment modalities aim at relieving stenoses. Yet, although revascularization often effectively relieves ischemia, it has proven disappointing in preventing myocardial infarction or prolonging life except in selected patient groups. Most acute coronary syndromes actually result from thrombotic occlusions that often associate with noncritical stenoses (7–9). On a per-plaque basis, fixed stenoses may cause fewer thrombotic complications than the more numerous noncritically stenotic lesions found in the same artery (Fig. 1) (9).

Simplified schema of diversity of lesions in human coronary atherosclerosis, depicting 2 morphologic extremes of coronary atherosclerotic plaques, to illustrate the challenge of molecular imaging of atherosclerosis. Stenotic lesions, well visualized by traditional angiographic approaches, tend to have smaller lipid cores, more fibrosis, and calcification; thick fibrous caps; and less compensatory enlargement (positive remodeling). They typically produce ischemia, detectable by perfusion scans, that is appropriately managed by combined medical therapy and often revascularization for symptom relief. Thus, traditional imaging modalities serve well to diagnose and aid the management of stenotic lesions. Nonstenotic lesions generally outnumber stenotic plaques, and they tend to have large lipid cores and thin fibrous caps, which are susceptible to rupture and thrombosis. These lesions often undergo substantial compensatory enlargement, leading to underestimation of lesion size by angiography. They do not cause perfusion defects on nuclear scans. Nonstenotic plaques may cause no symptoms for many years, but when disrupted, they can suddenly provoke an episode of unstable angina or acute myocardial infarction. Management of nonstenotic lesions should include lifestyle modification (and pharmacotherapy in high-risk individuals). Enlarged segments of this schematic show longitudinal section (left) and cross section (right). Many coronary atherosclerotic lesions may lie between these 2 extremes, produce mixed clinical manifestations, and require multipronged management. Because both types of lesions usually coexist in high-risk individuals, optimum management often requires both revascularization and systemic therapy. PTCA = percutaneous transluminal coronary angioplasty; CABG = coronary artery bypass graft. (Reprinted with permission of (51).)
Pathologic observations shed light on this disparity. Outward remodeling through compensatory enlargement permits the development of a considerable burden of atherosclerosis without luminal encroachment that causes stenoses. Indeed, a physical disruption of the atherosclerotic plaque, rather than preexisting high-degree stenosis, causes most fatal coronary events. Rupture of the plaque's fibrous cap causes most fatal coronary thrombi (Fig. 1). Plaques that have provoked fatal coronary thrombosis have thin fibrous caps, large lipid pools, abundant inflammatory cells, and relatively few smooth muscle cells (Fig. 1) (8,10).
Laboratory studies substantiate mechanisms that link inflammation to low levels of interstitial collagen, the prime constituent of the plaque's protective fibrous cap. Inflammatory mediators impair interstitial collagen synthesis by smooth muscle cells and boost production of collagen-destroying enzymes, including the matrix-metalloproteinase interstitial collagenases. Moreover, exposure to proinflammatory stimuli heightens monocyte recruitment and macrophage production of the potent procoagulant tissue factor. Thus, inflammation regulates not only the strength of the plaque's extracellular matrix but also its thrombogenicity (5).
Aspects of inflammation have therefore surfaced as 1 category of emerging major targets for functional imaging of atherosclerosis (Fig. 2). This vascular biology of atherosclerosis suggests several molecular processes that could serve as imaging targets (Table 1). The following sections will mention some of these biologic processes and provide examples of how molecular imaging might exploit them.

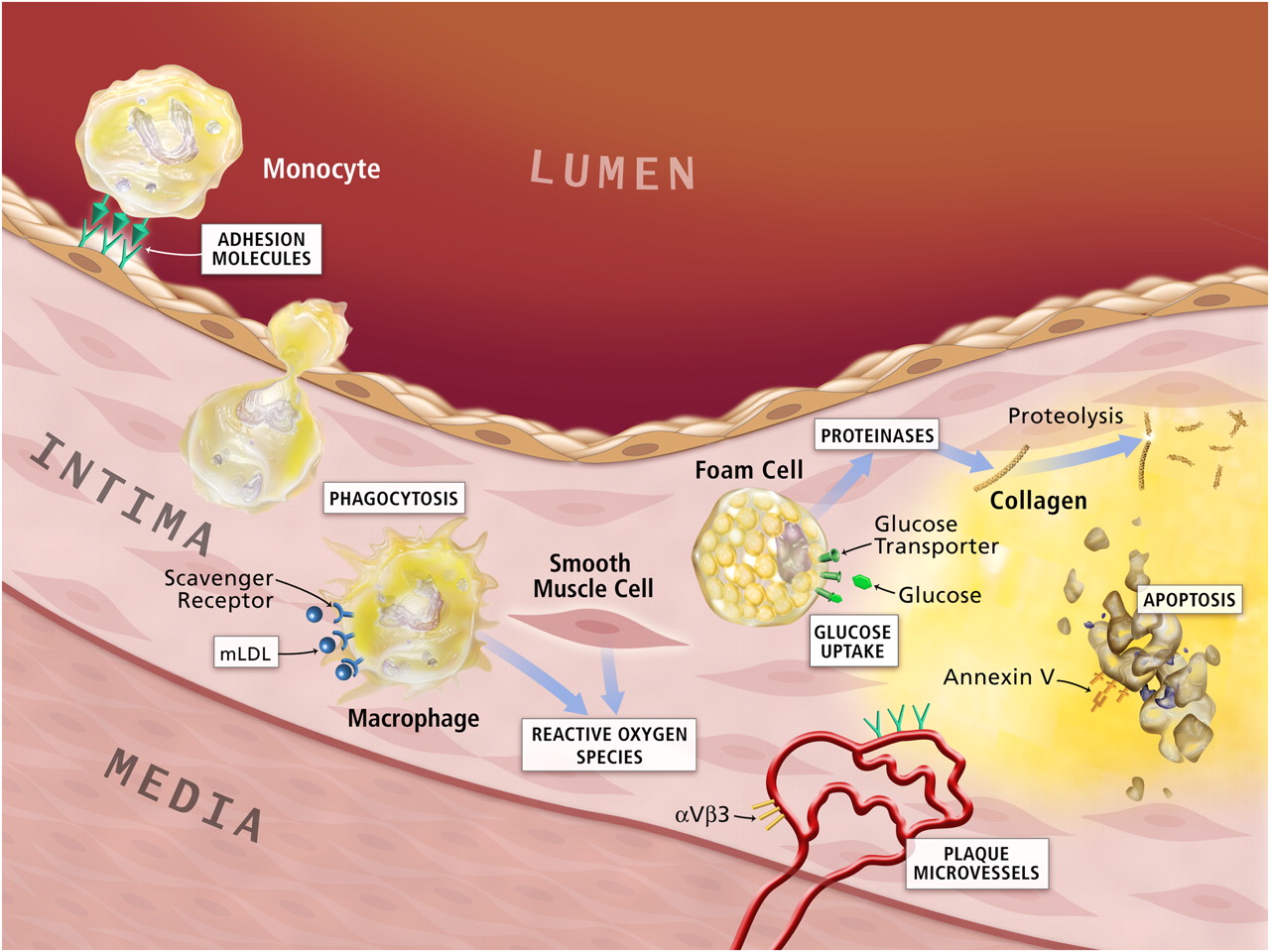
Potential molecular imaging targets in atherosclerosis. White boxes show putative targets for molecular imaging of atherosclerosis. Atherogenesis involves recruitment of inflammatory cells from blood, represented by the monocyte in the upper-left-hand corner of this diagram. Monocytes are the most numerous leukocytes in atherosclerotic plaque. Recruitment depends on expression of adhesion molecules on macrovascular endothelium, as shown, and on plaque microvessels. Once resident in the arterial intima, activated macrophages become phagocytically active, a process that provides another potential target for plaque imaging. Oxidatively modified low-density lipoprotein (mLDL)–associated epitopes that accumulate in plaques may also serve as targets for molecular imaging. Foam cells may exhibit increased metabolic activity, augmenting their uptake of glucose, a process already measurable in the clinic by 18F-FDG uptake. Activated phagocytes can also elaborate protein-degrading enzymes that can catabolize collagen in the plaque's fibrous cap, weakening it, and rendering it susceptible to rupture and hence thrombosis. Mononuclear phagocytes dying by apoptosis in plaques display augmented levels of phosphatidylserine on their surface. Probes for apoptosis such as annexin V may also visualize complicated atheromata. Microvessels themselves can express not only leukocyte adhesion molecules (shown in green) but also integrins such as αVβ3. Proof-of-principle experiments in animals support each process or molecule in white boxes as target for molecular imaging agents.
Functional Imaging Targets in Atherosclerosis
ENDOTHELIAL ACTIVATION
The healthy endothelial monolayer in arteries resists prolonged contact with blood leukocytes, produces endogenous vasodilator molecules, combats thrombosis, favors fibrinolysis, and expresses enzymes, such as superoxide dismutase, that can degrade reactive oxygen species. Laminar shear stress, as prevails in normal arteries, fosters these homeostatic endothelial functions (11,12). But endothelial cells become dysfunctional when exposed to disturbed flow, instead of laminar sheer stress, and to proatherogenic factors such as modified lipoprotein or proinflammatory cytokines. Among markers of endothelial dysfunction, leukocyte adhesion molecules have elicited special interest as a target for molecular imaging (Fig. 2). For example, vascular cell adhesion molecule-1 (VCAM-1) serves as a well-validated marker of endothelial activation. The monolayer of endothelial cells on the arterial intima provides a poor imaging target, but human atherosclerotic lesions harbor abundant microvessels rich in VCAM-1. VCAM-1 can internalize ligands and thus cause them to accumulate in activated endothelial cells and perhaps smooth muscle cells. Several VCAM-1–directed, peptide-based imaging agents have been developed (13), and at least 1 lead compound is in clinical development (14). Other adhesion molecules implicated in atherogenesis include intercellular initiation molecule 1 and P-selectin (CD62P) (15–17).
Microvessels in human atheromata abound, particularly in the plaque's base (18,19). These neovessels arise by angiogenesis, from the vasa vasorum or perhaps from the macrovascular luminal surface. Molecules involved in angiogenesis thus may also serve as targets for molecular imaging. Integrins expressed by neovessels in atherosclerotic arteries, such as αvβ3, may permit imaging of angiogenesis in plaques, and several 18F-labeled affinity ligands have been described (14). Studies of the flow in plaques, an index of microvascular functions, may also provide a novel imaging target in atherosclerosis (20).
MACROPHAGE RECRUITMENT AND ACTIVATION
After adhering to the endothelium via adhesion molecules such as VCAM-1, leukocytes can enter into the intima, drawn in by chemoattractant cytokines overexpressed in plaque. Mononuclear phagocytes, the most abundant leukocytes recruited to atheromata, enter the arterial wall as monocytes and mature into macrophages. Mononuclear phagocytes exhibit several functions that could serve as targets for molecular imaging. For example, one might simply monitor accumulation of these cells within the artery wall. Expression of activation markers, phagocytic function, or structures that identify subtypes of monocytes might go beyond mere accumulation of leukocytes to reveal their functional capacities as well (21,22).
Phagocytosis provides an attractive target for molecular imaging of macrophages, because it could lead to the capture and concentration of imaging agents. Imaging initiatives have taken advantage of several aspects of phagocytosis. Activated macrophages can internalize and concentrate nanoparticles—for example, those coated with dextran (23). Activated macrophages also express scavenger receptors, members of the broad pattern recognition receptor family. Oxidatively modified low-density lipoprotein can bind to certain scavenger receptors and thus undergo update and accumulation within macrophages (2,24). Epitopes associated with such modified lipoproteins may also serve as imaging targets (25–27).
Activated macrophages may use glycolysis more than other cells within plaques. 18F-FDG can measure glucose uptake and serves as an example of a metabolic marker, associated with inflammatory cells, that may also disclose functional information regarding plaques (Fig. 2) (28,29).
Macrophages within atheromata produce matrix-degrading proteinases implicated in weakening the fibrous cap and rendering plaques liable to rupture and thrombosis. The catalytic property of enzymes should amplify signals. Thus, proteinases represent promising targets for molecular imaging, and several active site binders and imaging prodrugs have been described (30–35). Because proinflammatory mediators boost the expression of these matrix-degrading enzymes in cells that elaborate them in atheromata, imaging proteinases may not only enable interrogation of a specific pathobiologic process (extracellular matrix digestion) but also may provide a window on inflammation in general.
The advanced atherosclerotic plaque contains a lipid core rich in cholesteryl esters, cholesterol monohydrate crystals, and cellular debris. Some refer to this compartment of the plaque as the necrotic core. Indeed, cells in atheromata can die by oncosis, by apoptosis (programmed cell death) (36–39). Smooth muscle cell apoptosis may disrupt repair mechanisms in the plaque that maintain the extracellular matrix required for structural integrity (40,41). Macrophage apoptosis may help generate the lipid core and, by the production of tissue-factor–rich apoptotic bodies, may generate thrombogenic microparticles whose release on plaque disruption can propagate thrombosis. Cells undergoing apoptosis exteriorize phosphatidylserine, a target for molecular imaging of dying cells and potentially for reporting on the biology associated with cell death in atheromata. Annexin V binds to phosphatidylserine, providing a ligand for imaging cell death (42,43).
Activated macrophages, smooth muscle cells, and endothelial cells heighten the production of reactive oxygen species that promote cellular damage or death and also serve as both a monitor of inflammation and a pathophysiologic mediator. Nicotinamide adenine dinucleotide phosphate hydrogen oxidases that produce superoxide anion, and myeloperoxidase that generates hypochlorous acid, have received considerable interest in the context of atherosclerosis (44–47). These 2 reactive oxygen species may represent attractive targets for molecular imaging (Fig. 2). Monitoring oxidative stress in plaques could provide an additional window on cellular functions that go beyond anatomy to reveal features of atheromata that influence their clinical importance (48).
The imaging of atherosclerosis is reaching beyond anatomy to encompass the assessment of aspects of plaque biology related to the pathogenesis and complication of the disease. Laboratory investigations that suggest the operation of inflammatory pathways from the inception through the complication of this disease have received considerable support from observations on human tissues and in human populations. Several pilot observations and experimental studies of atherosclerosis have served as proof of the principle that inflammatory processes during atherogenesis can provide a new window for imaging. Other recent biologic insights into atherosclerosis suggest imaging targets in addition to inflammatory processes. For example, targeting epitopes of modified lipoprotein may permit assessment of one of the triggering pathways thought to operate in the initiation of atherosclerosis. Indeed, constituents of oxidatively modified low-density lipoprotein may serve as a proximal inciting stimulus to the inflammatory response under way in the atheromatous plaque. Angiogenesis within plaques, another emerging target for atherosclerosis imaging, may result from inflammatory activation but can also amplify inflammation by providing a large surface area for ongoing leukocyte recruitment. Once resident in the plaque, activated phagocytic leukocytes can elaborate effector molecules of innate immunity, including proteinases and reactive oxygen species—aspects of atherogenesis also susceptible to molecular imaging. The harnessing of these biologic processes promises to provide a plethora of new targets for molecular imaging of atherosclerosis.
The goals for the years to come must include translation of the experimental work to the visualization of these appealing biologic targets in humans. This enterprise will require multidisciplinary teams and resources that extend beyond those available to most individual laboratories. In particular, molecular imaging probes will require scalable syntheses using good manufacturing processes for human use. The approval for human use of new imaging probes will necessitate toxicology studies. Bridging the gap between the animal laboratory and the clinic thus presents considerable challenges (49).
The dividends of realizing the promise of the molecular imaging of atherosclerosis should reward this investment. Evaluation of novel therapies and hence drug development in the atherosclerosis arena desperately need biomarkers that reflect biologic aspects of the disease. Molecular imaging strategies may well help to identify doses of new agents most likely to succeed in large clinical endpoint trials. Finally, molecular imaging of aspects of atherosclerosis may prove to be a valuable clinical tool in selected patients. Like any imaging modality, however, the clinical use of molecular imaging will require rigorous evaluation for added clinical benefit and cost-effectiveness (50).
Acknowledgments
We thank our many colleagues affiliated with the Donald W. Reynolds Clinical Cardiovascular Research Center at Harvard Medical School who contributed to our efforts to develop molecular imaging of atherosclerosis. We also acknowledge the National Institutes of Health and the American Heart Association for their support.
Footnotes
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication March 5, 2010.
- Accepted for publication March 12, 2010.





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies
- PET/MRI of Hypoxic Atherosclerosis Using 64Cu-ATSM in a Rabbit Model
- Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded "Cardiovascular Continuum"
- Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis
- Quantifying Plaque Burden and Morphology Using Coronary Computed Tomography Angiography to Predict Coronary Physiology: Helpful...But Is It Sufficient?
- 99mTc-cAbVCAM1-5 Imaging Is a Sensitive and Reproducible Tool for the Detection of Inflamed Atherosclerotic Lesions in Mice
- PET/CT Imaging of Chemokine Receptor CCR5 in Vascular Injury Model Using Targeted Nanoparticle
- PET Imaging of Chemokine Receptors in Vascular Injury-Accelerated Atherosclerosis
- Protein kinase C {delta} regulates the release of collagen type I from vascular smooth muscle cells via regulation of Cdc42
- Acute Coronary Events
- How Many PET Tracers Do We Need?
- Report of the National Heart, Lung, and Blood Institute Working Group on the Translation of Cardiovascular Molecular Imaging
- Disturbed Blood Flow Induces RelA Expression via c-Jun N-Terminal Kinase 1: A Novel Mode of NF-{kappa}B Regulation That Promotes Arterial Inflammation
- Visualizing the Vascular Glue: "Shearing" the Barriers to Atherosclerotic Plaque Imaging