


Abstract
Angiogenesis is a critical step in tumor development, in which vascular endothelial growth factor (VEGF) is a key growth aspect. Heat shock protein 90 (HSP90), a molecular chaperone, is essential for the activity of key proteins involved in VEGF transcription. Currently, no biomarkers to predict the effect of, or monitor, HSP90 inhibition therapy in individual patients exist. 89Zr-bevacizumab PET provides a noninvasive tool to monitor tumor VEGF levels. The aim of this study was to investigate 89Zr-bevacizumab PET for early antiangiogenic tumor response evaluation of treatment with the new HSP90 inhibitor NVP-AUY922. In xenografts of A2780 and its cisplatin-resistant CP70 human ovarian cancer subline, 89Zr-bevacizumab small-animal PET was performed before and after NVP-AUY922 treatment and verified with histologic response and ex vivo tumor VEGF levels. Compared with pretreatment values, 2 wk of NVP-AUY922 treatment decreased 89Zr-bevacizumab uptake by 44.4% (P = 0.0003) in A2780 xenografts, whereas tumor uptake was not affected in CP70 xenografts. The same pattern was observed in A2780 and CP70 tumor VEGF levels, measured with enzyme-linked immunosorbent assay, and mean vessel density after NVP-AUY922 treatment. These findings coincided with reduction in the proliferation rate, assessed by Ki67 staining, in A2780 tumor tissue only. Conclusion: 89Zr-bevacizumab PET was in line with the antiangiogenic response and direct antitumor effects after NVP-AUY922 treatment, supporting the specificity of 89Zr-bevacizumab PET as a sensitive technique to monitor the antiangiogenic response of HSP90 inhibition in vivo.
Angiogenesis, the formation of new blood vessels, is a critical factor involved in the development and growth of tumors (1). An important aspect involved in angiogenesis is vascular endothelial growth factor (VEGF). VEGF production by tumor cells is thought to be regulated by hypoxemia, growth factor signaling, cytokines, and cell differentiation (1). Overexpression of VEGF occurs in many human tumor types. Therefore, targeting angiogenesis is a rational treatment approach in many cancer types (2). Currently, this can be done by VEGF-neutralizing antibodies—small molecules blocking the VEGF receptor function—or by drugs inhibiting cellular tumor signaling pathways affecting angiogenesis. Inhibition of heat shock protein 90 (HSP90) is 1 way of affecting these pathways. HSP90 is a molecular chaperone, involved in maintaining the conformation, stability, cellular localization, and activity of several key oncogenic client proteins (3,4). HSP90 is constitutively expressed at 2- to 10-fold higher levels in cancer cells and present in active multichaperone complexes, conferring relative sensitivity to treatment with HSP90 inhibitors, compared with their normal counterparts (3,5). At this moment, several HSP90 inhibitors are being developed and investigated in preclinical and clinical studies. Of these, the geldanamycin analog 17-allylamino-17-demethoxygeldanamycin (17-AAG, or tanespimycin) has been studied most extensively and disease stabilizations and tumor responses have been observed in phase I–II clinical trials (6). Another class of synthetic HSP90 inhibitors is the resorcinolic pyrazole or isoxazole amide analogs, of which NVP-AUY922, an improved isoxazole resorcinol, is the most potent yet described (6).
Currently, no biomarkers to predict the effect of, or monitor, HSP90 inhibition therapy in individual patients exist. One potential way to monitor HSP90 therapy is to prove its effect via changes in client proteins of HSP90. Client proteins of HSP90 include receptors and transcription factors such as epidermal growth factor receptor 1 and 2 (EGFR and HER2), insulinlike growth factor type 1 receptor, and hypoxia-inducible factor 1α (3,7). However, most of these client proteins can be measured only by collecting tumor biopsies. Therefore, molecular imaging is an attractive alternative. It allows repetitive noninvasive follow-up of specific targets. A suitable candidate is VEGF, a downstream product of various HSP90 client proteins (1,4). Recently, it has been shown that HSP90-directed therapy leads to a reduction of VEGF secretion in tumor cell lines and a decreased mean vessel density (MVD) in tumor-bearing animal models (7–11). Because larger isoforms of VEGF remain in the extracellular tumor matrix, these isoforms are attractive as imaging targets (12,13). The noninvasive measurement of VEGF in the tumor might give insight about the effect of HSP90 inhibition and thus assist in tumor response prediction. Previously, we showed in a xenograft mouse model that bevacizumab, a monoclonal antibody against VEGF, radiolabeled with 89Zr and 111In can be used for in vivo VEGF visualization and quantification, presumably because of binding to cell- and matrix-bound VEGF (12,13). The advantage of 89Zr-bevacizumab is that it allows high resolution and quantitative PET. In addition, radiolabeled bevacizumab is currently used as a good manufacturing practice–produced tracer in clinical development (14).
The aim of this study was to evaluate 89Zr-bevacizumab PET for imaging of the early antiangiogenic tumor response after treatment with the HSP90 inhibitor NVP-AUY922. To this end, an in vivo mouse model was used with xenografts from ovarian cancer cell lines A2780 and its cisplatin resistant subline CP70. 89Zr-bevacizumab PET was performed before and after NVP-AUY922 treatment, and the imaging results were related to histologic response and ex vivo tumor VEGF levels.
MATERIALS AND METHODS
Cell Lines and In Vitro Experiments
A2780 and its 7-fold cisplatin-resistant subline CP70 were kindly provided by Dr. Thomas C. Hamilton (Fox Chase Cancer Center). All cells were cultured in RPMI 1640 (Invitrogen), supplemented with 10% heat-inactivated fetal calf serum (Bodinco BV) and 2 mM l-glutamine (Invitrogen) at 37°C in a fully humidified atmosphere containing 5% CO2. Cells were subcultured 3 times per week.
For in vitro experiments, NVP-AUY922 was dissolved in dimethyl sulfoxide at 10 mM and stored at −80°C. A2780 and CP70 cells were plated (3 × 105 cells per well in 3 mL) and treated with 50 and 100 nM NVP-AUY922, respectively; relevant plasma levels in mice; or control medium (n ≥ 3) (6,15). After 24 h of incubation, supernatant was collected and centrifuged at 180g for 15 min. Supernatant samples were stored at −20°C until analysis. VEGF levels were determined with the human VEGF enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) according to the manufacturer's protocol.
Conjugation and 89Zr Labeling of Bevacizumab
Conjugation and labeling of bevacizumab were performed as described previously (13,16). In short, the chelate desferrioxamine B (Desferal; Novartis) was succinylated (N-suc-desferrioxamine), temporarily filled with stable iron, and coupled to the lysine residues of bevacizumab by means of a tetrafluorophenol-N-suc-desferrioxamine ester. Conjugation was performed at room temperature for 30 min at pH 9.5–9.7. After conjugation, the mixture was set to pH 4.2–4.4 (0.1 M H2SO4), and 50 μL of 25 mg of ethylenediaminetetraacetic acid per milliliter (Calbiochem) were added to remove stable iron. The solution was incubated for 30 min at 35°C and purified by ultrafiltration, diluted in water for injection (5 mg/mL), and stored at −20°C. Labeling was performed with 89Zr (half life, 3.27 d) produced by Cyclotron BV. In brief, the 89Zr-oxalate solution was set at pH 3.9–4.2 and mixed for 3 min. With 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Sigma-Aldrich) buffer, the solution was adjusted to pH 6.7–6.9. N-suc-desferrioxamine-bevacizumab was added and incubated for 45 min at room temperature.
Animals
Tumor cells for xenografting were harvested by trypsinization and resuspended in RPMI 1640 and Matrigel (high-concentration Matrigel; BD Bioscience). In vivo imaging and ex vivo biodistribution experiments were conducted using male nude BALB/c mice (BALB/cOlaHSD-foxnnu) obtained from Harlan Nederland. At 6–8 wk of age, the mice were injected subcutaneously with 5 × 106 A2780 or CP70 cells mixed with 0.1 mL of Matrigel. When the tumor measured between 6 and 8 mm in diameter (±0.3 cm3, for A2780 at 2 wk after injection and for CP70 at 3–4 wk after injection) in vivo studies were started.
Small-Animal PET and Ex Vivo Biodistribution
89Zr-bevacizumab (4 ± 0.27 MBq, ±5 μg) was injected into the penile vein. During a scan sequence, images were made at 24 and 144 h after injection of the tracer. Two scan sequences were obtained, before and after NVP-AUY922 treatment. Animals were imaged using a microPET Focus 220 rodent scanner (CTI Siemens). Static images (30-min acquisition time) were obtained. After image reconstruction, in vivo quantification was performed with AMIDE (Amide's a Medical Imaging Data Examiner) software (version 0.9.1; Stanford University) (17). For the quantification of radioactivity within the tumor, 3-dimensional volumes of interest were manually drawn using the 3-dimensional Freehand tool, plane by plane, pixel by pixel, with no change in thresholds. The total injected dose was calculated by decay correction of the total activity present at 24 h after injection in the animal—the time at which the clearance of the injected antibodies is considered to be almost negligible, as described earlier (13). The data are presented as the percentage injected dose per gram of tissue (%ID/g), assuming a tissue density of 1 g/cm3. Animals were sacrificed after the last scan (144 h, second scan sequence), and organs and tissues were excised, rinsed of residual blood, and weighed. Samples and primed standards were counted for radioactivity in a calibrated well-type LKB-1282-Compu-γ-system (LKB Wallac) and corrected for physical decay. Tissue activity is expressed as %ID/g. Harvested tumors were divided, immediately frozen at −80°C, and paraffin-embedded for further analysis.
A2780 tumors grew rapidly, with a doubling time of 3–6 d (6,18–20). It was considered unachievable to maintain these tumors in the untreated animals for the duration of the drug treatment of the other animals, because the tumors would grow unacceptably large. Therefore, for ex vivo comparison nontreated control animals were sacrificed after the first administration of 89Zr-bevacizumab, comparable to the pretreatment scan of the animals treated with NVP-AUY922. Nontreated control animals (A2780, n = 6; CP70, n = 5) were injected with 89Zr-bevacizumab (4 ± 0.27 MBq, ±5 μg), and mice were sacrificed 144 h after injection of the tracer. Ex vivo biodistribution was performed as described above.
All animal experiments were performed with isoflurane inhalation anesthesia (induction, 3%; maintenance, 1.5%).
NVP-AUY922 Treatment
NVP-AUY922 was formulated in water containing 5% glucose and administered at 50 mg/kg twice weekly intraperitoneally (volume, 6.5 mL/kg). Treatment was started immediately after the first (pretreatment) scan sequence (A2780, n = 8; CP70, n = 6). After 2 wk of treatment, mice received a second administration of 89Zr-bevacizumab, and a second scan sequence was performed. NVP-AUY922 treatment was continued during this sequence.
All animal experiments were approved by the animal experiments committee of the University of Groningen.
Determination of Antiangiogenic Response
From frozen tumors, 3 random samples were lysed manually using mammalian protein extraction reagent (Pierce). Thereafter, mixtures were centrifuged at 20,000g for 15 min and subsequently stored at −20°C until analysis. The protein concentration of the samples was determined by a Bradford assay (21). VEGF levels were measured as described above. Data are presented as picogram of VEGF per milligram of protein.
Paraffin-embedded tumors were stained with antibodies against von Willebrand factor, Ki67 (Dako), and hematoxylin and eosin. The MVD was scored in 3 areas, defined as hot-spot areas with the maximum number of microvessels, as described earlier (13). The proliferation index was calculated by percentage of Ki67-positive cells in at least 3 high-power fields (×400) using a calibrated grid. Hematoxylin and eosin staining was performed to assess areas of necrosis. Areas were scored as percentage necrosis and vital tumor tissue per slide.
Statistical Analysis
Data are presented as mean ± SEM. Statistical analysis was performed using the Mann–Whitney test for nonparametric data and a paired sampled t test for paired data (SPSS, version 14; SPSS Inc.). A P value of 0.05 or less was considered significant.
RESULTS
NVP-AUY922 Decreases VEGF Secretion in A2780 Cells In Vitro
After 24 h of incubation, VEGF levels in the culture medium of nontreated A2780 cells were higher (1.8-fold) than those in the culture medium of nontreated CP70 cells (P = 0.042). Treatment of the A2780 cells with 50 nM NVP-AUY922 for 24 h resulted in a 43.2% (P = 0.038) reduction of VEGF levels in the culture medium, compared with the culture medium of nontreated cells (Fig. 1). VEGF levels in the culture medium of CP70 cells were not significantly decreased (namely 23.8% lower, P = 0.20). Increased doses of NVP-AUY922 up to 100 nM did not further reduce VEGF secretion by A2780 or CP70 cells.

Effect of HSP90 inhibition by NVP-AUY922 on VEGF secretion. A2780 and CP70 tumor cells were incubated for 24 h in 50 and 100 nM NVP-AUY922 or control medium. Control VEGF levels of A2780 cells were higher than those of control CP70. Data are presented as mean ± SEM. *P < 0.05. **P < 0.05.
89Zr-Bevacizumab Uptake Decreases in A2780 Tumors After NVP-AUY922 Treatment
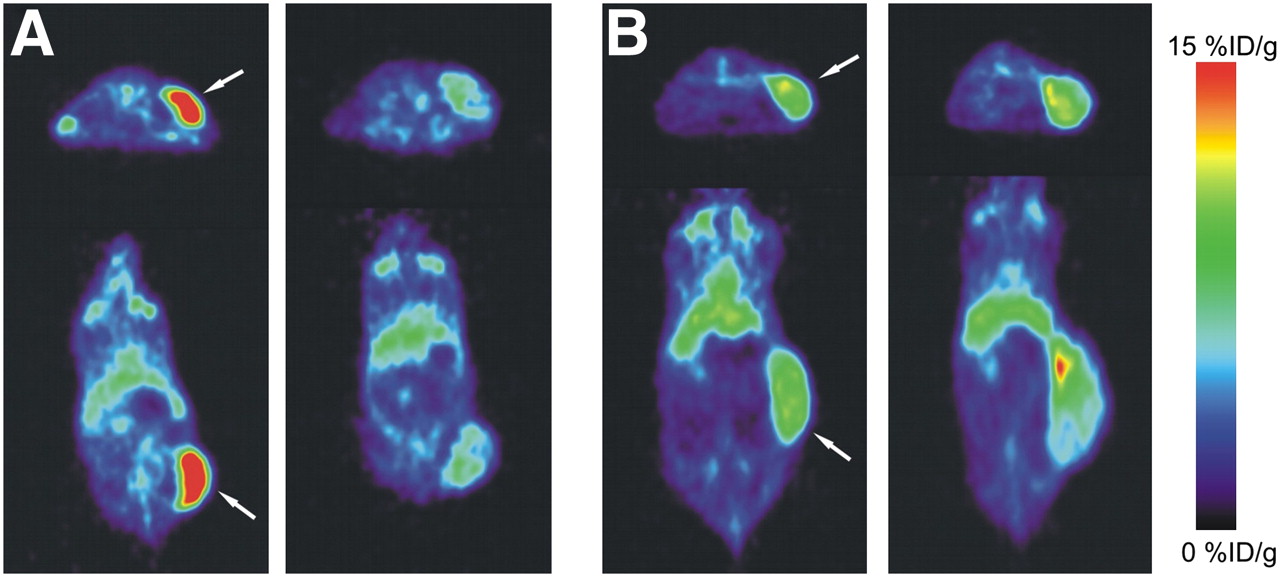
PET 144 h after injection showed clear tumor localization of 89Zr-bevacizumab in the A2780 and CP70 xenograft model (Fig. 2). 89Zr-bevacizumab uptake was homogeneous within the tumor. Before treatment, 89Zr-bevacizumab tumor uptake in A2780 xenografts (12.9 ± 2.2 %ID/g, 144 h after injection) was higher (P < 0.0001) than that in CP70 xenografts (6.9 ± 1.0 %ID/g). Twice weekly treatments with 50 mg of NVP-AUY922 per kilogram, compared with pretreatment, decreased 89Zr-bevacizumab uptake by 44% (P = 0.0003), as quantified by small-animal PET at 144 h after injection of 89Zr-bevacizumab (Fig. 3). Similar results were seen in ex vivo biodistribution experiments, in which 89Zr-bevacizumab uptake was 48% lower (P = 0.029) in NVP-AUY922–treated versus nontreated animals (Fig. 4A). A2780 tumors grew rapidly, with a doubling time of 3–6 d (6,18–20). In our study, volumes (assessed by small-animal PET, which correlated nicely with ex vivo tumor weight [r2 = 0.95]) of NVP-AUY922–treated A2780 tumors increased only moderately (2.2 ± 0.2-fold) during the study period (3 wk), indicating an effect of NVP-AUY922 on tumor growth.
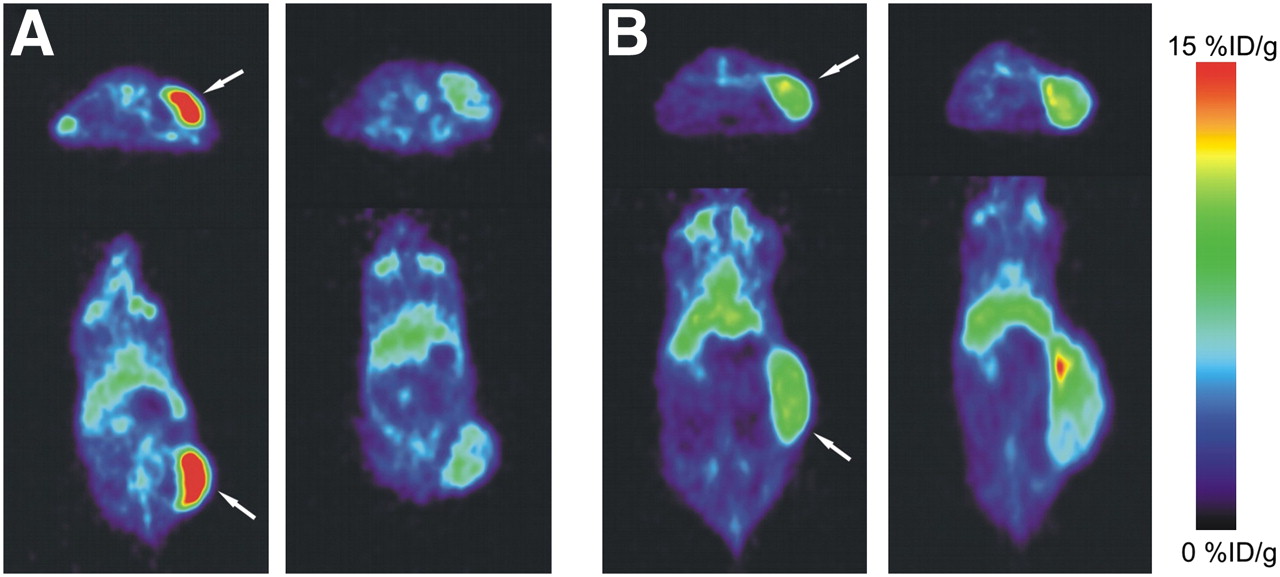
Representative transversal and coronal small-animal PET images of 89Zr-bevacizumab obtained before treatment of A2780 (A) and CP70 (B) xenografts (left) and after NVP-AUY922 treatment (right). Images were obtained at 144 h after injection of 89Zr-bevacizumab. Tumor is indicated by arrow.

Individual small-animal PET quantification of A2780 and CP70 xenografts before and after NVP-AUY922 treatment.

(A) Ex vivo tumor biodistribution of 89Zr-bevacizumab and tumor lysate VEGF levels in control and NVP-AUY922–treated A2780 and CP70 xenografts. (B) Small-animal PET quantification and ex vivo biodistribution of 89Zr-bevacizumab after NVP-AUY922 treatment, and ex vivo organ biodistribution (right) of 89Zr-bevacizumab in control (n = 11) and NVP-AUY922–treated (n = 14) animals. Data are presented as mean ± SEM. *P < 0.05.
In contrast to A2780, treatment with NVP-AUY922 did not change 89Zr-bevacizumab tumor uptake in CP70 xenografts, compared with pretreatment tumor uptake (namely an increase of 8%, P = 0.475), as quantified by small-animal PET (Fig. 3). Similar results were seen with ex vivo biodistribution studies of nontreated versus treated CP70 tumors (27% increase, P = 0.125). Tumor volumes increased 2.1 ± 0.5-fold during the study period (3 wk), which is comparable to the growth rate before the start of the study in that time. Treatment with NVP-AUY922 did not change normal-organ (nontumor) biodistribution of 89Zr-bevacizumab between treated and nontreated tumor-bearing mice (all organs, P > 0.05) (Fig. 4C).
89Zr-Bevacizumab Imaging Corresponds to Tumor VEGF Levels in A2780 and CP70 Xenografts Before and After NVP-AUY922 Treatment
To verify that changes in 89Zr-bevacizumab tumor uptake were VEGF-driven, tumor VEGF levels of nontreated control mice were compared with VEGF levels of NVP-AUY922–treated (50 mg/kg, twice weekly) mice in both A2780 and CP70 xenografts by quantitative ELISA. In A2780 xenografts, tumor VEGF levels were 69% lower (P = 0.041) after NVP-AUY922 treatment, whereas no significant change of VEGF levels was found in CP70 (20% reduction, P = 0.608). These results correspond to 89Zr-bevacizumab tumor uptake as assessed by biodistribution in these animals (Fig. 4A). Both 89Zr-bevacizumab tumor uptake and tumor VEGF levels are lower after NVP-AUY922 treatment in A2780 xenografts, compared with controls. However, this is not the case for CP70 xenografts. Furthermore, higher initial VEGF levels in A2780 versus CP70 are in line with the 89Zr-bevacizumab biodistribution findings in these animals. In the present study, small-animal PET quantification was comparable to ex vivo biodistribution (Fig. 4B), as described earlier (13). Therefore, these results demonstrate that changes in 89Zr-bevacizumab small-animal PET correspond to changes in tumor VEGF levels by NVP-AUY922–mediated HSP90 inhibition.
NVP-AUY922 Treatment Decreases MVD and Ki67 Staining in Responding A2780 Tumors
The MVD was 60% lower (P = 0.005) in NVP-AUY922–treated A2780 tumors than in nontreated tumors (Fig. 5). This lower MVD shows that in A2780 tumors a decrease in VEGF secretion is accompanied by an antiangiogenic effect in A2780, as determined histologically, after NVP-AUY922. Additionally, antiproliferative effects were seen in A2780 xenografts (proliferation index reduced by 35%, P = 0.023), indicating also that a direct tumor effect occurred parallel to antiangiogenic effects after NVP-AUY922 treatment (Fig. 5). In CP70 tumors, no differences in MVD (P = 0.720) and proliferation rate (P = 0.688) were observed, which is compatible with the findings on 89Zr-bevacizumab PET and tumor VEGF levels in this tumor cell line (data not shown).

Representative hematoxylin and eosin (HE) staining (A); percentage necroses, Ki67 staining, and MVD (right y-axis) (B); representative Ki67 staining (C); and representative von Willebrand factor staining for MVD of control and NVP-AUY922–treated A2780 xenograft (D). Data are presented as mean ± SEM. *P < 0.05.
Furthermore, in neither A2780 nor CP70 xenografts did 2 wk of NVP-AUY922 treatment result in increased tumor necrosis as shown by hematoxylin and eosin staining. In NVP-AUY922–treated and nontreated xenografts, at least 80% of the tumor consisted of vital areas (Fig. 5). Therefore, decreased 89Zr-bevacizumab tumor uptake in A2780 was not due to increased tumor necrosis.
DISCUSSION
In the present study, we have demonstrated that the antiangiogenic effect of HSP90 inhibition can be adequately monitored in vivo using 89Zr-bevacizumab PET. The specificity of the technique is illustrated by the fact that a decreased 89Zr-bevacizumab uptake is related to the presence of an antiangiogenic response (confirmed by means of decreased VEGF levels and MVD) in the A2780 model, whereas no change in 89Zr-bevacizumab uptake was observed in the nonresponsive CP70 model. Additionally, the extent of antiangiogenic response was related to the antiproliferative effect of the NVP-AUY922 treatment. Thus, 89Zr-bevacizumab PET allows in vivo visualization and quantification of early antiangiogenic tumor response to treatment with HSP90 inhibition. As such, it may be used as an early biomarker for the effect of HSP90 inhibition.
It is thought that reduction of VEGF secretion by tumor cells after HSP90 inhibition is mediated via multiple pathways. For example, HSP90 inhibition by 17-AAG reduced epidermal growth factor, insulinlike growth factor type 1, betacellulin, transforming growth factor α, and heregulin β1–induced VEGF secretion by tumor cells in vitro (7,9,11). In prostate cancer cells, geldanamycin treatment decreased hypoxia-inducible factor 1α levels, which corresponded to lower VEGF levels in vitro (8).
In addition, HSP90 inhibition has demonstrated antiangiogenic effects in vivo. We observed in our A2780 xenograft model a decreased MVD after NVP-AUY922 treatment. Similar effects were observed in a human glioblastoma xenograft model (6). Furthermore, it was shown that NVP-AUY922 inhibited endothelial cell function and migration, decreased VEGF-R2 levels on endothelial cells, and reduced hypoxia-inducible factor 1α levels in a human glioblastoma xenograft model (6). These results are comparable to the effects seen with other HSP90 inhibitors. Treatment with 17-AAG and 17-(dimethylaminoethyl-amino)-17-demethoxygeldanamycin (17-DMAG, or alvespimycin), a water-soluble geldanamycin analog, decreased MVD in gastric, colon, and hepatocellular carcinoma xenograft models (7,11,22,23). Additionally, 17-DMAG downregulated VEGF-R2 protein expression on endothelial cells, inhibited VEGF-A–induced Erk and Akt activation, and downregulated total Akt expression (24). Furthermore, treatment with 17-DMAG decreased platelet-derived growth factor–Rβ expression and function on vascular smooth muscle cells (24). All these results signify the antiangiogenic effect of HSP90 inhibition in cancer.
In the present study, we investigated whether this antiangiogenic effect by HSP90 inhibition could be monitored by 89Zr-bevavizumab PET. Previously, it was shown that NVP-AUY922 treatment induced HSP70 and decreased c-Raf and cyclin-dependent kinase 4 in A2780 cells, indicating sufficient sensitivity of this cell line to HSP90 inhibition (6). We treated A2780 and CP70 tumor–bearing mice with comparable dosing schemes that showed efficiency in other xenograft models (6,15). Daily dosing of 50 mg of NVP-AUY922 per kilogram during 7 d reduced tumor growth by 90% in A2780 xenografts, compared with nontreated animals (6). We showed with small-animal PET quantification a vast decrease of 89Zr-bevacizumab tumor uptake in A2780 xenografts, whereas no decrease in 89Zr-bevacizumab uptake was seen in CP70 xenografts. In an HER2 overexpressing xenograft model—in which we evaluated NVP-AUY922 treatment with 89Zr-trastuzumab—we did not observe changes in tumor uptake of radiolabeled control IgG after NVP-AUY922, suggesting that minimal changes in antibody uptake occur because of alterations in perfusion and extravasation (25). Previously, we showed that 89Zr-bevacizumab accumulates in tumor blood vessels and its extracellular matrix, presumably because of localization of large human VEGF isoforms. No binding takes place of mouse-derived VEGF (12,13). To verify whether changes in 89Zr-bevacizumab uptake also reflect changes in total tumor human VEGF levels, we assessed VEGF levels in tumor lysates. The ex vivo VEGF ELISA we used detects both extra- and intracellular human VEGF in tumor lysates. Most likely, equilibrium is present between extracellular and intracellular VEGF. VEGF levels in A2780 xenografts were higher than in CP70 xenografts and they decreased after HSP90 inhibition, whereas VEGF levels in CP70 did not change. These results are in line with our 89Zr-bevacizumab imaging results and demonstrate that 89Zr-bevacizumab small-animal PET can quantify noninvasively both absolute differences between VEGF levels in tumors and changes in VEGF levels after therapy.
These in vivo results mimic our in vitro experiments, in which initial CP70 VEGF secretion levels were lower than A2780 VEGF secretion levels, and no significant effect of HSP90 therapy was observed. It is not completely clear why CP70 did not respond to HSP90 inhibition and why initial VEGF levels were lower with A2780 than with CP70. Several studies have demonstrated a complex interaction between cisplatin and HSP90. Cisplatin can bind to the C-terminal domain of HSP90, promoting a conformational change in HSP90, whereas HSP90 inhibitors interact with the N-terminal domain of HSP90 (26). Combined treatment of cisplatin and HSP90 inhibition has shown additive and antagonistic effects in colon and ovarian tumor cell lines (18,27,28). For instance, in A2780 tumor cells concomitant administration of 17-AAG and carboplatin was antagonistic in vitro, whereas 17-AAG followed by carboplatin or vice versa was additive both in vitro and in vivo (18). Future studies should elucidate the interaction between cisplatin and HSP90 therapy in both cisplatin-sensitive and -resistant models. This complexity does, however, point to the relevance of an early predictive factor (29).
Other client proteins that have been used for noninvasive monitoring of HSP90 therapy are HER2 and EGFR. Smith-Jones et al. demonstrated that the early degradation of HER2 (−50%) during 17-AAG therapy could be monitored by small-animal PET in a BT474 breast cancer xenograft model, using 68Ga-DCHF, a F(Ab′)2 fragment of the HER2 antibody trastuzumab (30,31). Furthermore, decreased HER2 expression measured by small-animal PET corresponded to reduced tumor growth. In contrast, tumor uptake of 18F-FDG (the most widely used tumor tracer, which visualizes glucose uptake) did not change after HSP90 therapy (31). Likewise, 64Cu-DOTA-cetuximab was used to monitor EGFR degradation by 17-AAG in a PC-3 prostate cancer xenograft model (32). Uptake of 64Cu-DOTA-cetuximab in 17-AAG–treated animals was lower (−39%) than in nontreated animals at 48 h after the last administration of 17-AAG, which corresponded to a decreased EGFR staining in tumor samples. In contrast to EGFR and HER2—which are abundantly expressed in a selected group of cancer patients—VEGF plays an important, universal role in cancer; therefore, 89Zr-bevacizumab imaging is likely to be more widely applicable to the monitoring of HSP90 inhibition.
CONCLUSION
89Zr-bevacizumab PET was in line with the antiangiogenic response and direct antitumor effects after NVP-AUY922 treatment, supporting the specificity of 89Zr-bevacizumab PET as a sensitive technique to monitor the antiangiogenic response of HSP90 inhibition in vivo. Exploration of 89Zr-bevacizumab PET in phase II studies as an early predictive marker during HSP90 inhibition has been started.
Acknowledgments
This study was supported by a personal grant to one of the authors and grants RUG 2007-3739 and RUG 2009-4273 from the Dutch Cancer Society. Michael R. Jensen, Cornelia Quadt, and Carlos Garcia-Echeverria are employees and stockholders of Novartis Pharma AG.
Footnotes
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication September 24, 2009.
- Accepted for publication January 11, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Roadmap for the Development and Clinical Translation of Optical Tracers Cetuximab-800CW and Trastuzumab-800CW
- Near-infrared fluorescence molecular endoscopy detects dysplastic oesophageal lesions using topical and systemic tracer of vascular endothelial growth factor A
- Molecular Imaging of Ovarian Cancer
- Antibody Positron Emission Tomography Imaging in Anticancer Drug Development
- A Tyrosine Kinase Inhibitor-Based High-Affinity PET Radiopharmaceutical Targets Vascular Endothelial Growth Factor Receptor
- 89Zr-trastuzumab and 89Zr-bevacizumab PET to Evaluate the Effect of the HSP90 Inhibitor NVP-AUY922 in Metastatic Breast Cancer Patients
- Glypican-3-Targeted 89Zr PET Imaging of Hepatocellular Carcinoma: Where Antibody Imaging Dares to Tread
- In Vivo Visualization of MET Tumor Expression and Anticalin Biodistribution with the MET-Specific Anticalin 89Zr-PRS-110 PET Tracer
- A Phase 1, Dose-Escalation, Pharmacokinetic and Pharmacodynamic Study of BIIB021 Administered Orally in Patients with Advanced Solid Tumors
- Interrogating Tumor Metabolism and Tumor Microenvironments Using Molecular Positron Emission Tomography Imaging. Theranostic Approaches to Improve Therapeutics
- 89Zr-Bevacizumab PET Imaging in Primary Breast Cancer
- Measurement of Tumor VEGF-A Levels with 89Zr-Bevacizumab PET as an Early Biomarker for the Antiangiogenic Effect of Everolimus Treatment in an Ovarian Cancer Xenograft Model
- Advances in Immuno-Positron Emission Tomography: Antibodies for Molecular Imaging in Oncology
- Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes
- Nanobodies Targeting the Hepatocyte Growth Factor: Potential New Drugs for Molecular Cancer Therapy
- Molecular Imaging of Tumors with Radioactive Labeled Antibodies from Laboratory to the Clinic
- Toward Molecular Imaging-Driven Drug Development in Oncology