


Abstract
We investigated the effects of cisplatin and the hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG) in combination in a panel of human colon adenocarcinoma cell lines that differ in their p53 and mismatch repair status. Analysis of cytotoxicity after combined treatment revealed additive effects of cisplatin and 17-AAG in the HCT 116, DLD1, and SW480 cell lines and antagonism in HT-29 cells. Clonogenic assays demonstrated antagonism in HT-29, an additive effect in SW480, and synergism in HCT 116 and DLD1 cell lines. Analysis of signaling pathways revealed that cisplatin-induced activation of c-Jun N-terminal kinase (JNK) was fully blocked by 17-AAG in HT-29 and SW480 cells, whereas in HCT 116 and DLD1 cells it was inhibited only partially. The activation of caspases was also more pronounced in DLD1 and HCT 116 cell lines. These data suggested that a minimal level of apoptotic signaling through JNK was required for synergism with this combination. To test this hypothesis, we used the specific JNK inhibitor SP600125; when JNK was inhibited pharmacologically in HCT 116 and DLD1 cells, they demonstrated increased survival in clonogenic assays. Alternatively, sustained activation of JNK pathway led to an increase of the cytotoxicity of the cisplatin/17-AAG combination in HT-29 cells. Taken together, these data suggest that the synergistic interaction of this combination in colon cancer cell lines depends on the effect exerted by 17-AAG on cisplatin-induced signaling through JNK and associated pathways leading to cell death. An implication of that finding is that quantitative effects of signaling inhibitors may be critical for their ability to reverse cisplatin resistance.
The active search for drugs directed against novel molecular targets for the treatment of cancer led to the development and introduction into clinical trials of several classes of signaling inhibitors, among them the inhibitors of the molecular chaperone Hsp90 (Workman and Kaye, 2002). Geldanamycin and its analog 17-AAG act by binding to the ATP/ADP pocket of Hsp90 and cause the destabilization of its complexes with client proteins. Dissociation of hsp90-protein complexes triggers the proteosomal degradation of a number of important signaling proteins, including AKT, Raf-1, ErbB2 kinase, mutant p53, and others (Neckers, 2002; Workman and Maloney, 2002). The inhibitory effect of geldanamycin on signaling through the activator protein 1 and nuclear factor κB transcription factors has also been shown (Vasilevskaya and O'Dwyer, 1999; Lewis et al., 2000). 17-AAG has demonstrated antitumor activity both in preclinical models (Schulte and Neckers, 1998) and in phase I trials (Münster et al., 2001a; Wilson et al., 2001) as single agent. Recent studies have shown that 17-AAG enhances the therapeutic effects of doxorubicin and paclitaxel (Taxol) in several cellular models (Blagosklonny et al., 2001; Munster et al., 2001b) making it a promising candidate for use in combination with common therapeutic agents, including DNA-damaging compounds. One of the DNA-damaging drugs used for treatment of several tumor types is cisplatin [cis-diamminedichloroplatinum(II); DDP] (Rosenberg, 1999). Treatment with DDP or its analogs causes the formation of DNA-platinum adducts, followed by cell cycle arrest and cell death primarily through apoptosis (Johnson et al., 2001) in both p53-proficient and -deficient cell lines (Allday et al., 1995; Seki et al., 2001), although under certain circumstances DDP has been shown to cause necrosis (Gonzales et al., 2001). The role of MAP kinase cascades in mediating cisplatin effects is under active investigation: work to date has generated numerous conflicting reports implicating MAP kinases in both enhancement and inhibition of cisplatin cytotoxicity (Yang et al., 2003). These mechanisms may also be critical to an interaction with ansamycins, because we have previously shown that both geldanamycin and 17-AAG were able to inhibit MAPK pathways (Vasilevskaya and O'Dwyer, 1999; Vasilevskaya et al., 2003). The conflicting results in the literature and recent description of the importance of both duration and intensity of signaling as a source of response specificity (Marshall, 1995; Wolfman et al., 2002) prompted a more detailed evaluation of these interactions.
We showed recently that the DDP/17-AAG combination demonstrated additive effects in HCT 116 but antagonized each other in HT-29 cells (Vasilevskaya et al., 2003). Therefore, we postulated that p53 status could contribute to observed phenotype. The introduction of a transcriptionally inactive dominant-negative mutant of p53 into HCT 116 cells partially recapitulated antagonism of DDP/17-AAG combination in this cell line, mainly because of inhibition of cisplatin-induced signaling through Fas and caspases 8 and 3, rather than changes in MAPK signaling. To further investigate the role of p53 and stress pathways in these phenomena, we expanded our study to two more p53-deficient colon cancer cell lines, DLD1 and SW480. SW480 cells are mismatch repair-proficient and express high levels of inactive mutant p53 protein, similar to HT-29 cells; the DLD1 cell line is mismatch repair-deficient and has levels of nonfunctional p53 comparable with those of HCT 116 cells. We studied the cellular responses of this panel to the DDP/17-AAG combination with a focus on the role of JNK and apoptotic signaling in particular.
We show here that the DDP/17-AAG combination is additive or synergistic in all cell lines, irrespective of p53 and mismatch repair status, except for the HT-29 line. The cytotoxic effect of the combination is associated with the level of JNK activity reaffirming the pro-apoptotic role of the JNK pathway. In the HCT 116 cell line, down-regulation of JNK activity by the specific inhibitor SP600125 (Bennet et al., 2001) led to increased survival because of inhibition of apoptotic pathways; similar effects of JNK inhibition on clonogenic survival were observed for DLD1 cells. The activation of caspases was more pronounced in DLD1 and HCT 116 cell lines, compared with the less sensitive HT-29 and SW480 cell lines, which also demonstrated a stronger inhibitory effect of 17-AAG on cisplatin-induced JNK signaling. In a complementary fashion, when signaling through JNK was enhanced in HT-29 cells, a shift from antagonism to additivity for the DDP/17-AAG combination was observed in clonogenic assays. Taken together, these data suggest that the cytotoxic efficiency of cisplatin in combination with 17-AAG depends, at least in part, on the quantitative effect the latter drug exerts on cisplatin-induced signaling through JNK and associated pathways leading to cell death.
Materials and Methods
Cell Lines and Reagents. Colon cancer cell lines HT-29, HCT 116, DLD1, and SW480 were purchased from American Type Culture Collection (Manassas, VA). Cells were cultivated in Eagle's minimal essential media supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml), streptomycin (100 U/ml), and Fungizone (Invitrogen, Carlsbad, CA). Cultures were maintained in a humidified incubator at 37°C in 5% CO2-95% air. 17-AAG was kindly provided by Dr. Edward Sausville (Developmental Therapeutics Program, National Cancer Institute, Bethesda, MD). Stock solutions of 17-AAG and the JNK inhibitor SP600125 (Biomol, Plymouth Meeting, PA) were prepared in dimethyl sulfoxide, aliquoted, and stored at -20°C. Cisplatin (Sigma Chemical Co., St. Louis, MO) was dissolved in sterile PBS at a final concentration of 1 mg/ml (3.33 mM). The caspase 8 inhibitor, IETD-CHO, was purchased from BioSource International, Inc. (Camarillo, CA). A HA-tagged constitutively active mutant of SEK1 (Guan et al., 1998) cloned into pcDNA3 plasmid was kindly provided by Dr. Dennis Templeton (Case Western Reserve University, Cleveland, OH).
MTT Assay and Isobologram Analysis. MTT assays and isobologram analysis were carried out as described in (Vasilevskaya et al., 2003). Briefly, cells were plated in 96-well plates at a density of 2 × 103 (HT-29 and SW480) or 1.5 × 103 (HCT 116 and DLD1) cells per well and treated with various concentrations of DDP (six points ranging from 0 to 8 μM) and 17-AAG (seven points ranging from 0 to 100 nM) alone and in combination. Cytotoxicity was measured using the standard MTT assay after a 72-h exposure to drugs. After each experiment, survival curves were generated, two for DDP and 17-AAG alone, and 13 for the 42 pairs of drug combinations. The IC50 values for each drug in combination were determined, and IC50 units were derived as the ratio of IC50 for DDP and 17-AAG in this particular drug combination relative to the IC50 of each drug alone (which was designated as 1) for each cell line. The method of Tremblay et al. (2000) was used to determine combination indices at IC50, which were calculated as the sum of ratios of IC50 of each drug in DDP/17-AAG combination resulting in 50% of cell kill, compared with untreated control, to IC50 of this drug when used alone. Combination index values above 0.9 but less than 1.1 represent additive effect, and those less than 0.9 or above 1.1 represent synergism or antagonism, respectively.
Clonogenic Assays. For clonogenic assays, cells were plated in six-well plates at a density of 250 per well and after 24 h, DDP and 17-AAG were added at the concentrations corresponding to IC25 for each drug for an additional 72 h. After removal of drug-containing media, cells were cultivated for 7 to 10 days. Formed colonies were then fixed in 75% ethanol, stained with Coomassie Blue (Sigma), and counted manually. All experiments were performed at least three times in duplicate. The JNK inhibitor (10 μM of SP600125) was added 2 h before addition of 17-AAG and DDP. Assessment of survival after treatment with high concentrations of drugs (30 μM of CDDP and 500 nM of 17-AAG) was done also; cells were plated in 24-well plates 24 h before addition of drugs, incubated in the presence of 17-AAG and DDP for 48 h, fixed, and stained with Coomassie Blue.
Cell Treatment, Protein Extract Preparation, and Western Blotting. Cells were treated for 24 h with 0.01% DMSO (control), 500 nM of 17-AAG, or 30 μM of cisplatin alone and in combination. Protein extracts were prepared as described previously (Vasilevskaya and O'Dwyer, 1999). The protein concentration of cleared cellular extracts was measured using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Extracts were diluted with lysis buffer to obtain equal protein concentrations, aliquoted, and stored at -70°C. Western blotting was carried out according to the manufacturer's procedure, using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), and the ECL+Plus detection system (Amersham Biosciences, Piscataway, NJ). The antibodies used were: mouse monoclonal antibodies against phospho-ERK, phospho-c-Jun, and phospho-ATF2; rabbit polyclonal antibodies against c-Jun, ERK, JNK, p38, and SEK1; and goat polyclonal antibodies against β-actin and HA-tag from Santa Cruz Biotechnology; rabbit polyclonal antibodies against phosphorylated forms of JNK and p38 from Cell Signaling Technology (Beverly, MA). Mouse monoclonal antibodies raised against caspases 8, 9, and 3 were purchased from Oncogene Research Products (Boston, MA).
Cell Cycle Analysis. Cells were plated into 65-mm dishes (106 cells/dish), and 20 to 24 h later treated with DDP and 17-AAG alone or in combination as described above. After 24 h of drug exposure, samples for FACS analysis were prepared as described previously (Murphy et al., 1996). Briefly, cells were trypsinized, washed with PBS/0.5% FBS and fixed in 70% ethanol at -20°C. After subsequent washes, cells were resuspended in 1 ml of PBS/0.5% FBS, containing 40 U/ml of RNase A, and stained with propidium iodide (16 μg/ml) for 30 min at 37°C, left overnight at 4°C and subjected to analysis of DNA content using FACScan flow cytometer (BD Biosciences, San Diego, CA) and ModFit LT software (Verity Software House, Inc., Topsham, ME).
Caspase Assays. ApoTarget caspase colorimetric protease assay kits were purchased from BioSource International (Camarillo, CA). Cells were plated in 10-cm Petri dishes and subjected to drug treatment as described above. After 24 h, cellular extracts were isolated, and assays were carried out according to the manufacturer's recommendations. All experiments were performed at least three times in duplicate.
Transfections. FuGENE reagent (Roche Applied Science, Indianapolis, IN) was used to transfect HT-29 cells with empty vector or pcDNA3 expressing constitutively active SEK1 (HA-ASEK) according to the manufacturer's recommendations. After 48 h, cells were plated in selective media containing 0.75 mg/ml G418; after cultivation for 7 days, neomycin-resistant cells were pooled together and frozen in several batches. Only cells after one passage were used in experiments.
Statistical Analysis. Data presented were analyzed with unpaired Student's test; p values of less than 0.05 were accepted as a statistically significant difference compared with controls. In figure legends: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Results
Cisplatin and 17-AAG Demonstrate Additive Effects in DLD1 and SW480 Colon Cancer Cell Lines in Isobologram Analysis. The DLD1 and SW480 cell lines were treated as in experiments with HT-29 and HCT 116 lines (Vasilevskaya et al., 2003). Briefly, cytotoxicity after treatment with various concentrations of cisplatin and 17-AAG alone or in combination was assessed by MTT assay, followed by isobologram analysis and calculation of combination indices at IC50. We used the method described by Tsai et al. (1989) to create isobolograms at the IC50 level; IC50 unit values 17-AAG less than 1 were plotted against corresponding IC50 unit values of cisplatin. The distribution of dots along the line connecting values of 1 constitutes an additive effect of two drugs, while scattering below or above represents synergism and antagonism, respectively. Figure 1 shows that in both SW480 and DLD1 cell lines the combination of DDP and 17-AAG exerts at least additive effects despite the absence of functional p53. Table 1 presents combination indices at IC50 for all four cell lines studied, with values above 0.9 but less than 1.1 representing additive effect and those less than 0.9 or above 1.1 representing synergism or antagonism, respectively.

Effects of combined cisplatin and 17-AAG in colon cancer cell lines after simultaneous administration. Shown are the isobolograms at IC50 based on the results of MTT assays for cell lines treated with combinations of 17-AAG and DDP. Isobolograms for HT-29 and HCT 116 cell lines are reproduced from earlier publication (Vasilevskaya et al., 2003). Each isobologram combines results from at least three independent experiments.
Cytotoxic effect of cisplatin and 17-AAG combination against colon cancer cell lines Shown are the IC50 values for each drug based on the results of MTT assays, combinatorial indices at IC50 derived from MTT assays, and IC25 concentrations for DDP and 17-AAG established in clonogenic assays.
Cisplatin and 17-AAG Combinations Demonstrate Differential Cytotoxicity in Colon Cancer Cell Lines. Although the MTT assay is commonly used to evaluate the cytotoxicity of various agents, it is more accurate in assessment of growth inhibition as a result of cytotoxicity rather than true cell survival, because MTT methodology does not allow one to distinguish between the living cells and the ones already committed to die. We wished to confirm the results of the MTT assays and to evaluate the cytotoxicity of DDP and 17-AAG combination in colon cancer cell lines further by using two additional approaches: first, we used drugs at the concentrations of IC25 (shown in Table 1) in clonogenic assays and thus were able to define a correlation between results of MTT assays and cell survivability; second, after treatment with high concentrations of drugs (10× IC50, derived from MTT assays) for 24 h, we were able to assess the effects on signaling through stress pathways leading to apoptosis. The results of the clonogenic assays (Fig. 2A) revealed highly variable effects of the DDP/17-AAG combination: from synergism in both HCT 116 and DLD1 cell lines through elevated cell survival in SW480 cells to antagonism in HT-29 cell line. The results of clonogenic assays presented as combination indices are shown below the diagrams and were calculated as follows: CI25 = [(100% - x) + (100% - y)]/100% - z, where x, y, and z are percentages of surviving colonies (compared with untreated controls) after treatment with DDP alone, 17-AAG alone, or with DDP/17-AAG combination, respectively. Thus, the value of 1 represents additivity when the outcome of combination treatment equals cumulative effects of each drug alone used at IC25 concentrations. Values below 0.9 and above 1.1 represent the synergism and antagonism of combination, respectively. After treatment of cells with high doses of drugs, the HCT 116 and DLD1 cells again showed higher sensitivity to the DDP/17-AAG combination than HT-29 and SW480 cells (Fig. 2B).

Effect of cisplatin and 17-AAG combinations on survival of colon cancer cell lines. A, graph demonstrates the results of clonogenic assays with drugs used in combination at the concentrations of IC25 (average values from Table 1) for each drug alone: 17-AAG (□), DDP (▪), and DDP/17-AAG combination ( ). Combination indices are shown below the diagram. Graph shows average values from at least four independent experiments (in duplicate), bars represent standard deviation. B, cells were seeded into 24-well plates 24 h before drug treatment at high concentrations (30 μM DDP and 500 nM 17-AAG). After 48 h, cells were fixed and stained with Coomassie blue. The picture shown is representative of three independent experiments.
). Combination indices are shown below the diagram. Graph shows average values from at least four independent experiments (in duplicate), bars represent standard deviation. B, cells were seeded into 24-well plates 24 h before drug treatment at high concentrations (30 μM DDP and 500 nM 17-AAG). After 48 h, cells were fixed and stained with Coomassie blue. The picture shown is representative of three independent experiments.
Higher Resistance to DDP/17-AAG Combination Is Associated With G2/M Arrest. The antagonism of cisplatin and 17-AAG when added with a 24-h interval in HCT 116 cells (Vasilevskaya et al., 2003) suggested the importance of the impact of each drug on the cell cycle. To investigate whether addition of 17-AAG influences the effect of cisplatin on the cell cycle and thus decreases the toxicity of the drug combination, we evaluated the effects of each drug alone and in combination on cell cycle distribution in the cell lines studied. Asynchronously growing cells were treated as described above, followed by FACS analysis. Data in Table 2 demonstrate that DDP treatment of all cell lines, irrespective of p53 status, resulted in distribution of cells between G1 and S phases of cell cycle with virtually no cells entering G2/M phase, which points to S phase arrest. When HCT 116 cells were treated with 17-AAG alone, most of the cells were arrested in G1, probably because of the presence of functional p53 on the one hand and inhibition of Cdk4/Cyclin D complex by 17-AAG on the other (Srethapakdi et al., 2000). In the p53-deficient cell lines, cells were equally distributed between G1 and G2 phases with profound inhibition of entry into the S phase. This observation points to importance of functional p53 in maintaining 17-AAG-induced G1 arrest in HCT 116 model. The combination of two drugs alleviated differences in cell cycle distribution compared with those after treatment with each drug alone. The proportion of cells arrested in the G2/M phase varied from 8% in SW480 to the highest of 24% in HCT 116 cell line. Thus, higher sensitivity to the combination was associated with G2/M arrest; however, it is unclear whether this difference is significant enough to cause observed phenotypes.
Effect of cisplatin and 17-AAG combination on the cell cycle distribution in colon cancer cell lines Results shown reflect percentage of cells in each phase of cell cycle according to DNA content analysis by FACS. Cells were treated for 24 h with 30 μM DDP and 500 nM 17-AAG alone or in combination before FACS analysis. Data represent average values from two independent experiments.
Inhibition of Cisplatin-Induced Signaling through JNK Is Associated with Higher Resistance to the Combination of DDP and 17-AAG. The results of cytotoxicity assays (Fig. 2B) prompted us to concentrate on the stress signaling pathways leading to cell death to find the basis for different outcomes after DDP/17-AAG treatment. We showed earlier that geldanamycin and 17-AAG differentially affect the signaling through JNK pathway in HT-29 and HCT 116 cell lines (Vasilevskaya et al., 2003). The importance of duration and intensity of signaling through MAP kinase pathways was originally postulated for the ERK pathway (Marshall, 1995), and recently demonstrated for JNK and p38 apoptotic signaling (Wolfman et al., 2002). We hypothesized that quantitative differences in the effects of 17-AAG in these cell lines could be responsible, at least in part, for observed phenotypes, with a functional JNK pathway being an important part of cisplatin-induced signaling leading to cell death. We wished to determine whether the status of the MAP kinase pathways in DLD1 and SW480 cells also correlated with their responses to combined treatment. Figure 3 shows that when used in combination, 17-AAG blocks cisplatin-induced JNK activation, c-Jun phosphorylation, and c-Jun induction in SW480 cells, which mirrors its effects in HT-29 cells but exerts only partial inhibition of this pathway in the DLD1 cell line. It shows also that in SW480 and HT-29 cell lines, JNK is responsible for activation of ATF2, because cisplatin-induced activation of p38 MAPK was not augmented by 17-AAG. Thus, in cell lines that demonstrated relative resistance to combined treatment, cisplatin-induced JNK activation was impaired, whereas in other two, in which DDP/17-AAG combination was synergistic, the JNK pathway was affected to a lesser degree. These results supported the hypothesis that the synergism of cisplatin and ansamycins is associated with quantitative changes in signaling through JNK upon treatment.
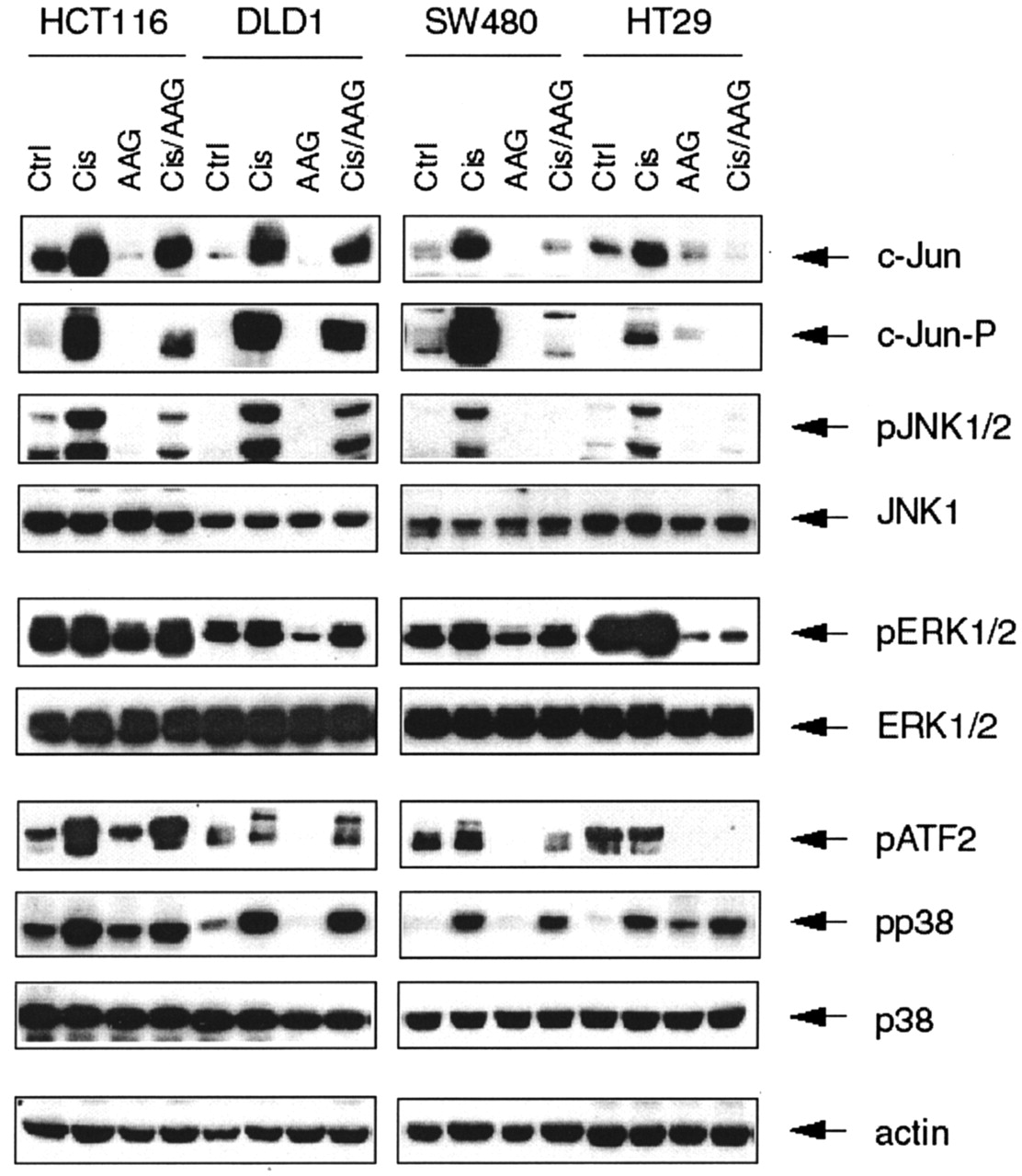
Differences in survival of colon cancer cell lines after treatment with cisplatin and 17-AAG correlate with the status of JNK pathway. Cells were treated with 30 μM DDP and 500 nM 17-AAG for 24 h. Total protein extracts were isolated and subjected to electrophoresis followed by Western blotting analysis. Western blotting with actin antibodies was performed to monitor protein content of each sample. The picture shown is representative of at least two independent experiments.
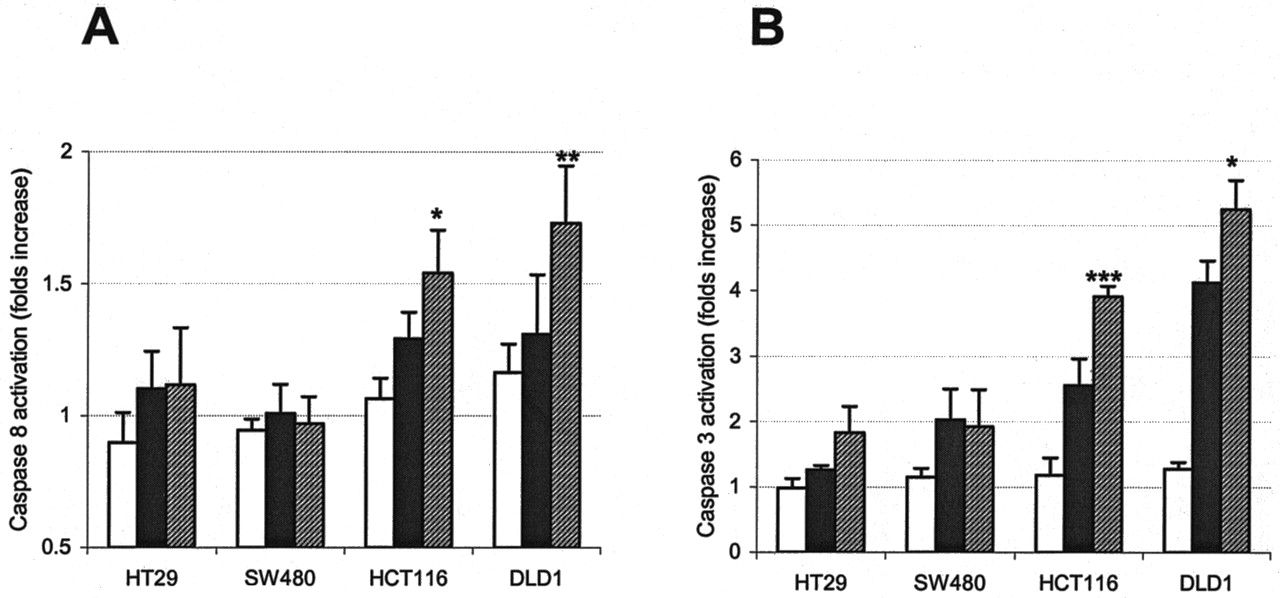
Activation of Caspases in Colon Cancer Cell Lines by the DDP/17-AAG Combination Correlates with the Impact on JNK Signaling. The correlation of DDP/17-AAG cytotoxicity in the panel of colon cancer cell lines with the activity of JNK is consistent with a pro-apoptotic role for this pathway. To further support this assumption, we assayed activation of caspases 8 and 3 in these cells after treatment with high concentrations of DDP and 17-AAG (Fig. 4). Again, cell lines retaining a higher level of JNK activation after drug exposure demonstrated more profound activation of both caspases, which could be represented as HT-29 = SW480 < HCT 116 < DLD1. We could detect neither a noticeable effect of the DDP/17-AAG combination on the activation of caspase 9 in any cell line except HCT 116 nor any significant fluctuation in the expression and/or phosphorylation status of several Bcl-2 family members, including Bcl-2, Bax, and Bcl-XL (not shown).

Differential effects of the cisplatin and 17-AAG combination in colon cancer cell lines correlate with activation of apoptotic pathways. Cells were treated with high concentrations of 17-AAG (□), DDP (▪), and DDP/17-AAG combined ( ) for 20 h, protein extracts were isolated and used in colorimetric caspase 8 (A) and caspase 3 (B) assays as described under Materials and Methods. Graphs represent the average values of three independent experiments in duplicate; bars represent S.D.
) for 20 h, protein extracts were isolated and used in colorimetric caspase 8 (A) and caspase 3 (B) assays as described under Materials and Methods. Graphs represent the average values of three independent experiments in duplicate; bars represent S.D.
Inhibition of JNK in HCT 116 and DLD1 Cells Increases Survival after Combined Treatment. If the pro-apoptotic function of JNK pathway is critical for a synergistic interaction between cisplatin and ansamycins, complete inhibition of the pathway should oppose the synergism. To study this interaction further, we used SP600125, an inhibitor of JNK shown to inhibit JNK1 and JNK2 activation in several cell lines (Bennet et al., 2001). We have shown (Fig. 3) that 17-AAG inhibits DDP-induced JNK activation with consequent decrease in phospho-c-Jun and, because of positive regulation of c-jun gene by c-Jun and ATF2 phosphorylation, total c-Jun level. However, because 17-AAG is not a specific JNK inhibitor, we opted to complement the inhibitory effect of 17-AAG on JNK with specific inhibition by SP600125. As Fig. 5A shows, pretreatment of cells with 10 μM SP600125 led to inhibition of both JNK and c-Jun phosphorylation in HCT 116 cells but did so to a lesser degree in DLD1 cells. The level of total c-Jun protein was reduced accordingly. Treatment of HCT 116 cells with DDP/17-AAG combination in the presence of SP600125 led to the profound inhibition of JNK pathway, with more significant decreases in both phosphorylated and total c-Jun. When SP600125 was used in clonogenic assays with cisplatin and 17-AAG, the resulting JNK inhibition led to increased survival of both the HCT 116 and DLD1 cells. Therefore, the use of a JNK inhibitor led to a transition from synergism to additivity in HCT 116 and DLD1 cells, as reflected in combination indices for clonogenic assays as shown (Fig. 5B). The addition of 17-AAG before cisplatin (to down-regulate basal and cisplatin-induced signaling through JNK) led to a similar shift from synergism to additivity for DLD1 cells in clonogenic assays (not shown). This result is in accord with our previous findings that the scheduling of these agents is critical to their positive interaction (Vasilevskaya et al., 2003).

Modulation of JNK activity affects the cytotoxicity of the DDP/17-AAG combination in HCT 116 and DLD1 cell lines. A, SP600125 inhibits cisplatin-induced JNK activation in HCT 116 cells and, to a lesser extent, in DLD1 cells. HCT 116 and DLD1 cells were treated with 10 μM of SP600125 for 2 h before addition of DDP and 17-AAG for 24 h as described for Fig. 3, followed by protein extract preparation and Western blot analysis. B, inhibition of JNK results in increased survival of HCT 116 and DLD1 cells in clonogenic assays. Graph demonstrates survival of cells treated with 17-AAG (□), DDP (▪), and DDP/17-AAG combination ( ) in the absence or presence (asterisk) of 10 μM SP600125. Combination indices are shown below the diagram. Graph represents the average of values from at least four independent experiments in duplicate; bars represent S.D.
) in the absence or presence (asterisk) of 10 μM SP600125. Combination indices are shown below the diagram. Graph represents the average of values from at least four independent experiments in duplicate; bars represent S.D.
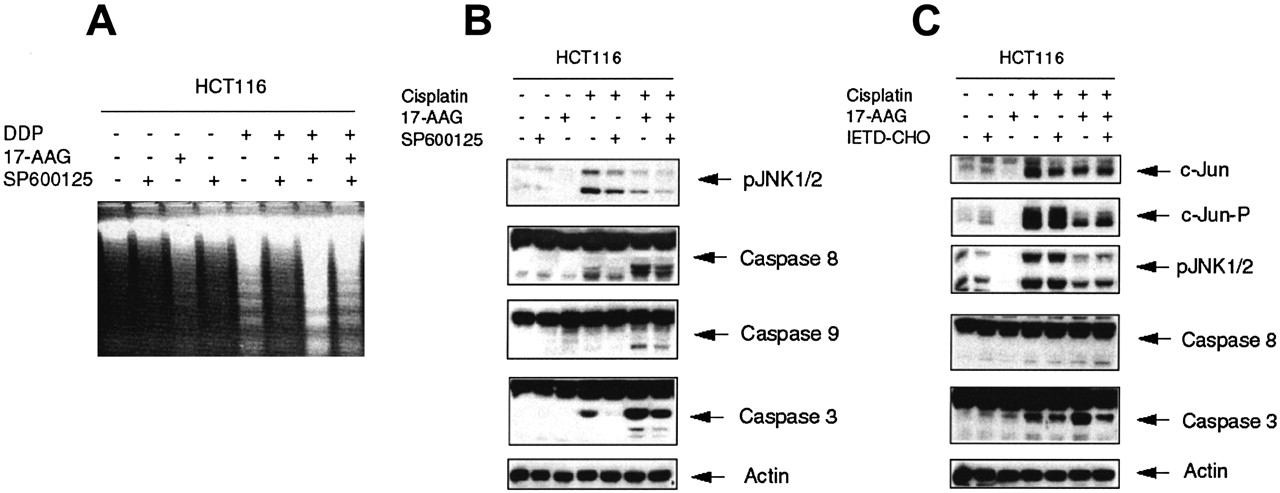
Inhibition of JNK in the HCT 116 Cell Line Interferes with Apoptosis Induced by Cisplatin Alone and in Combination with 17-AAG. Because cisplatin induces cell death mostly by apoptosis, it is likely that increased resistance of HCT 116 cells to the DDP/17-AAG combination after complete JNK inhibition could be the result of impaired apoptotic signaling by cisplatin. Indeed, DNA fragmentation analysis revealed that SP600125 caused inhibition of apoptosis in HCT 116 cells treated with high doses of cisplatin alone and, to a lesser degree, in combination with 17-AAG (Fig. 6A). These data correlated with inhibition of caspases in the presence of a JNK inhibitor (Fig. 6B): SP600125 blocked activation of caspases 8 and 3 after treatment with cisplatin alone and significantly inhibited activation of caspases 8, 9, and 3 caused by the DDP/17-AAG combination. Pretreatment of HCT 116 cells with a specific inhibitor of caspase 8, IETD-CHO, did not affect the level of JNK phosphorylation but led to diminished activation of caspase 3, indicating that JNK is activated upstream of caspase 8 during cisplatin-induced apoptosis in the HCT 116 cell line (Fig. 6C). These data demonstrate again that inhibition of JNK in HCT 116 cells effectively interferes with caspase 8-mediated apoptosis caused by cisplatin alone and in combination with 17-AAG.

Inhibition of JNK in HCT 116 cells interferes with apoptosis caused by cisplatin alone and in combination with 17-AAG. Inhibition of JNK leads to impaired apoptosis in HCT 116 cells. Cells were treated as described for Fig. 5A, followed by DNA fragmentation analysis (A) or protein extract preparation and Western blot analysis (B) as described under Materials and Methods. C, cisplatin-induced JNK activation is independent of caspase 8 activation. HCT 116 cells were treated with 10 μM of caspase 8 inhibitor (IETD-CHO) for 2 h before drug treatment (as described for Fig. 3), followed by Western blot analysis. All pictures are representative of at least two independent experiments.
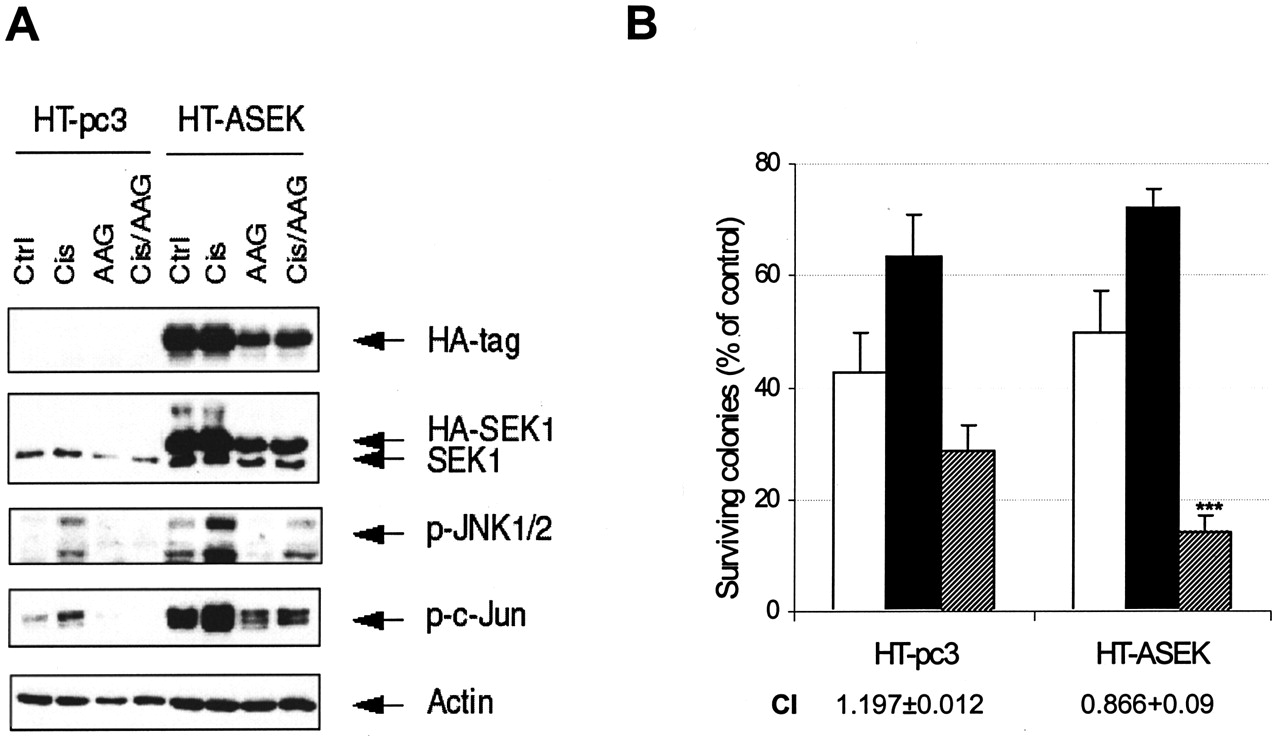
Persistent Activation of the JNK Pathway by a Constitutively Active Form of Sek1 Leads to Enhanced Cytotoxicity of the DDP/17-AAG Combination in HT-29 cells. Finally, we attempted to attenuate the strong inhibitory effect of 17-AAG on the JNK pathway in HT-29 cells by introducing the constitutively active form of SEK1 into this cell line. We expected that this would lead to a persistent activation of the JNK pathway and incomplete inhibition of JNK by 17-AAG, which in turn would result in additive interaction of DDP and 17-AAG in HT-29 cells. Cells were transfected with empty vector or pcDNA3 with cloned HA-tagged construct (HA-ASEK). We used a neomycin-resistant pool instead of transiently transfected cells because the HT-29 cell line demonstrates rather low efficiency of transfection. Figure 7A demonstrates the expression of mutant SEK1 in the pool of neomycin-resistant cells (HT-ASEK) compared with control cells (HT-pc3), and the enhancement of cisplatin-induced signaling through JNK in the presence of 17-AAG. Both control and HA-ASEK-expressing cells were subjected to clonogenic assays according to the scheme described above. Results of clonogenic assays demonstrated enhanced cytotoxicity of DDP/17-AAG combination in HT-29 cells expressing mutant SEK1, with a shift in combination indices (Fig. 7B). These data serve as additional confirmation of the pro-apoptotic role of JNK in cisplatin signaling and importance of its activity for cytotoxicity of the DDP/17-AAG combination.

Restoration of JNK activity in HT-29 cells enhances cytotoxicity of DDP/17-AAG combination. A, HT-29 cells were transfected with empty vector (HT-pc3) or a constitutively active form of SEK1 (HT-ASEK); neomycin-resistant cells were pooled and subjected to treatment with DDP and 17-AAG alone and in combination (as described for Fig. 3). Western blot analysis demonstrates expression of the mutant form of SEK1 and enhanced signaling through JNK in the presence of 17-AAG. B, clonogenic assays demonstrate shift from antagonism to additivity in HT-ASEK cells upon combined treatment. Graph demonstrates survival of cells treated with 17-AAG (□), DDP (▪), and DDP/17-AAG combination ( ) compared with control—the amount of colonies formed in untreated wells. Combination indices are shown below the diagram and reflect the enhanced cytotoxicity of DDP/17-AAG combination. Plating efficiency for all control experiments was in excess of 85%. Graph represents the average values of three independent experiments in duplicate; bars represent S.D.
) compared with control—the amount of colonies formed in untreated wells. Combination indices are shown below the diagram and reflect the enhanced cytotoxicity of DDP/17-AAG combination. Plating efficiency for all control experiments was in excess of 85%. Graph represents the average values of three independent experiments in duplicate; bars represent S.D.
Discussion
Platinum compounds are routinely used in treatment of several types of tumors, but acquired resistance often diminishes the efficacy of these drugs. Accumulated evidence supports a role for altered signaling in mediating resistance. One of the ways to enhance the cytotoxicity of cisplatin in sensitive cancers, and to overcome drug resistance in unresponsive tumors, is to target signaling pathways with specific inhibitors in combination with cisplatin. In this study, we investigated the cytotoxic effects of cisplatin, combined with the Hsp90 inhibitor 17-AAG, in a panel of colon cancer cell lines. This panel of colon carcinomas represents a useful model by virtue of their extensive molecular characterization with respect to p53 and DNA repair mechanisms. They provide a model for the application of ansamycin therapy to other solid tumors.
The major role of inhibition of PI3K/AKT and Raf-1/MEK/MAPK pathways in the mechanism of 17-AAG antitumor activity as a single agent and in combination with paclitaxel was demonstrated in various cellular models and in xenograft tumors (Schulte and Neckers, 1998; Hostein et al., 2001; Münster et al., 2001b; Basso et al., 2002; Calabrese et al., 2003; Solit et al., 2003). Interaction with these pathways may form the basis for the synergistic interaction of 17-AAG and UCN01, protein kinase C, and Chk1 inhibitor (Jai et al., 2003). In this study, we demonstrated that 17-AAG can be successfully combined with cisplatin, but the synergism of this combination depends on the extent of JNK pathway inhibition by 17-AAG.
Biological characteristics of cancer cells that have been proposed to influence cisplatin cytotoxicity include p53 status, DNA repair (including mismatch repair), cell cycle distribution, and signaling to apoptosis (Rosenberg, 1999; Manic et al., 2003). Our model permits us to draw conclusions regarding roles of each of these factors in the interaction of 17-AAG and cisplatin. We showed previously that modulation of p53 function led to the reversal of synergism and partial expression of an antagonistic phenotype in an HCT 116-derived cell line when treated with the DDP/17-AAG combination, as a result of impaired Fas-mediated apoptosis in this particular cellular model (Vasilevskaya et al., 2003). However, in the panel of colon cancer cell lines presented here, the outcome of combinatorial treatment did not correlate with p53 status; only the HT-29 cell line demonstrated antagonism of the DDP/17-AAG combination in both MTT and clonogenic assays. Mismatch repair has been shown to contribute to cisplatin sensitivity (Branch et al., 2000). But again, we did not observe a strong correlation between mismatch repair status and the sensitivity of colon cancer cell lines either to cisplatin, which was in accord with previous reports (Branch et al., 2000; Sergent et al., 2002) or to 17-AAG as a single agent (Table 1). We assume, therefore, that mismatch repair status cannot be considered a major factor in responses of colon cancer cell lines to the treatment with DDP/17-AAG combination. The status of nucleotide excision repair as a possible factor in DDP/17-AAG interaction was not explored in this study. The effects of DDP and ansamycins on cell cycle distribution are cell specific; for each drug, the ability to cause both G1/S and G2/M arrest was reported (Knudsen et al., 2000; Srethapakdi et al., 2000; Blagosklonny et al., 2001). Our data (Table 2) pointing to association of sensitivity to combination treatment with higher proportion of cells in G2/M phase of cell cycle is in accord with data reported recently for cell lines treated with combination of cisplatin and farnesyl transferase inhibitors (Smalley and Eisen, 2003). However, more detailed studies are required to determine the relationship of this affect to cytotoxicity.
The role of the JNK pathway in cellular responses to cisplatin is the focus of numerous studies yielding controversial results (reviewed in Vasilevskaya and O'Dwyer, 2003). The majority of publications supporting an antiapoptotic role of JNK in cells treated with cisplatin are based on the results of experiments in which overexpression of dominant-negative forms of c-Jun or ATF2 was considered to be equivalent to inhibition of JNK (Potapova et al., 2001; Pan et al., 2002; Hayakawa et al., 2003). In addition to the fact that protective role of c-Jun against cisplatin cytotoxicity through enhancement of DNA repair has been shown in these studies, the ability of c-Jun and, most importantly, JNK itself to mediate divergent apoptotic responses to DNA damage should be taken into account. Examples include activator protein 1-dependent induction of Fas-L and JNK-dependent activation of p53, Bad, and Bax or inhibition of Bcl-2 and Bcl-XL (Vasilevskaya and O'Dwyer, 2003). Our data supporting a pro-apoptotic role of the JNK pathway are in accord with reports stressing the importance of this pathway for cisplatin-induced apoptosis in various cellular models (Sanchez-Perez et al., 1998, 2000; Mansouri et al., 2003). For colon cancer cell lines used in this study, the cytotoxicity of the DDP/17-AAG combination correlated with the level of JNK activity that could be sustained in the presence of 17-AAG; partial inhibition was compatible with synergistic effects in HCT 116 and DLD1 cell lines, and full inhibition resulted in antagonism in HT-29 cell line (Fig. 1C and 2). Activation of caspases followed the same pattern (Fig. 3), supporting the JNK-dependent nature of DDP/17-AAG induced apoptosis. The necessity for functional JNK was also demonstrated in colony-forming assays, when both drugs were used at low concentrations, and in which the inhibition or sustained activation of JNK resulted in shifts to higher or lower resistance to the combination, respectively (Fig. 4B and 6B). Thus, complete abrogation of signaling through the pathway is associated with resistance to entry into apoptosis as a consequence of treatment with cisplatin, whereas a small amount of residual activity of JNK permits such a consequence, and promotes additivity or synergy. In a study exploring the role of N-Ras in JNK signaling, a minimal level of activity of JNK was both necessary and sufficient to permit activation of apoptosis in response to tumor necrosis factor-α (Wolfman et al., 2002). These data are consistent with a recently published study of ovarian cancer cells in which abrogation of JNK signaling was a marker of resistance (Mansouri et al., 2003).
In conclusion, the extent of the inhibition 17-AAG exerts on JNK signaling that leads to cell death is a key factor in differential responses of colon cancer cell lines to combination of cisplatin and 17-AAG. However, other features of 17-AAG, such as depletion of established and/or yet unknown antiapoptotic hsp90 client proteins, could contribute to the synergistic cytotoxicity of this combination and shift the balance toward cell death. In this regard, the inhibition of nuclear factor κB function might be especially important, because several of its transcription targets have been shown to play an antiapoptotic role (reviewed in Pahl, 1999). This aspect of action of 17-AAG and platinum compounds combination in colon cancer cell lines is currently under investigation. Combination studies of ansamycins and cytotoxic drugs in cancer treatment trials will need to take these interactions into account.
Footnotes
-
ABBREVIATIONS: hsp90, 90-kDa heat-shock protein; 17-AAG, 17-allylamino-17-demethoxygeldanamycin; DDP or cisplatin, cis-diamminedichloroplatinum (II); MAP, mitogen-activated protein; MAPK, mitogen-activated protein kinase; JNK, c-Jun NH2-terminal kinase; SP600125, anthra[1,9-cd]pyrazol-6(2H)-one 1,9-pyrazoloanthrone; FBS, fetal bovine serum; PBS, phosphate-buffered saline; HA, hemagglutinin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium; FACS, fluorescence-activated cell sorting; ERK, extracellular signal-regulated kinase (same as MAPK); IETD-CHO, Ile-Glu-Thr-Asp-aldehyde; HA-ASEK, hemagglutinin-tagged active SEK1; SEK1, stress-activated protein kinase/ERK kinase; ATF2, activating transcription factor 2.
-
This work was supported in part by National Cancer Institute grant CA49820.
- Received July 24, 2003.
- Accepted October 10, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}