


Abstract
Phosphodiesterases are enzymes that inactivate the intracellular second messengers 3′,5′-cyclic adenosine-monophosphate and/or cyclic guanosine-monophosphate. Of all 11 known phosphodiesterase families, phosphodiesterase-10A (PDE10A) has the most restricted distribution, with high expression in the striatum. PDE10A inhibitors are pursued as drugs for treatment of neuropsychiatric disorders. We have synthesized and evaluated 18F-JNJ41510417 as a selective and high-affinity radioligand for in vivo brain imaging of PDE10A using PET. Methods: The biodistribution of 18F-JNJ41510417 was evaluated in rats. Rat plasma and perfused brain homogenates were analyzed by high-performance liquid chromatography to quantify radiometabolites. Dynamic small-animal PET was performed in rats and in wild-type and PDE10A knock-out mice and compared with ex vivo autoradiography. Blocking and displacement experiments were performed using the nonradioactive analog and other selective PDE10A inhibitors. Results: Tissue distribution studies showed predominant hepatobiliary excretion, sufficient brain uptake (0.56 ± 0.00 percentage injected dose at 2 min after tracer injection), and continuous accumulation of the tracer in the striatum over time; rapid washout of nonspecific binding from other brain regions was observed. Polar radiometabolites were detected in plasma and brain tissue. Dynamic small-animal PET showed continuous tracer accumulation in the striatum, with rapid decline in the cortex and cerebellum. Pretreatment and chase experiments with PDE10A inhibitors showed that the tracer binding to PDE10A was specific and reversible. Imaging in PDE10A knock-out and wild-type mice further confirmed that binding in the striatum was specific for PDE10A. Conclusion: Experiments in rats and PDE10A knock-out mice indicate that 18F-JNJ41510417 binds specifically and reversibly to PDE10A in the striatum, suggesting that this new fluorinated quinoline derivative is a promising candidate for in vivo imaging of PDE10A using PET.
Phosphodiesterase-10A (PDE10A) belongs to a family of cyclic nucleotide phosphodiesterases, enzymes that hydrolyze adenosine and/or guanosine 3′,5′-cyclic monophosphates (cAMP and cGMP, respectively). These cyclic nucleotides play an important role as second messengers in the central nervous system, serving to regulate a wide variety of neuronal functions. The human genome encodes 21 phosphodiesterase genes that are categorized into 11 distinct families. The PDE10A enzyme is the single member of the PDE10 family (1). Of all known phosphodiesterase families, PDE10A has the most restricted distribution, with high expression in the brain and testes only (2,3). In the brain, PDE10A messenger RNA and protein are highly abundant in the medium spiny neurons of the striatum, the principal input site of the basal ganglia which is involved in the regulation of motor, appetitive, and cognitive processes (3,4). Studies using papaverine (a relatively selective PDE10A inhibitor with a half-maximal inhibitory concentration of 36 nM) and behavioral experiments with PDE10A knock-out (KO) mice have indicated that PDE10A inhibition results in activation of the medium spiny neurons (5–7). These findings suggest that PDE10A inhibitors may be a novel therapeutic approach to the treatment of diseases characterized by a reduced activity of these neurons, such as schizophrenia, Huntington's disease, Parkinson's disease, obsessive-compulsive disorder, and addiction (1,8–10). Noninvasive imaging of PDE10A using PET would allow the distribution of this enzyme to be studied in vivo in these diseases. Furthermore, noninvasive imaging would be useful for the clinical development of PDE10A inhibitors, by giving direct insight into the relationship between enzyme activity and administered dose of the candidate drug (11–14). A first attempt to visualize PDE10A has recently been made by Tu et al. (15), who successfully radiolabeled the relatively selective PDE10A inhibitor papaverine with 11C. Although 11C-papaverine showed selective PDE10A binding in vitro, it failed in vivo because of rapid washout of the tracer from the striatum, as was observed in rat biodistribution studies and small-animal PET brain imaging in monkeys (15).
The aim of this work was to synthesize and evaluate a specific and selective radioligand for imaging of PDE10A in the living brain using PET. Initial in vitro PDE10A inhibition studies, performed by Johnson & Johnson Pharmaceutical Research and Development, identified the quinoline JNJ41510417 as a potent PDE10A inhibitor. JNJ41510417 is a structure derived from the chemical series that includes PF-2545920 (Fig. 1, also known as MP-10), a compound developed by Pfizer (16–18). In vitro studies demonstrated that JNJ41510417 is selective for PDE10A (>1,000-fold over PDE2A, PDE3A, PDE4D3, PDE5A3, PDE7A, PDE8A1, PDE9A, PDE6, and PDE1B1) and has a half-maximal inhibitory concentration of 0.5 nM for rat PDE10A (Johnson & Johnson, unpublished data, 2009). In general, passive diffusion across the cerebral endothelium is considered to be possible for molecules with medium lipophilicity (logarithm of octanol/bufferpH7.4 distribution coefficient [log D7.4], 0.9–3.5), a molecular mass less than 600 Da, and a low capacity to form hydrogen bonds (19,20). JNJ41510417 is a small molecule with a molecular mass of 424, a low polar surface area of 0.53 nm2 (53 Å2) (21,22), a measured log D7.4 of 4.7, and a plasma protein binding of 99.5% (both in humans and in rats) (Johnson & Johnson, unpublished data, 2009). Despite its rather high lipophilicity and plasma protein binding, JNJ41510417 seems to be potent, with an in vivo occupancy (median effective dose) of 0.48 mg/kg (Johnson & Johnson, unpublished data, 2009). Because of these characteristics, JNJ41510417 was radiolabeled with 18F (Fig. 1) and evaluated as a potential PET ligand to visualize PDE10A activity in vivo. This article reports the in vivo evaluation of 18F-JNJ41510417 in rats and PDE10A KO mice. The synthesis of the precursor and authentic reference material and the radiochemistry will be published in detail elsewhere.

Chemical structure of PF-2545920 (MP-10; Pfizer), TP-10, and 18F-JNJ41510417, a potential PET ligand for in vivo imaging of PDE10A in brain.
MATERIALS AND METHODS
General
High-performance liquid chromatography (HPLC) analysis was performed on a LaChrom Elite HPLC system (Hitachi) connected to an ultraviolet spectrometer set at 254 nm. For the analysis of radiolabeled compounds, the HPLC eluate (after passage through the ultraviolet detector) was led over a 7.62-cm (3-in.) NaI(Tl) scintillation detector connected to a single-channel analyzer (GABI box; Raytest). Quantification of radioactivity measurements in biodistribution studies and in vivo stability analyses was performed using an automated γ-counter equipped with a 7.62-cm (3-in.) NaI(Tl) well crystal coupled to a multichannel analyzer (1480 Wizard; Wallac). The results were corrected for background radiation and physical decay during counting.
Animals were housed in individually ventilated cages in a thermoregulated (∼22°C), humidity-controlled facility under a 12 h–12 h light–dark cycle, with access to food and water ad libitum. All animal experiments were conducted according to the Belgian code of practice for the care and use of animals, after approval from the university ethics committee for animals.
Biodistribution Studies
The biodistribution study of 18F-JNJ41510417 was performed in healthy male Wistar rats (body weight, 270–340 g) at 2, 30, and 60 min after injection (n = 3/time point). Anesthetized rats (2.5% isoflurane in O2 at a flow rate of 1 L/min) were injected with about 1.1 MBq of the tracer via a tail vein and sacrificed by decapitation at the specified times points. Blood and major organs were collected in tared tubes and weighed. The radioactivity in blood, organs, and other body parts was counted using an automated γ-counter. For the calculation of total radioactivity in blood, blood mass was assumed to be 7% of the body mass.
Plasma Radiometabolite Analysis
After intravenous administration of about 59 MBq of 18F-JNJ41510417 via a tail vein of anesthetized rats (2.5% isoflurane in O2 at a flow rate of 1 L/min), blood was collected via the contralateral tail vein at 2, 30, and 60 min after injection (from the same animal) in lithium heparin–containing tubes (4.5-mL lithium heparin PST tubes, BD Vacutainer; BD) and stored on ice. Next, the blood was centrifuged for 10 min at 3,000 rpm to separate the plasma. Plasma (0.1 mL) was spiked with 10 μg of authentic JNJ41510417 and 10 μg of JNJ41797444. Plasma was then analyzed with HPLC (Chromolith C18, 3 × 100 mm; Merck) eluted with gradient mixtures of 0.05 M sodium acetate (pH 5.5) (A) and CH3CN (B) (0–4 min: isocratic 0% B and flow rate of 0.5 mL/min; 4–14 min: linear gradient 0% B to 90% B and flow rate of 1 mL/min; and 14–17 min: isocratic 90% B and flow rate of 1 mL/min). After passing through an inline ultraviolet detector (254 nm), the HPLC eluate was collected as 1-mL fractions. The radioactivity in all fractions was measured using an automated γ-counter.
Perfused Brain Radiometabolite Analysis
For each studied time point, 2 rats were injected with about 37 MBq of 18F-JNJ41510417. At 30 or 60 min after injection, the rats were sacrificed by an overdose of pentobarbital (200 mg/kg intraperitoneally; Nembutal [CEVA Santé Animale]). The rats were perfused by injection of saline into the right ventricle until the liver turned pale. The brain was isolated; the cerebrum and cerebellum were separated and homogenized in 3 and 2 mL of CH3CN, respectively, for about 2 min. A volume of 1 mL of this homogenate was diluted with an equal volume of water, and 1 mL of the supernatant was filtered through a 0.22-μm filter (Millipore). About 0.5 mL of the filtrate was diluted with 0.1 mL of water and spiked with 10 μg of authentic JNJ41510417 and 10 μg of JNJ41797444. A volume of 0.5 mL of the homogenate extracts was injected onto an HPLC system, consisting of an analytical XBridge column (C18, 5 μM, 3 × 100 mm; Waters) eluted with a mixture of 0.05 M sodium acetate (pH 5.5) and CH3CN (65:35 v/v) at a flow rate of 0.8 mL/min. The HPLC eluate was collected as 1-mL fractions after passing through the ultraviolet detector (254 nm), and radioactivity in the fractions was measured using an automated γ-counter.
Radiometabolite Analysis of Rat Blood, Plasma, and Perfused Brain Homogenate After In Vitro Incubation with 18F-JNJ41510417
Rat blood, plasma, and homogenated perfused brain were incubated with about 7.4 MBq of 18F-JNJ41510417 at 37°C. After 60 min of incubation, the samples were cooled on ice. Blood and brain homogenate samples were processed and analyzed onto reversed-phase HPLC as described earlier in the previous two sections of this article.
Small-Animal PET Studies
Imaging experiments were performed on a Focus 220 microPET scanner (Concorde Microsystems) using male Wistar rats and wild-type (WT) and PDE10A KO mice. During all scan sessions, animals were kept under gas anesthesia (2.5% isoflurane in O2 at a flow rate of 1 L/min). Dynamic 120-min scans were acquired in list mode. Acquisition data were Fourier rebinned in 31 time frames (4 × 15 s, 4 × 1 min, 5 × 3 min, 8 × 5 min, and 10 × 6 min) and reconstructed with filtered backprojection. A summed image (frames 1–31) of the reconstructed data was spatially normalized to a 11C-raclopride template of the rat brain in Paxinos coordinates created in-house (23). The affine transformation was then used to normalize all time frames of the dynamic dataset to allow automated and symmetric volume-of-interest analyses. Time–activity curves were generated for the striatum, cerebral visual and retrosplenial cortex, and cerebellum for each individual scan, using PMOD software (version 3.1; PMOD Technologies Ltd.). The radioactivity concentration in the different brain regions was expressed as standardized uptake value (SUV) as a function of time after injection of the radiotracer by normalization for body weight of the animal and injected dose. Because PDE10A is expressed at a low level in the cerebellum (3), (striatum − cerebellum)/cerebellum (S/C-1) ratios were calculated for the transient equilibrium phase (32–75 min after tracer injection). These ratios were determined in order to provide the relative difference in tracer uptake between cerebellum and striatum and to have a preliminary approximation of binding potential values for the striatum.
Anesthetized rats (2.5% isoflurane in O2 at a flow rate of 1 L/min) were injected with about 37–74 MBq of a high-specific-activity formulation of 18F-JNJ41510417 via the tail vein. For pretreatment and displacement experiments, JNJ41510417, MP-10, and TP-10 were dissolved and administered in a vehicle containing 10% dimethylsulfoxide and 20% (2-hydroxypropyl)-β-cyclodextrine. MP-10 and TP-10 (chemical structure in Fig. 1) are specific PDE10A inhibitors, with subnanomolar potency (1,16,24). Pretreatment studies were performed by subcutaneous administration at approximately 60 min before radiotracer injection. A displacement study was performed by intravenous injection at 60 min after radiotracer injection. Rats were pretreated with authentic JNJ41510417 (self-blocking, 2.5 mg/kg, n = 1), TP-10 (5 mg/kg, n = 1), and several doses of MP-10 (0.5, 1, and 5 mg/kg, n = 2 per dose). Doses and time of pretreatment were based on the results obtained by the study of Schmidt et al. (24). A washout period of at least 4 d was maintained between different pretreatment studies. Also, 2 PDE10A KO mice and 1 WT mouse (J&J PRD, Neuroscience) were injected with 13 MBq of 18F-JNJ41510417 via the tail vein and were scanned dynamically for 60 min using small-animal PET.
Ex Vivo Autoradiography in WT and PDE10A KO Mice
After the small-animal PET scan, the mice were sacrificed, brain was removed, and cerebrum was separated from cerebellum and rapidly frozen in 2-methylbutane (−40°C). Frontal sections (30 μm) from the cerebrum were obtained using a cryotome (Shandon cryotome FSE; Thermo Fisher), mounted on adhesive microscope slides (Superfrost Plus; Thermo Fisher Scientific), and exposed to a phosphor storage screen film (super-resolution screen; Perkin Elmer) for about 24 h. The screens were read using a Cyclone Plus system (Perkin Elmer) and analyzed using Optiquant software (Perkin Elmer). The results are expressed as digital light units/mm2, normalized for body weight of the animal and injected dose ([DLU/mm2] × [body weight/injected dose]).
RESULTS
Biodistribution Studies
Table 1 shows the percentage injected dose (%ID) at 2, 30, and 60 min after injection of the radiotracer. At 2 min after injection of the tracer, about 4.0% of the injected dose was present in the blood and cleared to 2.1% by 60 min after injection. The total brain uptake of the tracer at 2 min after injection was 0.56%. At 60 min after tracer injection, 55% of the injected dose was present in the liver and intestines, whereas urinary excretion of the tracer was minimal (2.2 %ID at 60 min after injection).
Biodistribution of 18F-JNJ41510417 in Normal Rats at 2, 30, and 60 Minutes After Tracer Injection
Table 2 presents the SUVs for the studied brain regions and blood. At 2 min after tracer injection, the radioactivity concentration in the striatum was highest of all brain regions. The striatum was the only brain region in which the tracer concentration increased as a function of time after injection. At 2 min after injection of the tracer, the striatum-to-cerebellum ratio was about 1.3, and this ratio increased to 7.0 by 60 min after injection. Striatum-to-cortex and striatum-to-hippocampus ratios were also 5.8 or more at 60 min after tracer injection.
18F-JNJ41510417 Concentration in Different Brain Regions and Blood at 2, 30, and 60 Minutes After Tracer Injection
Radiometabolite Analysis of 18F-JNJ41510417 in Plasma of Rats
Figure 2A shows the reconstructed radiochromatograms from rat plasma analysis at 2, 30, and 60 min after tracer injection, with the peak corresponding to intact 18F-JNJ41510417 eluting with a retention time of approximately 13 min. The small peak eluting just before (retention time, ∼11 min) the intact tracer was identified as 18F-JNJ41797444 (Fig. 2B). Unidentified polar radiometabolite(s) eluting around 2 min were also detected in the brain (see below). An overview of the results from the plasma radiometabolite analysis is presented in Table 3. At 2 min after injection of the radiotracer, almost all the recovered radioactivity in plasma was in the form of intact tracer. At 30 min after tracer injection, 11% of polar metabolites were found in plasma, and this amount increased to 27% at 60 min after injection. No apolar metabolites were detected. The recovery of the HPLC and Chromolith column–injected radioactivity was 89% (n = 2).

(A) Reconstructed radiochromatograms of rat plasma analysis at 2 (blue), 30 (pink), and 60 (yellow) min after injection of 18F-JNJ41510417. (B) Chemical structure of radiometabolite 18F-JNJ41797444. Retention time was approximately 11 min in radiochromatogram (A). no. = number.
Relative Percentages of Intact Tracer and Radiometabolites in Rat Plasma at 2, 30, and 60 Minutes After Injection of 18F-JNJ41510417
Radiometabolite Analysis of 18F-JNJ41510417 in Perfused Cerebrum and Cerebellum of Rats
An overview of the results from the perfused rat brain radiometabolite analysis is presented in Table 4. The fraction of polar radiometabolites detected in the cerebellum was higher than that in the cerebrum. At 30 min after tracer injection, about 95% of the recovered radioactivity was present as intact tracer in the cerebrum; in the cerebellum, this recovered fraction was approximately 90%. After 60 min, the amount of intact tracer in the cerebrum decreased to approximately 86%; in the cerebellum, this fraction was about 74%. The recovery of the HPLC and XBridge column–injected radioactivity was 79% (n = 2).
Relative Percentages of Intact Tracer and Radiometabolites in Perfused Rat Cerebrum and Cerebellum at 30 and 60 Minutes After Injection of 18F-JNJ41510417
Radiometabolite Analysis of Rat Blood, Plasma, and Perfused Brain Homogenate After In Vitro Incubation with 18F-JNJ41510417
18F-JNJ41510417 was shown to be metabolically stable in rat whole blood, rat plasma, and rat brain homogenate in vitro. The fraction of polar metabolites observed after 60 min of incubation at 37°C was negligible (<2.2%).
Small-Animal PET
Baseline Scans
A high-intensity signal was observed in the striatum with only background radioactivity in cortical regions and the cerebellum (Fig. 3). High uptake was also present in the Harderian glands. After the high initial brain uptake, radioactivity cleared from the cortical regions and cerebellum, whereas the radiotracer activity increased in the striatum (Fig. 4A).
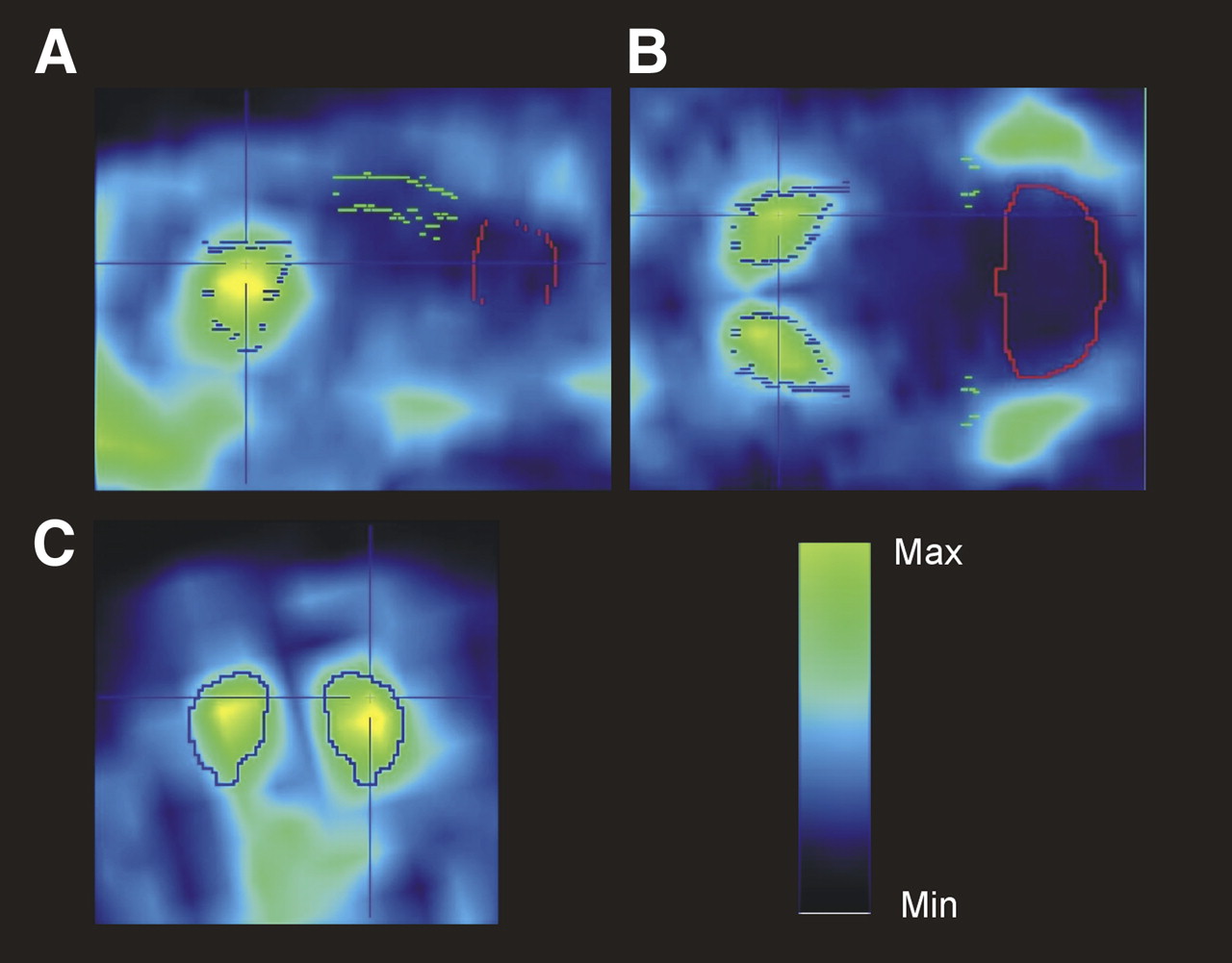
Sagittal (A), transversal (B), and coronal (C) sections of rat brain through striatum, cortex, and cerebellum. These are averaged images (60–120 min after tracer injection scaled on SUV 1) of a representative rat injected with 56 MBq of 18F-JNJ41510417. Max = maximum; Min = minimum.

Small-animal PET time–activity curves for 18F-41510417 in striatum, cortex, and cerebellum. (A) Untreated baseline scan. (B) Pretreatment experiment: MP-10, 5 mg/kg, was injected subcutaneously 60 min before tracer injection. (C) Chase experiment: MP-10, 3 mg/kg, was injected intravenously (arrow) 60 min after tracer injection. (D) Transversal images corresponding to the chase experiment: averaged image (40–60 min after tracer injection) before chase injection (left) and averaged image (96–120 min after tracer injection) after chase injection (right). Max = maximum; Min = minimum.
Pretreatment Blocking Studies
Pretreatment with the authentic reference material and with PDE10A inhibitors at a dose of 5 mg/kg injected subcutaneously at 60 min before tracer injection resulted in a significant decrease of the radioactivity concentration in the striatum. Figure 4B shows this decrease for MP-10 (5 mg/kg). At baseline, a maximum average S/C-1 ratio of 2.9 (n = 2) was reached at about 32 min after tracer injection, which stayed constant until about 75 min after injection (Table 5). Self-blocking at a dose of 2.5 mg/kg and pretreatment with TP-10 and MP-10 at a dose of 5 mg/kg resulted in a significant reduction of the S/C-1 ratios, from 2.9 at baseline to 1.4 or less after pretreatment (Table 5). Predosing of rats with MP-10 (1 mg/kg) reduced tracer uptake in the striatum by about 14% relative to the tracer uptake in the cerebellum. At a dose of 0.5 mg/kg, the decrease in tracer uptake was about 7%.
Summary of Average S/C-1 Ratios
Displacement Study
One hour after injection of 18F-JNJ41510417 (40.7 MBq), MP-10 was injected intravenously at a dose of 3 mg/kg in a rat. The intensity of the signal in the striatum significantly decreased after injection of the chase compound (Fig. 4D). 18F-JNJ41510417 continuously accumulates in the striatum up to 60 min after tracer injection. After injection of the chase compound, the radioactivity decreased to the level of the cortex and cerebellum (Fig. 4C). Injection of the chase resulted in a significant reduction of the S/C-1 ratio from 2.9 before MP-10 injection to 1.0 after injection of the chase.
Small-Animal PET and Ex Vivo Autoradiography in PDE10A KO Mice
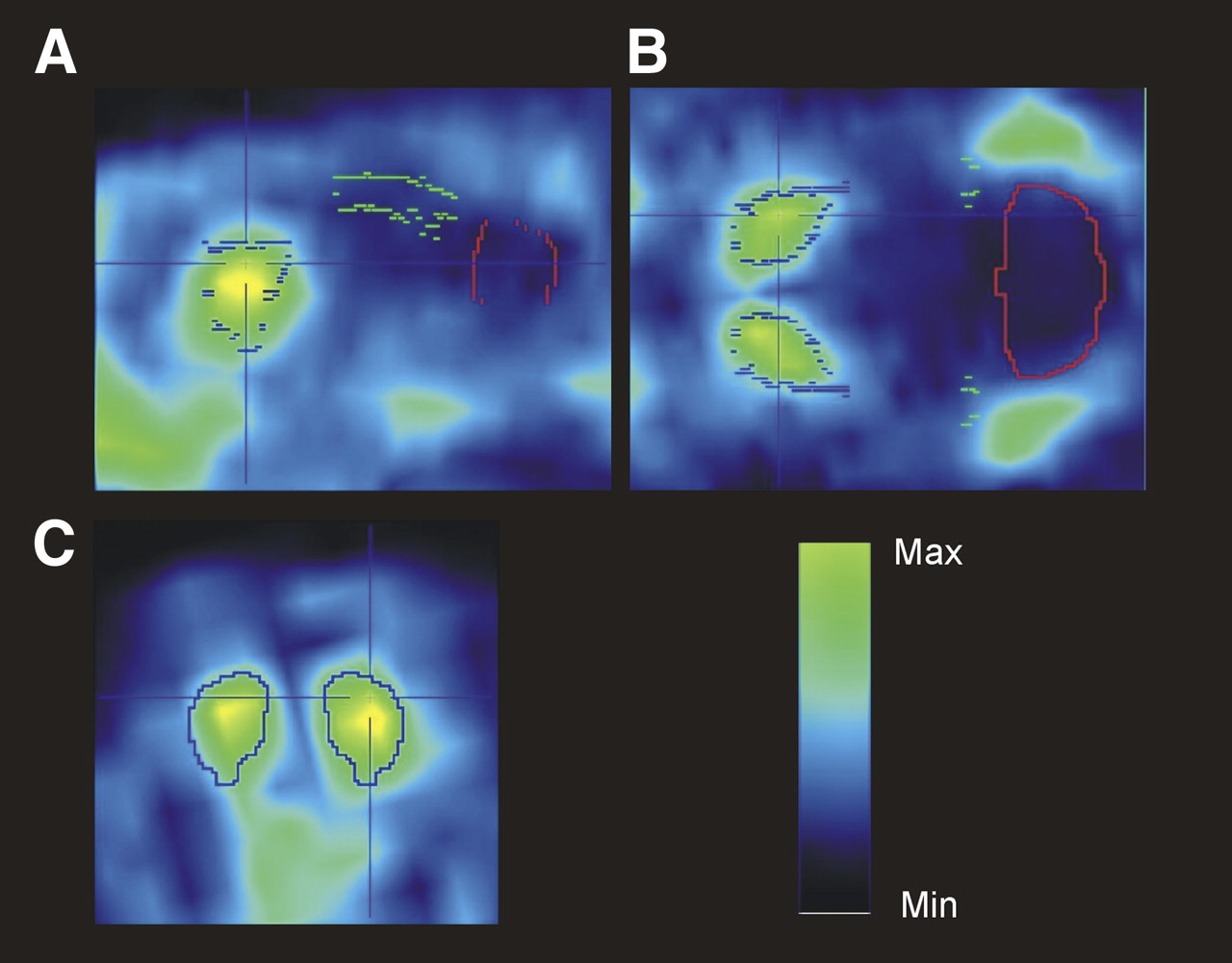
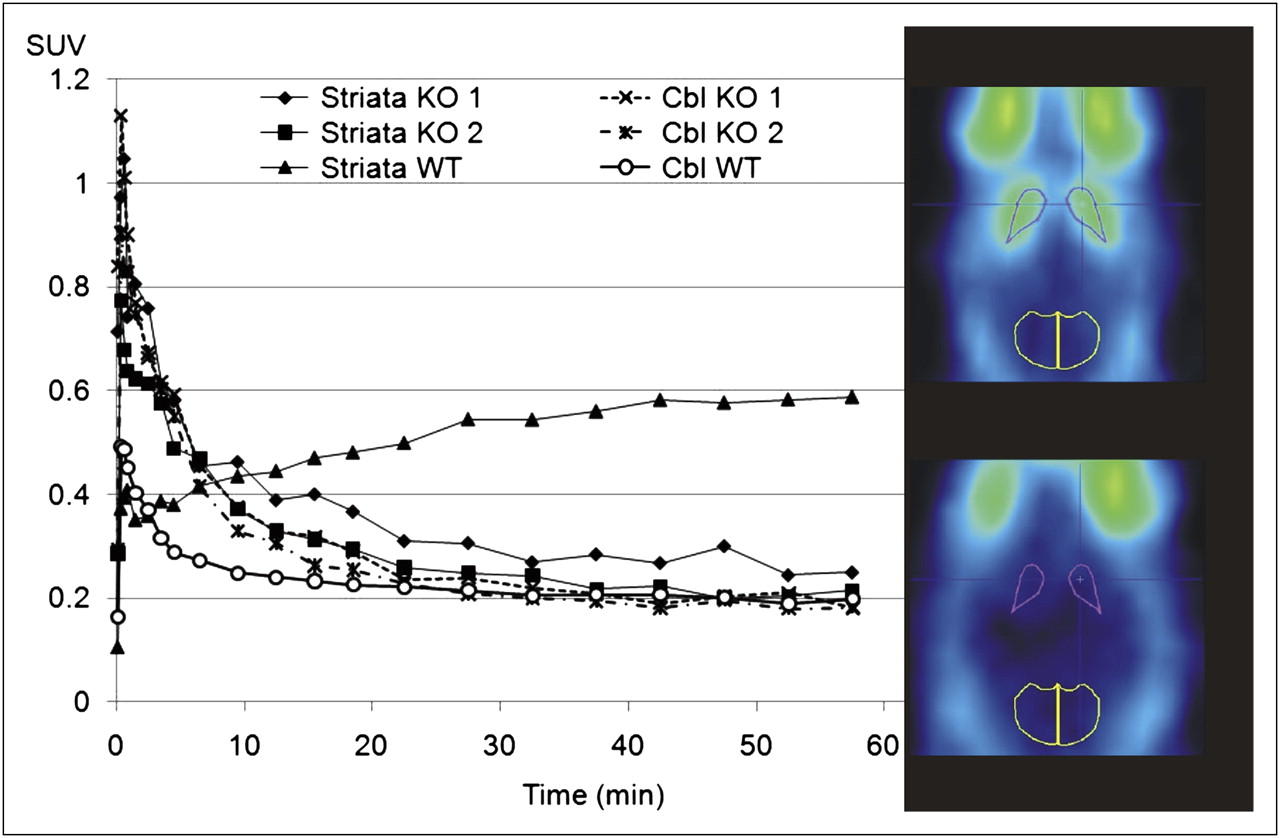
After injection of 18F-JNJ41510417, only background radioactivity was observed in the brain of the PDE10A KO mice (Fig. 5, lower image). The WT mouse, on the other hand, showed clear brain uptake with a high radioactivity concentration in the striatum (Fig. 5, upper image). High uptake was also present in the Harderian glands. After an initial peak in all regions, radioactivity was washed out from the striatum of the KO mice and the cerebellum of KO and WT mice, whereas a continuous accumulation of 18F-JNJ41510417 was observed in the striatum of the WT mouse (Fig. 5, graph).

Small-animal PET transversal sections of WT (upper image) and PDE10A KO (lower image) mouse brain injected with 18F-JNJ41510417 (summed images from 40 to 60 min after tracer injection). Regions of interest were drawn on areas corresponding to striatum and cerebellum to obtain time–activity curves of 18F-JNJ41510417 in striatum and cerebellum of 2 PDE10A KO mice and 1 WT mouse. Cbl = cerebellum.
Subsequent to the small-animal PET study, ex vivo autoradiography was performed (75 min after tracer injection) on cerebral coronal sections (Fig. 6). Analysis of the autoradiograms showed high tracer uptake in the striatum of the WT mouse. The binding to WT striatum was about 24 times higher than that to KO striatum. There was also some uptake in the cortex of the WT mouse brain, with a striatum-to-cortex ratio of about 14 and a WT cortex–to–KO cortex ratio of about 1.9 (Fig. 6, Table). The uptake in the WT cortex could be partly due to partial-volume effects of the highly radioactive striatum.

Ex vivo autoradiograms of coronal cerebral sections (30 μM) from WT mouse brain (left) and PDE10A KO mouse brain (right) 75 min after injection with 18F-JNJ41510417. Binding is expressed as digital light units per mm2 (DLU/mm2) for brain regions of interest. Values are corrected for injected dose and weight of animal. Max = maximum; Min = minimum.
DISCUSSION
The aim of this study was to evaluate a potential PET ligand for in vivo imaging of the PDE10A enzyme in the brain. From several MP-10 analogs, JNJ41510417 was selected as the most promising candidate. Therefore, this compound was radiolabeled and evaluated in vivo in rats and PDE10A KO mice. Rat biodistribution studies showed a significant accumulation of the tracer in the striatum over time, whereas there was a clear washout from the other studied brain regions (cerebellum, cortex, hippocampus) in accordance with the reported intracerebral distribution of PDE10A (2–4). The highest radioactivity concentration in the striatum was 2.6 (SUV, at 60 min after tracer injection; Table 2), with a maximum striatum-to-cerebellum ratio of 7.0. Compared with other brain radioligands (e.g., 18F-fallypride (25): SUV striatum at 60 min after injection, ∼4; striatum-to-cerebellum ratio, ∼42), the observed maximal brain concentration is low. In view of the rather high lipophilicity and associated high plasma protein binding of the tracer, the brain uptake is still surprisingly high, suggesting rapid dissociation of the protein-bound fraction on passage of the blood through the brain capillaries. The high plasma protein binding can, however, be a potential limitation of this radioligand because the plasma-free fraction may vary in disease conditions and is an important parameter in kinetic imaging analysis when the specific distribution volume needs to be determined. However, if further studies show that the simplified reference tissue model can be used for kinetic model analyses, normalization to the reference region will effectively correct for intersubject variations in plasma-free fraction and thus avoid the need to measure this parameter.
Two polar radiometabolites of 18F-JNJ41510417 have been detected in rat plasma. The minor metabolite 18F-JNJ41797444, resulting from the cleavage of the ether bond and loss of the methyl-quinoline group, was not detected in the brain. The major polar metabolite, on the other hand, was detected in the cerebrum and cerebellum (Table 4). 18F-JNJ41510417 probably undergoes oxidative metabolism, generating N-dealkylated 18F-JNJ41510417. The polar radiometabolite eluting with the void volume likely is 18F-fluoroethanol or its oxidation products 18F-fluoroacetaldehyde or 18F-fluoroacetic acid. Because 18F-JNJ41510417 is stable in vitro in rat brain homogenates, its metabolism presumably occurs in the periphery. Polar radiometabolites entering the brain could complicate the quantification of PDE10A (26). The contribution of the radiometabolite to the brain signal is, however, relatively small and is not reflected in the in vivo time–activity curves of regions with low activity of PDE10A such as the cerebellum. The fraction of polar radiometabolites detected in cerebellum is comparable to that observed in plasma and higher than that observed in cerebrum. The latter can be explained by the large amount of specifically bound tracer in the striatum, thus enriching the total fraction of intact tracer in the cerebrum. Radiometabolite analysis of the cerebrum and cerebellum of PDE10A KO mice—thus in the absence of specific binding—showed that the fraction of radiometabolites in the cerebellum is identical to that of the cerebrum (results not shown). These findings, together with the fact that PDE10A is expressed at a low level in the cerebellum, suggest that a reference tissue kinetic model (27) could be used for kinetic model analyses and estimation of binding potential values for the striatum. However, a validation of this model by sampling and measuring metabolite-corrected arterial input functions is needed to ascertain the validity of a reference tissue model and to determine whether even a simplified reference tissue model is appropriate. Because no skull uptake was observed in small-animal PET experiments, the tracer is probably not defluorinated in vivo in rats. Defluorination would result in binding of free 18F-fluoride to bone and skull, resulting in inaccurate quantification of radiotracer binding in the brain due to partial-volume effects of the high amount of radioactivity in the skull. Species differences in susceptibility to radiodefluorination exist for many tracers. Although less extensive metabolism is generally expected in higher species (20), further in vivo evaluation in humans will be necessary to check the extent of defluorination.
Baseline small-animal PET studies in normal rats confirmed the results of the biodistribution studies. Time–activity curves showed increasing tracer accumulation in the striatum and clear washout from other brain regions. The tracer shows relatively slow kinetics, which may require longer acquisitions in clinical applications to obtain robust distribution volumes. Relatively high striatum-to-cerebellum ratios resulted in good signal-to-noise images in vivo. Self-blocking studies and blocking studies with the PDE10A-specific inhibitors TP-10 and MP-10 (1,16–18,24) showed a dose-dependent decrease of the in vivo binding of 18F-JNJ41510417. A displacement experiment with MP-10 demonstrated that binding of 18F-JNJ41510417 to PDE10A is reversible. The experiment using PDE10A KO and WT mice confirmed that 18F-JNJ41510417 binds specifically to PDE10A in the striatum.
CONCLUSION
To our knowledge, this is the first report of a successful radioligand for in vivo imaging of the PDE10A enzyme using PET. Biodistribution studies and small-animal PET in rats and PDE10A KO mice demonstrated that 18F-JNJ41510417 shows reversible and specific binding to PDE10A in the striatum. Further clinical evaluation of this radioligand is warranted to investigate its potential use as a PDE10A PET ligand for human brain imaging.
Acknowledgments
We thank Peter Vermaelen and Ann Van Santvoort from the Department of Nuclear Medicine (K.U. Leuven) for their assistance in the small-animal PET studies and Julie Cornelis (Laboratory for Radiopharmacy, K.U. Leuven) for her skillful help with the animal experiments. This research was funded by Johnson & Johnson Pharmaceutical Research and Development.
- © 2010 by Society of Nuclear Medicine
REFERENCES
- Received for publication March 9, 2010.
- Accepted for publication July 14, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A Rationally Designed Fully Human EGFRvIII:CD3-Targeted Bispecific Antibody Redirects Human T Cells to Treat Patient-derived Intracerebral Malignant Glioma
- Preclinical Evaluation of 18F-JNJ64349311, a Novel PET Tracer for Tau Imaging
- A novel thermoregulatory role for PDE10A in mouse and human adipocytes
- Change in PDE10 across early Huntington disease assessed by [18F]MNI-659 and PET imaging
- AMG 580: A Novel Small Molecule Phosphodiesterase 10A (PDE10A) Positron Emission Tomography Tracer
- Discovery and Development of 11C-Lu AE92686 as a Radioligand for PET Imaging of Phosphodiesterase10A in the Brain
- In Vivo Assessment and Dosimetry of 2 Novel PDE10A PET Radiotracers in Humans: 18F-MNI-659 and 18F-MNI-654
- Phosphodiesterase 10A PET Radioligand Development Program: From Pig to Human
- Quantification of 18F-JNJ-42259152, a Novel Phosphodiesterase 10A PET Tracer: Kinetic Modeling and Test-Retest Study in Human Brain
- Patterns of Brain Glucose Metabolism Induced by Phosphodiesterase 10A Inhibitors in the Mouse: A Potential Translational Biomarker