


Abstract
Dedifferentiation of thyroid carcinoma is accompanied by increased accumulation of the PET tracer 18F-FDG. The molecular mechanisms responsible for this phenomenon are poorly understood. Therefore, we studied the regulation of 18F-FDG uptake by the human follicular thyroid carcinoma cell line ML-1 and the as-yet-unknown oncogene expression of that cell line. The data obtained in ML-1 were compared with those of a well-differentiated thyroid cell line of rat origin (FRTL-5). Methods: The expression of the thyroid-stimulating hormone (TSH) receptor was investigated by immunocytochemistry, and the expression of the glucose transporters (GLUTs) was determined by Western blotting. Mutation analysis of ML-1 was performed for K-ras codons 12 and 13. The effect of TSH on intracellular cAMP levels was determined by a competitive enzyme immunoassay. Cells were incubated with 18F-FDG (0.5–1.0 MBq/mL) for 1 h, and tracer uptake was related to protein concentration. The effects of bovine TSH, the cAMP analog (Bu)2cAMP, and the phosphatidylinositol-3-kinase (PI3-kinase) inhibitor LY294002 on 18F-FDG uptake were investigated. Results: The TSH receptor was present in both cell lines. FRTL-5 clearly expressed GLUT-1 and also GLUT-4. In ML-1 only, the expression of GLUT-3 was detected. TSH and (Bu)2cAMP had a significant effect on 18F-FDG uptake or GLUT-1 expression in FRTL-5, but not in ML-1 cells. PI3-kinase inhibition by LY294002 downregulated 18F-FDG uptake in FRTL-5 by 58% ± 9% (n = 6) and in ML-1 by 26% ± 5% (n = 42, both P < 0.05). Mutation analysis of ML-1 cells revealed a Gly12Ser point mutation at codon 12 of the K-ras gene. Conclusion: 18F-FDG uptake in the thyroid carcinoma cell line ML-1 is no longer regulated by TSH or cAMP or mediated by GLUT-1. However, in this cell line, this variable is still governed to some extent by PI3-kinase located downstream to the constitutively active K-ras in the Ras-PI3-kinase-Akt pathway. These data suggest that increases in 18F-FDG uptake in thyroid carcinomas observed in vivo by PET may reflect activation of intracellular signal transduction cascades by oncogenes.
A significant proportion of metastases of well-differentiated thyroid carcinomas have increased glucose metabolism (1) and may thus be localized using PET with 18F-FDG. It is generally believed that an increase in 18F-FDG uptake reflects dedifferentiation of these tumors. This view is supported by the observation that radioiodine-negative metastases exhibit increased glucose uptake more frequently than radioiodine-positive neoplasms (1). Furthermore, the survival of patients harboring 18F-FDG–positive tumor deposits is significantly shorter than that of subjects with 18F-FDG–negative metastases (2).
In normal thyroid tissue, primary thyroid cultures, and well-differentiated thyroid cell lines, glucose uptake can be stimulated by thyroid-stimulating hormone (TSH) (3–8). The action of TSH on thyroid cells comprises stimulation of the adenylate cyclase, leading to an increase in the intracellular concentration of cAMP. Consequently, analogs of cAMP such as (Bu)2cAMP that are able to cross the cell membrane have also been shown to stimulate glucose metabolism in the well-differentiated rat cell line FRTL-5 (4). However, in contrast to iodine transport, the TSH-induced increase of 18F-FDG accumulation does not depend on protein kinase A (PKA) but on phosphatidylinositol-3-kinase (PI3-kinase) in FRTL-5 cells (7,8).
The molecular mechanisms causing upregulation of glucose metabolism in thyroid cancer are as yet not completely understood. It has been repeatedly demonstrated that 18F-FDG uptake can be stimulated by TSH also in thyroid cancer tissue in vivo (9–11). However, a higher sensitivity of 18F-FDG PET to detect thyroid cancer tissue at higher TSH levels has as yet not been proven in clinical studies: So was the sensitivity of 18F-FDG PET to detect thyroid tumor tissue even higher at lower levels of TSH in a large and heterogeneous group of 222 subjects with thyroid cancer examined (1). Wang et al. also did not report a significantly higher sensitivity of 18F-FDG PET at TSH stimulation, compared with that at TSH suppression (12).
The aim of this publication was to study the regulation of 18F-FDG uptake by the human follicular thyroid carcinoma cell line ML-1 and the as-yet-unknown oncogene expression of that cell line. ML-1 cells have been derived from a dedifferentiated recurrence of follicular thyroid carcinoma that had progressed despite 2 courses of radioiodine therapy (13). The identity and purity of this cell line was previously verified by 2 independent DNA profiling analyses that eliminated a possibility of misidentification or contamination of original ML-1 cells (14). The data obtained in ML-1 were compared with those of a well-differentiated thyroid cell line of rat origin (FRTL-5).
MATERIALS AND METHODS
Reagents
18F-FDG was purchased from PET Net GmbH. Dibutyryl cyclic AMP ((Bu)2cAMP), 2-(4-morpholino)-8-phenyl-4H-1-benzopyran-4-one (LY294002), bovine thyroid-stimulating hormone (bTSH), and Coon's modified Ham's F-12 medium and supplements were supplied by Sigma-Aldrich. Recombinant human TSH (rhTSH; Thryogen) was obtained from Genzyme and quantitated by reversed-phase high-performance liquid chromatography. Fetal calf serum (FCS), phosphate-buffered saline (PBS), and trypsin/ethylenediaminetetraacetic acid were obtained from Invitrogen/Gibco. The rat thyroid cell line FRTL-5 was obtained from the European Collection of Cell Cultures (no. 91030711) and from the American Type Culture Collection (CRL 8305). The human follicular thyroid carcinoma cell line ML-1 (ACC464) was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ).
Cell Culture
FRTL-5 cells were grown in Coon's modified Ham's F-12 medium supplemented with 5% FCS and a mixture of 6 hormones (6H medium) including insulin (10 mg/L), human transferrin (5 mg/L), somatostatin (10 μg/L), hydrocortisone (10 nM), glycyl-l-histidyl-l-lysine acetate (10 μg/L), and bTSH (1 IU/L) in a humidified incubator at an atmosphere of 5% CO2 at 37°C (15). FRTL-5 cells were routinely subcultured every 3–4 d. The cell viability of FRTL-5 cells was proven by trypan blue staining. ML-1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 2 mM l-glutamine, 100 μM sodium pyruvate, and 10% FCS.
Immunocytochemical TSH Receptor Staining
As described in detail previously (8), TSH receptor expression was determined by immunocytochemical staining using the DAKO-APAAP Kit (DAKO) and a monoclonal TSH receptor antibody (clone 4C1/E1/G8; Lab Vision Co.). Primary human thyreocytes, which were prepared from thyroid tissue as described previously (3,16), were used as a positive control and compared with staining results obtained from ML-1 cells in these experiments. In brief, approximately 150,000 cells were seeded in 8-well chamber slides (Nunc). After 24 h, cells reached 80%−90% confluence, were chilled on ice, washed with Tris-buffered saline (TBS, pH 7.6), and fixed in cold methanol for 1.5 min. The staining protocol was performed according to the specifications of the manufacturer. Cells were incubated with the TSH receptor antibody (ready-to-use solution, without dilution) for 30 min at room temperature. Slides were washed (TBS, 5 min), and a rabbit immunoglobulin fraction to mouse immunoglobulins (DAKO) was applied as secondary antibody for 30 min at room temperature. Negative control slides were obtained by omitting the primary antibody. Cells were examined at ×20 using an Olympus Corp. light microscope.
cAMP Stimulation of FRTL-5 and ML-1 Cells
FRTL-5 cells were initially cultivated in 6H medium for at least 3 d and then were shifted to medium containing no TSH (5H medium) for at least 5 d before use, allowing 2 feedings of 5H medium to remove any trace of TSH. FRTL-5 and ML-1 cells were plated in 96-well plates (Corning) 24 h before TSH stimulation. The cells were then incubated with different concentrations of rhTSH, diluted in respective medium supplemented with the phosphodiesterase inhibitor isobutylmethylxanthine (IBMX; 1 mM) to prevent degradation of cAMP to AMP, for 2 h at 37°C. Medium was then collected, and the cAMP concentration was measured by cAMP enzyme-linked immunosorbent assay (cAMP EIA Kit; Cayman Chemical).
18F-FDG Uptake Experiments
The methodology used to determine 18F-FDG uptake was identical to that described in detail previously (8). Cells were seeded at a concentration of 60,000/mL (ML-1) or 200,000/mL (FRTL-5) in multiwell plates (12-well). Before 18F-FDG uptake experiments, TSH deprivation was performed on 70%−80% confluent cell layers by changing to a deprivation medium (FRTL-5: Coon's F12 medium containing 5 mg/L transferrin, 0.5% FCS; ML-1: minimum essential medium containing 1 g/L glucose, 1% sodium pyruvate). Cells were cultured in the deprivation medium for 3 d. Subsequently, the cells were either treated with TSH (1 mU/mL) or (Bu)2cAMP (1 mM) for 24 h or maintained in the deprivation medium in the quiescent phase for a further 24 h (untreated cells). The effect of LY294002 (selective inhibitor of PI3-kinase) on 18F-FDG uptake was studied by adding the inhibitor in the final concentration of 10 μM to the incubation medium 30–45 min before stimulation at 37°C. 18F-FDG (0.1–0.5 MBq, 10 μL) was added to each culture well (volume = 1.0 mL), and incubation was continued for 45 min at 37°C. An aliquot of each well (50 μL) was withdrawn and used for radioactivity measurements. The incubation period was terminated by rapid aspiration of the medium and rinse of the cell layer with cold PBS. NaOH (0.5 mL, 0.1 M) was used to dissolve the cells. All samples were counted in a γ-counter (Wallac Wizard; Perkin Elmer), and the protein content in each well was measured by the method of Bradford (17). 18F-FDG uptake was determined as percentage of whole 18F-FDG radioactivity divided by total protein mass (%/mg).
Western Blot Analysis of GLUTs
Total cellular membrane fractions were isolated from cells using the ProteoExtract Kit (Merck Biosciences), according to the manufacturer's instructions. Briefly, the medium of control and treated cells was aspirated, and the cultures were washed with cold PBS. The cell layers remained adherent during the extraction procedure. The membrane fraction containing solutions were diluted with 0.9% NaCl solution (1:100) for measuring protein concentration by the QuantiPro BCA assay kit (Sigma) using bovine serum albumin as a standard. Electrophoresis of membrane preparations was performed on aliquots of each sample on a 10% polyacrylamide gel (precast NuPAGE Novex Bis-Tris gel; Invitrogen) and transferred to nitrocellulose membranes by using a Hoefer miniVE blot module (Amersham Biosciences). A prestained standard (5 μL, SeeBlue; Invitrogen) and a protein molecular weight reference (Magic Marker; Invitrogen) were loaded on each gel to allow visual control of blotting results and molecular weight determination. Nitrocellulose membranes were incubated in blocking solution (5% nonfat dry powdered milk in TBS buffer containing 50 mM Tris and 120 mM NaCl, pH 7.5) for 2 h at room temperature. Membranes were incubated with either a rabbit polyclonal antihuman GLUT-1 antibody (FabGennix Inc.), a rabbit antihuman GLUT-3 antiserum (GT32-S; α-Diagnostic Int.), or a rabbit antimouse GLUT-4 antiserum (GT41-S; α-Diagnostic Int.), each at a concentration of 1:1,000, overnight at 4°C. After the blots were washed in TBS buffer (0.1% polysorbate-20) for 3 × 15 min, they were incubated with a horseradish peroxidase–conjugated anti-rabbit secondary antibody (1:1,000 in TBS) for 1 h at room temperature. After repeated washing (3 × 15 min) in TBS buffer (0.1% polysorbate-20), blots were developed with a chemoluminescence reagent (ECL Plus; Amersham Biosciences) and analyzed by a Fluor-S MultiImager (Bio-Rad) using the software Quantity One (version. 4.6.3; Bio-Rad).
Gene Mutation Analysis
K-ras gene mutations were examined following the procedure previously described by Feng et al. (18). Briefly, exon 1 containing the mutation hot spots at codons 12 and 13 was amplified and sequenced in both directions using polymerase chain reaction primers. Identified sequence alterations were verified by sequencing a second, independently generated amplicon of exon 1. As a wild-type control, DNA isolated from human peripheral blood was used. The B-raf V600E mutation was analyzed using allele-specific polymerase chain reaction (PASA) as described previously (19).
Coefficient of Variation of Tracer Uptake Experiments and Statistics
Statistical analysis was performed using SPSS software (version 15.0; SPSS Software GmbH). All data are expressed as mean ± SD. The uptake values of control cells in inhibition experiments with 18F-FDG were set to 100% to allow for pooling of data from individual experiments. In addition, the average and the range of uptake values expressed in percentage per milligram of protein (%/mg) is also provided, usually within the figure legends. All statistical results are based on the nonparametric Mann–Whitney test. The number of samples used for statistical analysis and the number of independent experiments are given in the figure legends. Probability values less than 0.05 (P < 0.05) were considered to be significant.
RESULTS
The thyroid cell lines FRTL-5 and ML-1 clearly demonstrated expression of the TSH receptor as determined by immunocytochemical staining. A representative staining result for the human follicular thyroid carcinoma cell line ML-1 cells is shown in Figure 1 and compared with the staining result for normal human thyroid cells as a positive control (Fig. 1). Positive staining results for FRTL-5 cells using the identical staining protocol have been published previously (8).

Immunocytochemical staining of TSH receptor. Primary human thyreocytes (A, negative control; B, primary antibody), ML-1 (C, negative control; D, primary antibody). Staining results for FRTL-5 cells have been published previously (8).
In FRTL-5 and ML1 cells, activation of the TSH receptor using increasing concentrations of rhTSH led to increased intracellular accumulation of cAMP as determined by an enzyme immunoassay after stimulation in DMEM (5% FBS, 1 mM IBMX) for 3 h. The half-maximally effective concentration values of rhTSH-induced cAMP levels were 395.5 ng/mL for ML-1 cells and 5,489 ng/mL for FRTL-5 (n = 3, Fig. 2).

cAMP stimulation by increased rhTSH concentration, as determined by an enzyme immunoassay (Cayman Chemical) after stimulation for 2 h in DMEM, 10% FCS, and 1 mM IBMX for ML-1 cells and 5H medium, 5% FCS, and 1 mM IBMX for FRTL-5 cells. Values are expressed as mean ± SEM (n = 3). Mean value of medium only was <10 pmol/L and was printed on logarithmic axis at −1 for definition of background levels.
After TSH deprivation, TSH induced a 2.4-fold increased uptake of 18F-FDG in FRTL-5 cells (n = 23, P < 0.01) that was mimicked by a 2.6-fold increased uptake induced by (Bu)2cAMP (n = 23, P < 0.01, Fig. 3A). This finding is in accordance with our previous study (8), reproducing TSH responsiveness of the FRTL-5 cell batch used within the present study. In contrast, the human carcinoma cell line ML-1 did not show a significant increase of 18F-FDG uptake after treatment with TSH (n = 8, P > 0.05, Fig. 3B) or (Bu)2cAMP (101.1% ± 15.4%, n = 6). The observed 18F-FDG uptake in ML-1 cells was specific, because it could be blocked to background values by increasing the glucose content in the incubation medium from 5.5 to 200 mM glucose (9.41% ± 1.87%/mg, n = 6 vs. 3.04% ± 0.74%/mg, n = 3). Western blot analysis disclosed moderate expression of GLUT-3, but not of GLUT-1 or GLUT-4, in the membrane fraction of ML-1 cells. FRTL-5 cells expressed GLUT-1 and GLUT-4 but not GLUT-3 (Fig. 3C).

(A) TSH increased 18F-FDG uptake in FRTL-5 cells by factor of 2.4 (*P < 0.01, n = 23), also mimicked by pretreatment with cAMP enhancer (Bu)2cAMP. 18F-FDG uptake of untreated FRTL-5 cells was 2.03% ± 0.35%/mg (control). (B) 18F-FDG uptake in follicular thyroid carcinoma cell line ML-1 did not indicate TSH responsiveness. Mean 18F-FDG uptake of untreated ML-1 cells was 14.1%/mg, ranging between 9.2%/mg (minimum) and 20.1%/mg (maximum). Data are expressed as mean ± SD of 4 independent experiments, each performed in quadruplicate. (C) Representative Western blot analysis of GLUT subtype expression in membrane fractions of FRTL-5 and ML-1 cells.
Using the selective PI3-kinase inhibitor LY294002, which is significantly more stable than wortmannin in long-term incubation periods, we showed that TSH-induced 18F-FDG uptake is mainly mediated through the PI3-kinase pathway in FRTL-5 cells. LY294002 inhibited TSH-induced 18F-FDG uptake by 58.1% ± 8.7%, as referred to untreated control cells (P < 0.01, n = 6; Fig. 4A). As determined by Western blot analysis, GLUT-1 levels of membrane fractions obtained from LY294002-treated FRTL-5 cells also correlated with decreased 18F-FDG uptake (Fig. 4B).
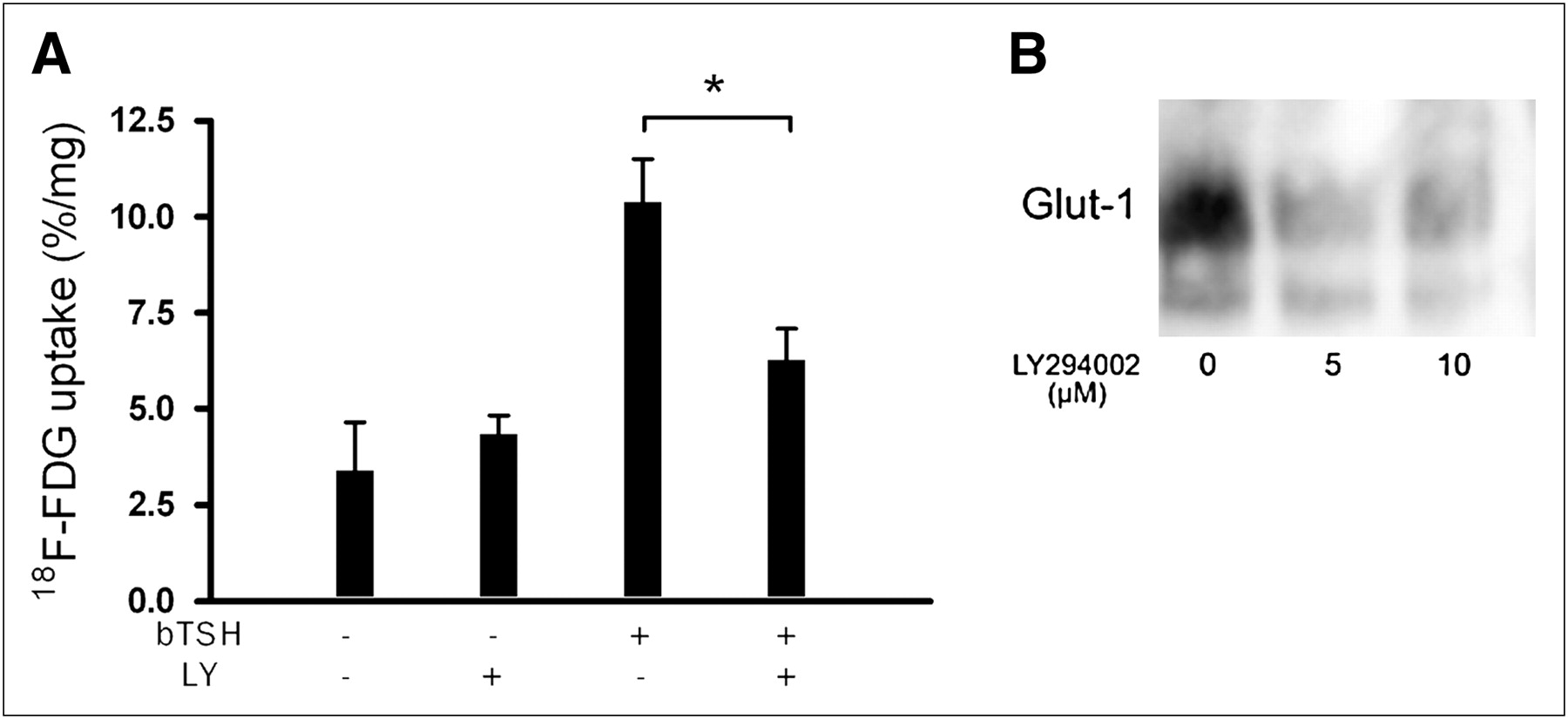
(A) FRTL-5 cells were treated with 10 μM LY294002 (LY) for 30 min before treatment with bTSH. After 48 h, 18F-FDG uptake studies were performed. LY294002 inhibited TSH-induced 18F-FDG uptake by 58.1% ± 8.7%, as referred to untreated control cells (*P < 0.01, n = 6). Values are expressed as mean ± SD of 2 independent experiments, each performed in triplicate. (B) Representative Western blot analysis showing decreased GLUT-1 expression in membrane fraction of LY294002-treated FRTL-5 cells.
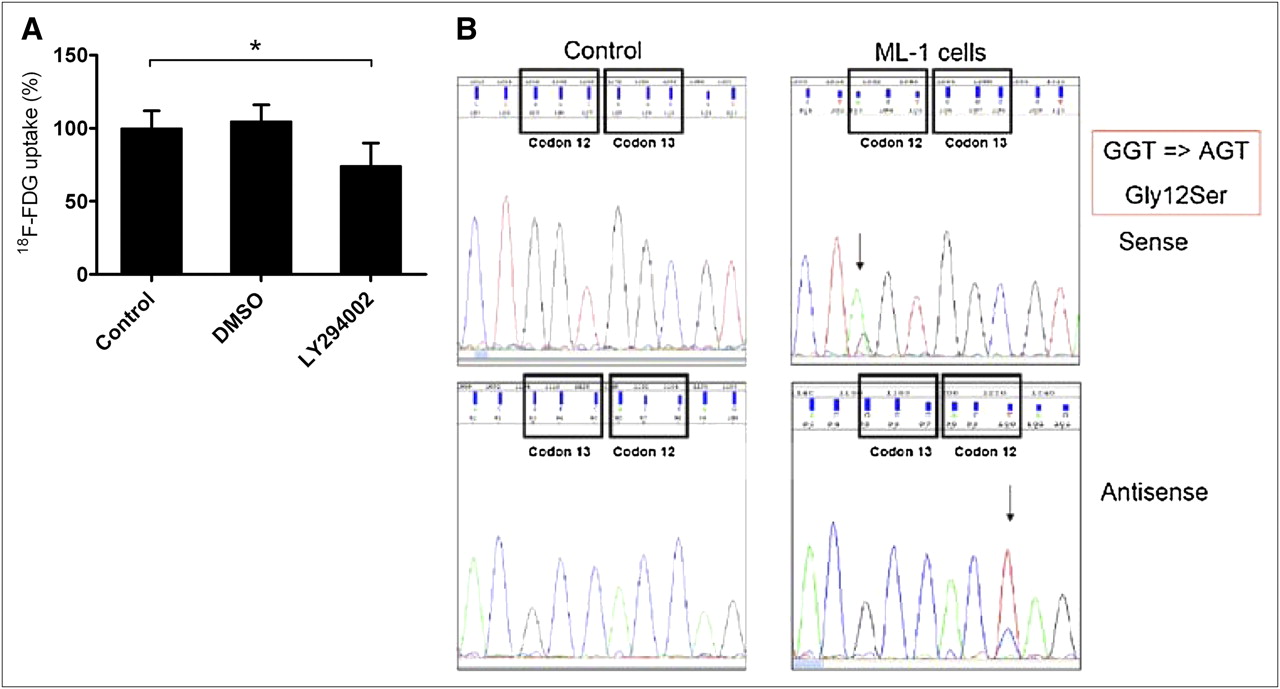
Assuming that the PI3-kinase–dependent signal transduction pathway is persistently upregulated in thyroid carcinoma cells, we examined the influence of LY294002 treatment on 18F-FDG uptake in ML-1 cells. As shown in Figure 5A, treatment of ML-1 cells with the PI3-kinase inhibitor led to significantly decreased 18F-FDG uptake by 25.8% ± 5.4% (*P < 0.01, n = 22 for control, n = 42 for LY294002).
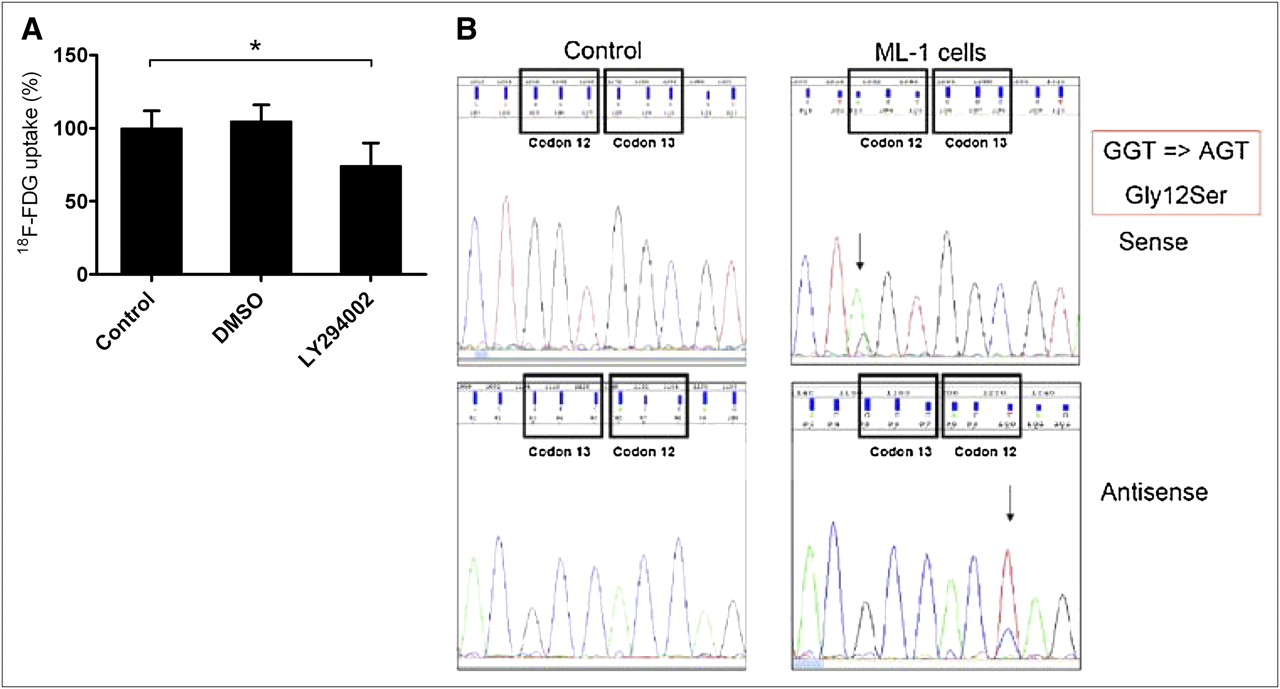
(A) ML-1 cells were treated with LY294002 for 45 min at 37°C, revealing significantly decreased 18F-FDG uptake by 25.8% ± 5.4% (*P < 0.01, n = 22 [control], n = 42 [LY294002]). Mean 18F-FDG uptake of untreated ML-1 cells was 6.73%/mg, with range of 4.07%/mg (minimum) to 8.89%/mg (maximum). (B) Mutation analysis of ML-1 cells revealed Gly12Ser point mutation at codon 12 of K-ras gene, resulting in oncogenic activation of K-Ras-PI3K-Akt pathway. AGT = trinucleotide sequence adenine (A), guanine (G), thymine (T) (codon 12, ML-1); DMSO = dimethyl sulfoxide; GGT = trinucleotide sequence guanine (G), guanine (G), thymine (T) (codon 12, control).
Mutation analysis of ML-1 cells demonstrated the Gly12Ser point mutation at codon 12 of the K-ras gene, providing evidence for oncogenic activation of the K-ras-PI3K-Akt pathway in ML-1 cells (Fig. 5B), whereas no mutation could be detected in the B-raf gene (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org).
DISCUSSION
18F-FDG uptake in the human follicular thyroid cancer cell line ML-1 can be competitively inhibited by nonradioactive glucose, as has been demonstrated for a wealth of other tumor cell lines as well (20,21). It is well known that the specific uptake of this tracer is governed by the various isoforms of GLUT as well as by those of hexokinase.
Normal thyroid tissue has been shown to express messenger RNA for GLUT-1, GLUT-3, GLUT-4, and GLUT-10 (22). In most tumor cells GLUT-1 is the major protein mediating the specific transport of 18F-FDG via the cell membrane (22). However, our data show that ML-1 cells do not express GLUT-1—but GLUT-3 instead—in their membranes. Ninety-five percent of human follicular carcinomas are deficient for GLUT-1 messenger RNA (23). This observation is not completely unexpected and also agrees with results obtained by Ciampi et al., who reported an overexpression of GLUT-3 in other well-differentiated follicular thyroid carcinoma cell lines (24).
An important prerequisite to the intracellular trapping of 18F-FDG is its phosphorylation, which is catalyzed by hexokinase. Using immunohistochemical staining of surgical specimens, Hooft et al. have demonstrated that 18F-FDG accumulation in thyroid cancer tissue as detectable in vivo by PET is associated with the expression of hexokinase I (25). It may, therefore, be hypothesized that also ML-1 as a cell line derived from a dedifferentiated follicular tumor expresses this isoenzyme. However, a direct proof of this hypothesis is still lacking.
18F-FDG uptake of the human follicular thyroid carcinoma cell line ML-1 could not be stimulated by TSH. The loss of TSH responsiveness demonstrated in our batch of ML-1 is particularly intriguing in view of the expression of the TSH receptor demonstrated immunocytochemically by ML-1 cells. One explanation of this phenomenon may be a defective TSH receptor or nonfunctional G-proteins. Our data rule out this explanation, however, because TSH stimulation increased intracellular cAMP levels significantly also in this cell line. Furthermore, the cAMP analog (Bu)2cAMP did not affect 18F-FDG accumulation. Therefore, disturbances of effector proteins or the signal transduction cascade further downstream must be considered as explanations of this observation.
In FRTL-5 cells, TSH stimulation of 18F-FDG uptake is mediated by the PI3-kinase and not by PKA as is the case for iodine transport (7,8). Also in ML-1 cells, 18F-FDG uptake decreased significantly on inhibition of the PI3-kinase by LY294002. This demonstrates some integrity of the signal transduction cascade governing glucose metabolism downstream of cAMP, still prevailing despite loss of responsivity to TSH and cAMP of the carcinoma cell line, compared with the normal thyroid tissue.
The stimulation of the PI3-kinase by TSH and cAMP in differentiated thyrocytes documents a cross-talk between the signal transduction cascades originating at the thyroid 7 transmembrane receptor for TSH and the tyrosine receptor kinase stimulated by insulin growth factors (IGFs) such as IGF-1 and IGF-2. This cross-talk seems organ-specific and has been demonstrated for glucose uptake (7,8) and cell proliferation (26,27).
The molecular mechanism mediating the interaction of these 2 signal transduction cascades has also been investigated. A direct stimulation of the PI3-kinase by cAMP or the PKA has as yet not been proven (27,28). Recently, evidence on the possible role of Ras in mediating the cross-talk between the 2 signal transduction cascades has emerged. Ras proteins have a central role in the control of cell growth and differentiation (29). Studies in transfected rat thyroid cells have shown that cAMP may activate ectopically expressed Ras (29), so that Ras may be considered a PKA-independent effector of TSH receptor stimulation. PI3-kinase activation has been described to be necessary for ras-induced proliferation in human thyrocytes (30). Activation of PI3-kinase by ras may therefore be the mechanism responsible for the TSH- and cAMP-induced increase in 18F-FDG uptake observed in our subclone of FRTL-5 cells, although direct proof of this effect is still lacking in the literature. The loss of responsiveness to TSH and also to cAMP of the ML-1 cells could therefore be explained by disturbances either on the level of the effectors linking cAMP to PI3-kinase, for example, Ras, or on the level of the PI3-kinase itself (Fig. 6).
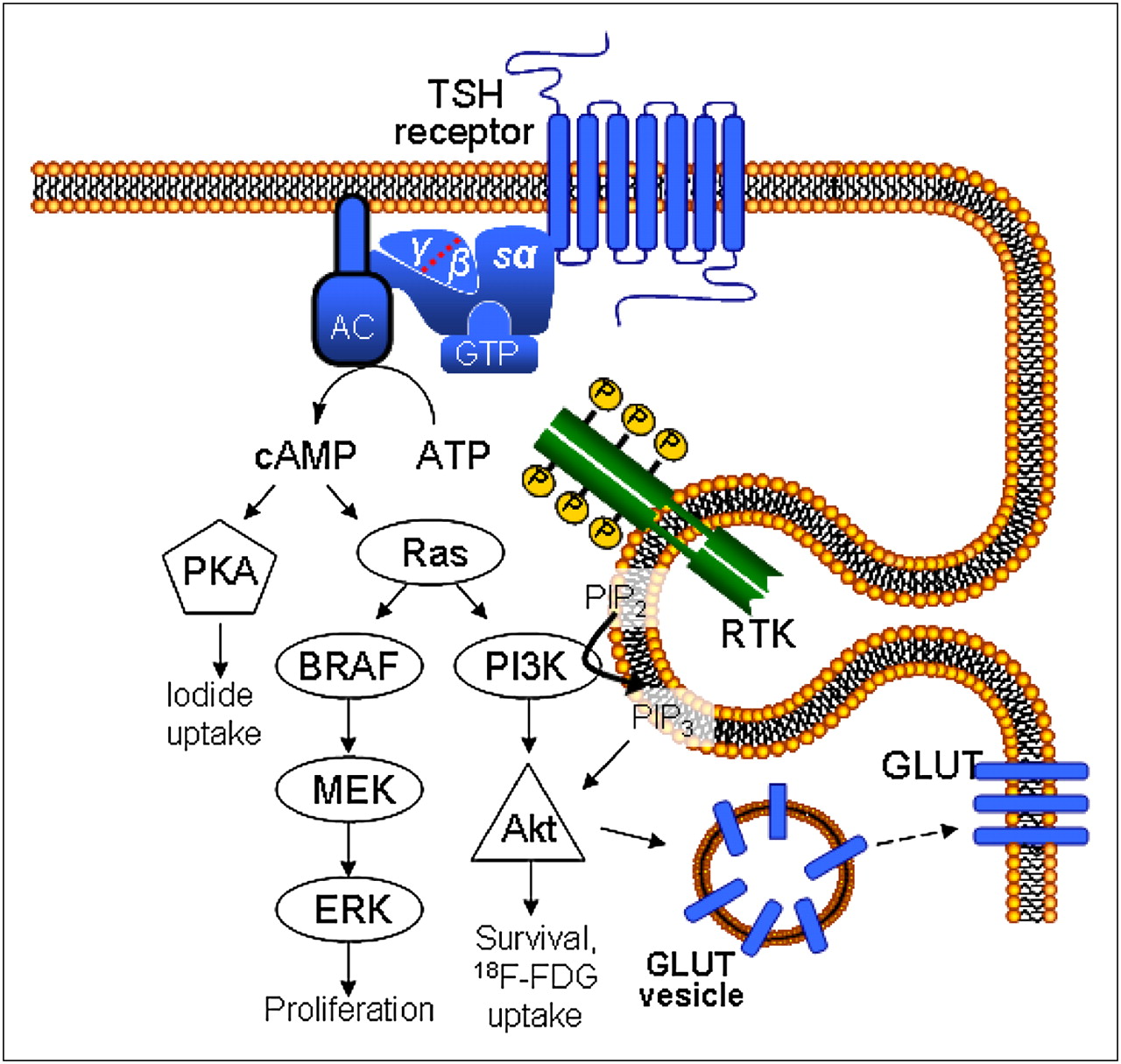
Putative scheme of signal transduction in thyreocyte according to Riesco-Eizaguirre and Santisteban and Rivas and Santisteban (31,37). TSH-stimulated 18F-FDG uptake is mediated via adenylate cyclase (AC) and cAMP, ras, PI3K, and Akt. Iodide uptake is regulated by PKA. Mitogen-activated protein (MAP) kinase pathway involving B-type Raf (BRAF), extracellular signal–regulated kinase, and mitogen-activated protein kinase kinase is supposed to steer cell proliferation. The Gly12Ser point mutation of K-ras oncogene as detected in ML-1 cells (Fig. 5B) results in constitutive activation of K-ras-PI3K-Akt pathway in this cell line, leading to enhanced glucose transport and use.
Using mutation analysis, we identified a mutation of the ras gene in our cell line, the K-ras oncogene. This mutation results in constitutive activation of this protein and has been found in 7%−62% of human follicular carcinomas (31). Therefore, the lack of TSH-induced increase in 18F-FDG uptake in ML-1 is due to this constitutive ras activation that is no longer dependent on intracellular cAMP levels.
Clearly, studying the effect of Ras inhibition on glucose uptake could directly prove this hypothesis. However, effectors reliably inhibiting Ras are lacking (32): Thus, the widely used inhibitors of the farnesyltransferase lack sensitivity because their action may be overruled by other enzymes with the capacity to prenylate Ras such as the geranylgeranyltransferase. Furthermore, these compounds also target a variety of other signaling molecules such as ρ-B and -E. Therefore, a more sophisticated approach capitalizing on, for example, RNA interference would have to be used for this purpose.
Most other oncogenes found in thyroid cancer involve signal-transduction proteins downstream of Ras. These are, in particular, mutations concerning the catalytic subunit of the PI3-kinase, occurring in 23% of anaplastic carcinomas but also in 8% of follicular cancers (33) and mutations of raf such as BRAFV600E, which is frequent in papillary carcinomas and unique to this tumor subtype. Also the RET/PTC oncogenes, which involve the rearrangement between the gene encoding the tyrosine kinase receptor RET with various heterologous genes (34), probably activate the Ras/RAF/MEK/MAP kinase pathway, although their oncogenic action is currently less well defined than that of the other oncogenes mentioned. RET/PTC oncogenes occur in sporadic papillary cancer and, in particular, in radiation-induced papillary carcinomas (35).
Our in vitro data suggest that 18F-FDG uptake determined in vivo with PET in thyroid tumor tissue harboring the K-ras mutation is not TSH-dependent. Patients carrying this oncogene could, therefore, be studied without prior cessation of l-thyroxine or without prior injections of human rhTSH. However, some caution should be exercised with regard to extrapolating data obtained in vitro to the situation encountered in vivo. Therefore, at present, the above statement should not be taken as a recommendation; it represents a hypothesis awaiting confirmation in a clinical study.
The effects of Ras stimulation are pleiotropic, involving at least 5 distinct further pathways, each regulating distinct cellular functions. In principle, activating or inactivating mutations involving a particular station along these pathways leads to loss of responsiveness of the particular cellular function in question to signals upstream. The K-ras mutation detected in ML-1 would, therefore, lead to a loss of TSH responsiveness mediated by all its effectors. Human tumors carrying this mutation would thus be expected to lose not only the TSH responsiveness of glucose metabolism but also that of other functions such as cell proliferation. Furthermore, in tumors exhibiting mutations in only 1 of the pathways governed by Ras, the TSH responsiveness of the other pathways would be expected to be preserved. On the basis of this framework, hypotheses on how small-molecule inhibitors of the signal transduction proteins influence accumulation of radioiodine and 18F-FDG in thyroid tumor tissue could be derived and then subjected to in vitro and in vivo verification. These hypotheses will, however, have to take into account that the small molecule inhibitors of signal transduction proteins currently being studied are not specific to one of these macromolecules but usually address several of these (36).
CONCLUSION
18F-FDG uptake in the thyroid carcinoma cell line ML-1 is no longer regulated by TSH or cAMP but still depends on enhanced activity of PI3-kinase resulting from the constitutive activation of mutated K-ras oncogene. The increase of 18F-FDG uptake in dedifferentiated thyroid cancer demonstrated by PET may therefore be due to increased activity of the second messenger PI3-kinase potentially caused by oncogenes such as mutated ras implicated in the pathogenesis of thyroid malignancies.
Acknowledgments
We are grateful to Ulrike Ittstein for skillful technical assistance.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication January 19, 2009.
- Accepted for publication April 28, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}