


Abstract
Folic acid was linked regioselectively through its α- and γ-carboxyl groups to 4-fluorobenzylamine (FBA), and the α- and γ-FBA-folate regioisomers were evaluated for their ability to bind to folate receptor–positive cells. The 18F-labeled α/γ-FBA-folate counterpart was examined for in vivo tumor targeting efficiency in nude mice bearing folate receptor–positive tumor cells. Methods: 18F-α/γ-FBA-folate was prepared in a 4-step reaction sequence starting from folic acid. The relative binding affinities of the α- and γ-FBA-folates to the folate receptor with respect to parent folic acid were determined in cultured KB-31 cells (nasopharyngeal epidermal carcinoma cell line) overexpressing the folate receptor using 3H-folic acid. Tumor accumulation of the 18F-labeled α/γ-FBA-folate and 18F-FDG was analyzed in vivo by high-resolution PET. Biodistribution and PET studies were performed under baseline and blockage conditions. Results: The radiochemical yield of the coupling step ranged from 15% to 44%, and the maximum specific radioactivity was 24 GBq/μmol. The in vitro binding affinities of the α- and γ-isomers and folic acid were 71, 62, and 41 nmol/L, respectively. PET revealed heterogeneous uptake of the radioligand, with the highest activity concentrations found in the tumor rim. In contrast, 18F-FDG uptake in a nude mouse bearing KB-31 folate receptor–positive tumors was negligible. Radioligand uptake in tumors at 125 min after injection amounted to 6.56% of the injected dose per gram of tissue (%ID/g) in control animals, whereas radioactivity accumulation in the tumors of folic acid–treated animals was significantly reduced by more than 80%—to 1.07 %ID/g (P = 0.001). Conclusion: This new 18F-labeled folic acid derivative is a promising tool for PET imaging of folate receptor–positive tumors.
Folic acid, also known as vitamin B9, is an essential dietary vitamin required by eukaryotic cells for 1-carbon metabolism, DNA synthesis, and spinal canal and brain development during early pregnancy (1). Since the early 1990s, folic acid has become a tool for targeting tumor cells. The folate receptor, or folate-binding protein (FBP), is overexpressed in a variety of epithelial carcinomas (2). Folate receptor is a glycoprotein receptor anchored to the cell membranes and exhibits a high affinity (dissociation constant, ∼10−9 nmol/L) for folic acid (3–5). Upon binding to folate receptor, folic acid is transferred into the cells via endocytosis (6,7). Folate receptor is restricted in normal human tissues (8) but has significant levels of expression in thyroid, choroid plexus, and kidneys (9). In contrast, the so-called reduced folate carrier is a low-affinity membrane-spanning protein and is expressed ubiquitously in normal tissues (10).
A number of 111In-, 66/67/68Ga-, and 99mTc-folate–based radiopharmaceuticals have been synthesized and successfully evaluated as diagnostic agents for imaging folate receptor–positive tumors. The most widely studied derivatives were labeled either with 111In or with 99mTc (11,12). A folic acid radiopharmaceutical labeled with 18F would be of great interest because, compared with other radionuclides, 18F has excellent imaging characteristics. The half-life of 18F, 110 min, also allows for syntheses that are more complex and satellite distribution to PET centers with no radiochemistry facilities. The structure of folic acid (compound 1; Fig. 1) does not lend itself to direct radiolabeling with 18F; therefore, we sought to functionalize folic acid using 4-fluorobenzylamine as a prosthetic group. Here, we report the synthesis, radiolabeling, and in vitro and in vivo evaluation of the new 18F-labeled folic acid derivative in nude mice bearing folate receptor–positive tumors.

Native folic acid (compound 1), γ-FBA-folate (compound 2), and α-FBA-folate (compound 3).
MATERIALS AND METHODS
General
Solvents were purchased from Merck and Fluka and were used without further purification. Chemicals were obtained from Fluka and Bachem. N2-N,N-dimethylaminomethylene-10-formylpteroic acid methyl ester was kindly provided by Merck Eprova AG. Unlabeled α- and γ-FBA-folates were synthesized in our laboratory (the synthesis will be reported elsewhere). An Extralut 3 column was obtained from Merck. High-performance liquid chromatography (HPLC) analysis and purification were performed on a Merck-Hitachi L-6200A provided with a γ-counter. The HPLC system used for the semipreparative purification consisted of a Waters 510 pump, a Knauer ultraviolet detector, and a Geiger Müller LND 714 counter with an Eberlein RM-14 instrument. 3H-Folic acid potassium salt (37 MBq/mL, 888 GBq/mmol) was purchased from Amersham Biosciences. The scintillation solution, Ultima Gold high-flash-point liquid scintillation cocktail, was purchased from Packard Co. KB-31 cells (CCL-17) were purchased from American Type Culture Collection. Special RPMI cell culture medium (without folic acid, vitamin B12, phenol red) was purchased from Cell Culture Technologies GmbH. Radioactivity (β-radiation of 3H) was measured with a β-counter (TRI-CARB, 1900 TR, liquid scintillation analyzer; Packard). Protein concentrations for the in vitro experiments were measured with a microplate reader (model 550; Bio-Rad), using a Micro BCA protein assay kit (Socochim). Experimentally acquired raw data were transformed using Prism software (GraphPad).
Synthesis of Compound 5
4-(Dimethylamino)benzonitrile (2 g, 13.7 mmol) and methyl trifluoromethane triflate (2.15 mL, 19.0 mmol) were refluxed for 6 h under an argon atmosphere. The orange solid residue was isolated by filtration and dissolved in 250 mL of water. The aqueous solution was extracted with CH2Cl2 (2 × 200 mL) and thereafter concentrated to dryness. Compound 5 was crystallized from CH3OH/AcOEt (87% yield). Analytic data for 5: 1H nuclear magnetic resonance (dimethyl sulfoxide [DMSO]-d6): δ: 3.63 (s, 9 H), 8.19 (s, 4 H). Mass spectrometry (matrix-assisted laser desorption/ionization): m/z: (161.4) [M]+.
Radiochemistry
Production of 18F−.
18F− was obtained via the 18O(p,n)18F reaction on 98% enriched 18O-water. Aqueous 18F− was transferred into a tightly closed 10-mL Reacti-Vial (Pierce Biotechnology, Inc.) containing 19−20 mg of Kryptofix K222 (Merck) and 2−3 mg of K2CO3 dissolved in MeCN (0.5 mL) and 1.5 μL of bidistilled water.
Synthesis of 18F-α/γ-FBA-Folate (Compounds 7 and 8).
The preparation of the 18F-Kryptofix-K222 complex was performed under standard conditions (13). Briefly, aqueous 18F− solution was concentrated to dryness at 115°C under a nitrogen stream for 10−15 min. The residue was dissolved in anhydrous MeCN (1 mL), and the solvent was evaporated to dryness at 115°C under a nitrogen stream. This procedure was repeated twice. Five to 6 mg of compound 5 that had been dissolved in anhydrous DMSO (300 μL) were added to the Reacti-Vial containing the 18F-Kryptofix-K222 and heated to 115°C for 20 min. The reaction mixture was quenched by adding 3 mL of bidistilled water, and the resulting solution was eluted through a t-C18 Sep-Pak cartridge (Millipore Corp.). The cartridge was washed with 0.5 mL of water and dried with N2 flow over 10 min. 18F-4-Fluorobenzonitrile was eluted from the cartridge with tetrahydrofuran (THF) (3 mL) and collected in a 10-mL Reacti-Vial containing a 0.6-g molecular sieve (3- pore diameter) named RVB. After 5 min at room temperature, the THF solution was transferred to a 10-mL Reacti-Vial named RVC. RVB was washed with THF (2 × 0.5 mL), and the washing fractions were also transferred to RVC. A saturated THF solution of LiAlH4 (0.9 mL) was added dropwise to RVC, which was heated at 120°C for 20 min. After cooling to room temperature, 0.5 mL of bidistilled water was added. After the addition of THF (1 mL), the suspension in RVC was transferred to an Extralut 3 cartridge. RVC was washed twice with THF (1 mL), and the washings were loaded to the same cartridge. 18F-4-Fluorobenzylamine (compound 6) was eluted from the cartridge with CH2Cl2 (6 mL) and collected in a 10-mL Reacti-Vial named RVD. The solvent was evaporated at room temperature under a weak N2 flow. In a separate vial, a suspension of folic acid (3 mg, 6.8 μmol) and 1-hydroxybenzotriazole (HOBt) (0.9 mg, 6 μmol) in DMSO (300 μL) and N-(3-dimethylaminopropyl)-N-ethyl-carbodiimide (1.5 mg, 7.8 μmol) in DMSO (300 μL) was added 5 min before the final coupling reaction. The DMSO solution containing the activated folic acid was added to RVD, and the coupling reaction was performed by heating at 100°C for 30 min. The reaction mixture was quenched with 1 mL of HPLC eluent (23% CH3OH, 77% NH4HCO3 [pH 7]). 18F-labeled compound was purified using the following semipreparative HPLC conditions: Spherisorb 5 ODS 1 (Phenomenex) column, 250 × 10 mm; isocratic elution (flow, 3 mL/min); eluent, 23% CH3OH, 77% NH4HCO3 (pH 7). The fraction containing the product was collected. After evaporation, the product was formulated using a 0.15 mol/L concentration of phosphate buffer and filtered through a 0.22-μm Millipore filter.
pore diameter) named RVB. After 5 min at room temperature, the THF solution was transferred to a 10-mL Reacti-Vial named RVC. RVB was washed with THF (2 × 0.5 mL), and the washing fractions were also transferred to RVC. A saturated THF solution of LiAlH4 (0.9 mL) was added dropwise to RVC, which was heated at 120°C for 20 min. After cooling to room temperature, 0.5 mL of bidistilled water was added. After the addition of THF (1 mL), the suspension in RVC was transferred to an Extralut 3 cartridge. RVC was washed twice with THF (1 mL), and the washings were loaded to the same cartridge. 18F-4-Fluorobenzylamine (compound 6) was eluted from the cartridge with CH2Cl2 (6 mL) and collected in a 10-mL Reacti-Vial named RVD. The solvent was evaporated at room temperature under a weak N2 flow. In a separate vial, a suspension of folic acid (3 mg, 6.8 μmol) and 1-hydroxybenzotriazole (HOBt) (0.9 mg, 6 μmol) in DMSO (300 μL) and N-(3-dimethylaminopropyl)-N-ethyl-carbodiimide (1.5 mg, 7.8 μmol) in DMSO (300 μL) was added 5 min before the final coupling reaction. The DMSO solution containing the activated folic acid was added to RVD, and the coupling reaction was performed by heating at 100°C for 30 min. The reaction mixture was quenched with 1 mL of HPLC eluent (23% CH3OH, 77% NH4HCO3 [pH 7]). 18F-labeled compound was purified using the following semipreparative HPLC conditions: Spherisorb 5 ODS 1 (Phenomenex) column, 250 × 10 mm; isocratic elution (flow, 3 mL/min); eluent, 23% CH3OH, 77% NH4HCO3 (pH 7). The fraction containing the product was collected. After evaporation, the product was formulated using a 0.15 mol/L concentration of phosphate buffer and filtered through a 0.22-μm Millipore filter.
Analytic HPLC
Quality control was performed using a SphereClone 5μ ODS(1) (250 × 4.60 mm; Phenomenex) column and applying a linear gradient of HPLC eluents A and B. Eluent A consisted of 90% NH4HCO3 (0.05 mol/L [pH 7]) and 10% CH3OH; eluent B consisted of 100% CH3OH. A typical HPLC analytic gradient was 20%−50% B over 30 min, at a flow of 0.8−1 mL/min. The γ- and α-isomers of the 18F-labeled folate conjugate were separated using the following analytic HPLC conditions: SphereClone 5μ ODS(1) (250 × 4.60 mm; Phenomenex) column; eluent A, 95% trimethylammonium phosphate [pH 3.5] and 5% CH3OH; eluent B, 100% CH3OH; linear gradient, 0%−50% B or 0%−80% B over 30 min; flow, 0.8−1 mL/min.
Cell Cultures
KB-31 cells (human nasopharyngeal epidermal carcinoma cell line overexpressing the folate receptor) were cultured continuously as monolayers in 75-cm2 flasks at 37°C in a humidified atmosphere containing 7.5% CO2. The cells were folate starved using a folate-deficient RPMI 1640 medium (modified RPMI 1640 without folic acid, vitamin B12, and phenol red) supplemented with 10% heat-inactivated fetal calf serum (as the only source of folate) containing l-glutamine and antibiotics (penicillin, 100 IU/mL; streptomycin, 100 μg/mL; amphotericin B, 0.25 μg/mL). Cell culture media such as fetal calf serum–supplemented folate-deficient RPMI 1640 are known to feature a final folate concentration of approximately 3 nmol/L, that is, a value at the low end of the physiologic concentration in human serum (14).
Preparation of Cells for In Vitro Experiments.
Eighteen to 20 h before each experiment, the cells were seeded into 12-well plates (6.5 × 105 cells per well) to form subconfluent monolayers overnight. Experiments were performed in triplicate for each concentration.
Preparation of Cells for In Vivo Experiments.
For subcutaneous inoculation into the mice, subconfluent cells were harvested by treatment with ethylenediaminetetraacetic acid (1 mmol/L) in phosphate-buffered saline (PBS; 1× [pH 7.4]). The cells were then washed once with PBS and pelleted by being spun at 1,000g for 5 min at 20°C. The cells were resuspended in PBS for inoculation.
Determination of Inhibitory Concentration of 50% (IC50)
Cell-binding experiments using 3H-folic acid were performed according to the following general procedure: The monolayers were rinsed twice with ice-cold PBS (pH 7.4). Pure, ice-cold folate-deficient RPMI 1640 medium (without fetal calf serum, l-glutamine, and antibiotics) (475 μL) was added to each well. Five hundred microliters of the corresponding ice-cold solution of native folic acid (20 concentrations or, for compounds 2 and 3, 14 concentrations) were added. The well plates were preincubated at 4°C for 40 min. This incubation temperature was chosen to minimize endocytosis of folate receptor. A solution of 3H-folic acid (25 μL, ∼0.74 kBq/μL) was added and the well plates incubated again on ice at 4°C for 2 h. Preliminary experiments showed that 2 h is an optimal incubation time and that saturation of cell binding is almost reached at that time point. Each well was rinsed 3 times with ice-cold PBS (pH 7.4). The monolayers were dissolved in 1N NaOH (1,000 μL), transferred in 4-mL tubes, and homogenized by a vortex mixer. Scintillation solution (4 mL) was added to each sample, homogenized, and transferred into scintillation flacons to be counted for radioactivity using a β-counter. The IC50 values were calculated directly from the corresponding data using the Prism software. For native folic acid, the mean IC50 was determined in 2 independent experiments, whereas for compounds 2 and 3, 3 independent experiments were performed. The Student t test (unpaired, unequal variance) was used to determine whether there was a significant difference between IC50 values.
In Vivo Studies
Animals.
Animal studies complied with Swiss laws on animal protection, and housing and animal husbandry complied with local laws on animal protection. Five- to 6-wk-old female nude mice (CD1-Foxn1nu) were purchased from Charles River. The mice were housed at a controlled temperature (26°C), humidity (68%), and daily light cycle (12 h light/12 h dark) and were maintained on a folate-deficient rodent diet (to reduce their serum folate to a level near that of human serum) (15). After a 7- to 14-d acclimatization period, 0.1 mL of KB-31 tumor cell suspension (50 × 106 cells/mL) was inoculated into the subcutis of the right and left axilla of each mouse. The animal experiments were performed 10−12 d after the tumor cell inoculation, when the tumors had reached weights of 200–500 mg.
Biodistribution Study by Dissection.
The mice received an injection of 18F-α/γ-FBA-folate (0.33−0.81 MBq; maximum volume, 150 μL) via a lateral tail vein. Ten minutes before tracer administration, the blockage group (n = 4) received an intravenous injection of 200 μg of folic acid (dissolved in 100 μL of phosphate buffer), whereas the control group (n = 4) received an intravenous injection of 100 μL of phosphate buffer. At 125 min after injection, the animals were sacrificed by decapitation and dissected. Tumors and organs were removed, rinsed with phosphate buffer, dried with a piece of tissue paper, and measured together with an aliquot of the injected solution in a γ-counter (Wizard; PerkinElmer) using an energy window of 300–700 keV. Results were expressed as percentage injected dose per gram of tissue (%ID/g; ((cpm organ/g organ)/cpm injected) × 100).
PET Imaging.
PET experiments were performed with the 16-module variant of the quad-HIDAC tomograph (Oxford Positron Systems), the performance of which has been reported elsewhere (16,17). A most important characteristic of this dedicated small-animal PET system is an ultrahigh resolution of less than 0.9 mm3. The animals, which were awake, were lightly restrained and intravenously injected with the radioligand via a lateral tail vein. At various times after injection, the animals were anesthetized with isoflurane (Abbott) in an air/oxygen mixture and positioned in the PET camera as described previously (18).
Initial PET studies with the 18F-labeled folic acid derivative were performed to determine the PET protocol that would most clearly show tumors in nude mice (n = 4) bearing KB-31 tumor xenografts. Visual inspection of PET images obtained at various times after injection (30, 75, 120, and 165 min) revealed that tumor accumulation was highest when data acquisition began at 75 min after injection.
For a comparison of 18F-FDG uptake with 18F-α/γ-FBA-folate uptake, a single animal was initially injected with 16.9 MBq of 18F-FDG and scanned from 30 to 60 min after injection. Two days later, the same animal was imaged again 75 min after injection of 13.3 MBq of the 18F-folate derivative (scan duration, 45 min).
For a demonstration of the in vivo binding specificity of the radioligand, a single mouse was scanned first under control conditions (100 μL of phosphate buffer injected 10 min before 16.9 MBq of the radioligand) and again 24 h later under blockage conditions (200 μg of folic acid injected 10 min before 13.9 MBq of radioligand). Both PET scans were initiated 75 min after injection of the tracer and lasted for 45 min.
PET data were acquired in list mode and reconstructed in a single time frame using the fast, accurate iterative reconstruction algorithm with a 0.5-mm bin and a 200 × 200 × 240 matrix (19). Reconstruction did not include correction for scatter, randoms, or attenuation. Image files were normalized to the injected dose per body weight and analyzed using the dedicated software PMod (20).
RESULTS
Radiochemistry
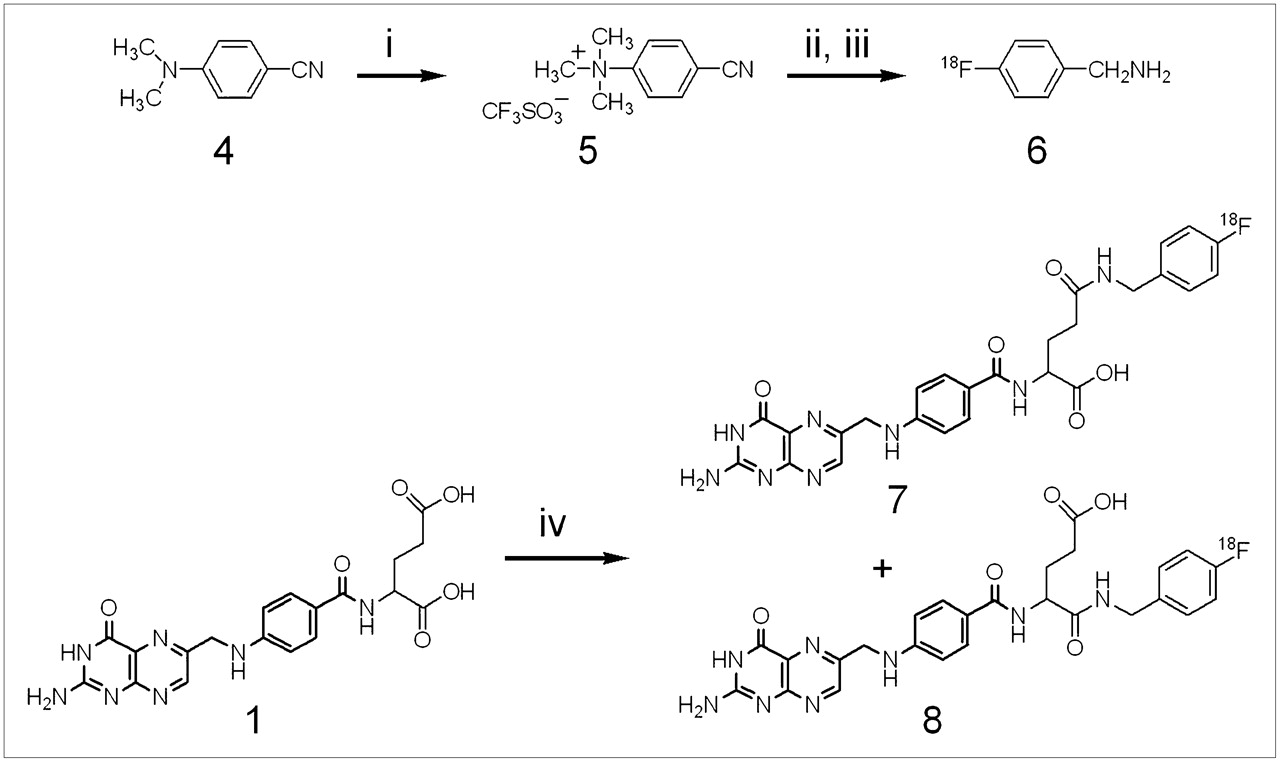
18F labeling was performed by no-carrier-added nucleophilic radiofluorination on 4-cyano-N,N,-trimethylanilinium trifluoromethane sulfonate (Fig. 2) using the same conditions as those reported by Dolle et al. (21). 4-18F-Benzonitrile was purified over a C18 Sep-Pak cartridge, and the nitrile functionality was reduced to that of the corresponding amine using a saturated THF solution of LiAlH4 instead of dry, powdered LiAlH4. The 18F-labeled prosthetic group was isolated by extraction with an Extralut 3 cartridge with a radiochemical yield (decay corrected) ranging from 8% to 13%. The final step of the synthesis, an in situ generation of activated folic acid, was accomplished using N-(3-dimethylaminopropyl)-N-ethyl-carbodiimide and HOBt 5 min before initiation of the coupling reaction. The yield of the coupling step after HPLC purification ranged from 15% to 44% (decay corrected), and the specific radioactivity ranged from 7 to 24 GBq/μmol. The product (a mixture of α- and γ-isomers) eluted as a broad peak with a retention time of 25 min.
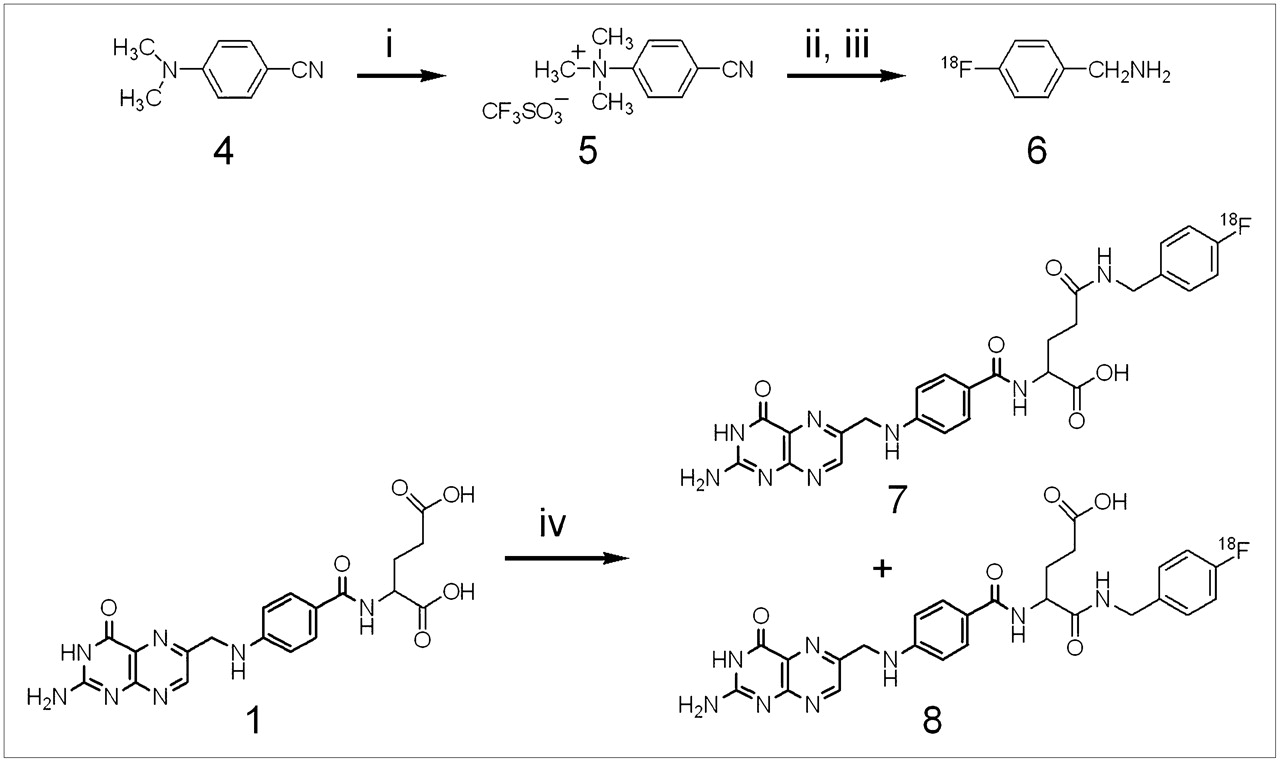
Radiosynthesis of 18F-labeled α- and γ-FBA-folate: (i) methyl trifluoromethane triflate, argon atmosphere, reflux, 6 h; (ii) 18F-Kryptofix-K222, DMSO 120°C, 20 min; (iii) LiAlH4, THF, 120°C, 12 min; (iv) 1, N-(3-dimethylaminopropyl)-N-ethyl-carbodiimide, HOBt, DMSO, 100°C, 30 min.
18F-labeled FBA-folate was characterized by analytic HPLC by coinjection with a reference compound (γ-FBA-folate) using an eluent buffered to pH 7. Under these conditions, only 1 radioactive peak was observed (data not shown). In contrast, at pH 3.5, 2 distinct radioactive peaks corresponding to the γ- and α-isomers were identified (Fig. 3). The γ/α-isomeric ratio was 4:1.

Analytic HPLC of 18F-labeled folic acid derivative as obtained after semipreparative HPLC purification and formulation. Column: SphereClone 5μ ODS(1) (250 × 4.6 mm; Phenomenex); eluent A: 95% trimethylammonium phosphate (pH 3.5) and 5% CH3OH; eluent B: 100% CH3OH; linear 0%−80% B over 30 min; flow: 0.8 mL/min.
Binding Assays
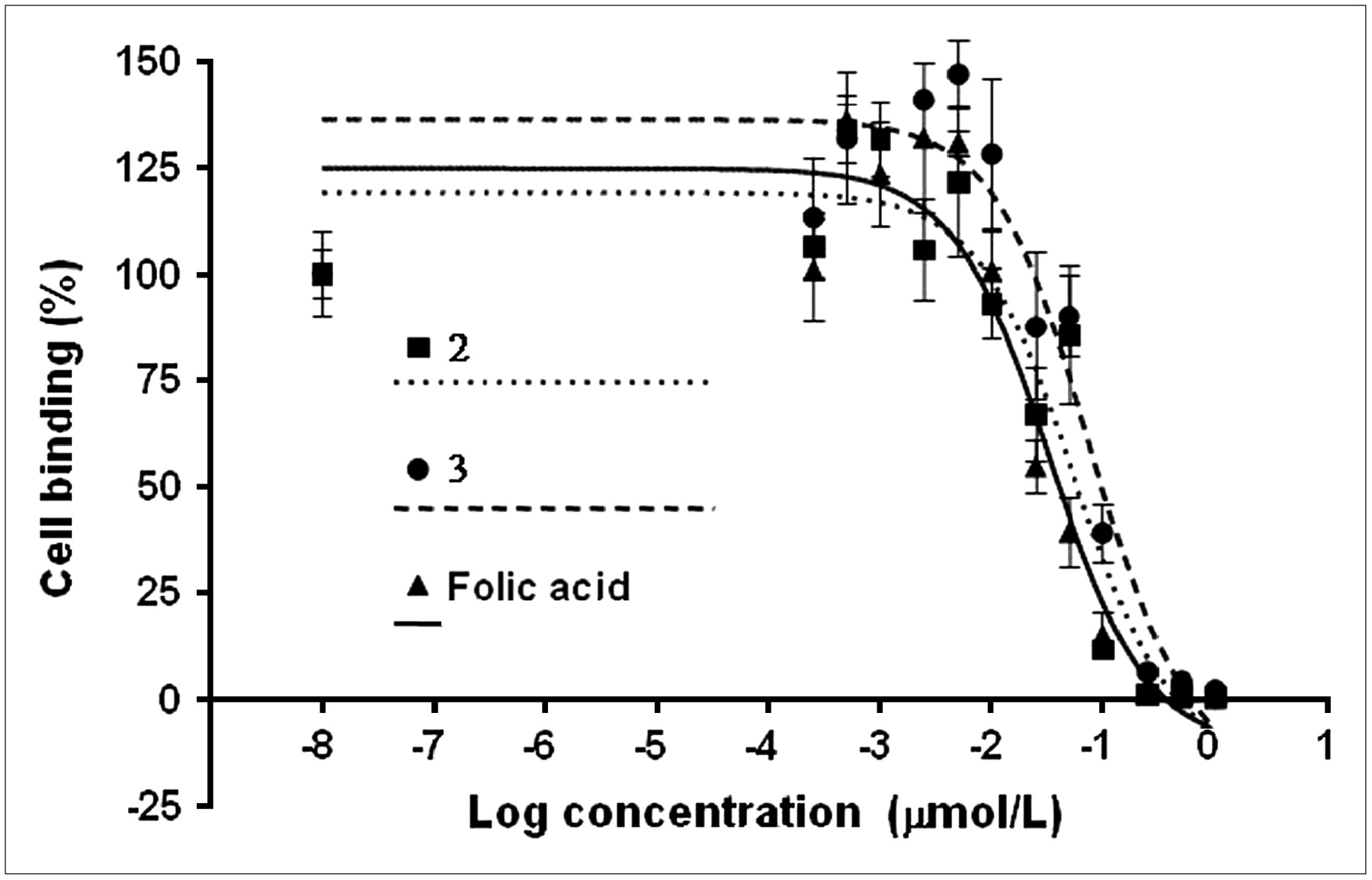
As reported in a previous investigation (12), the cells were grown in folate-deficient medium to reproduce a folic acid concentration similar to that of human plasma. The IC50 values were obtained by inhibition experiments using 3H-folic acid. Three representative binding curves are shown in Figure 4. The mean IC50 values for the α- and γ-regioisomers were 71 ± 8 and 62 ± 6 nmol/L, respectively, suggesting that the binding affinities of the α- and γ-regioisomers do not significantly differ (P > 0.05) and are comparable to that of native folic acid (IC50 = 41 nmol/L; P > 0.05).
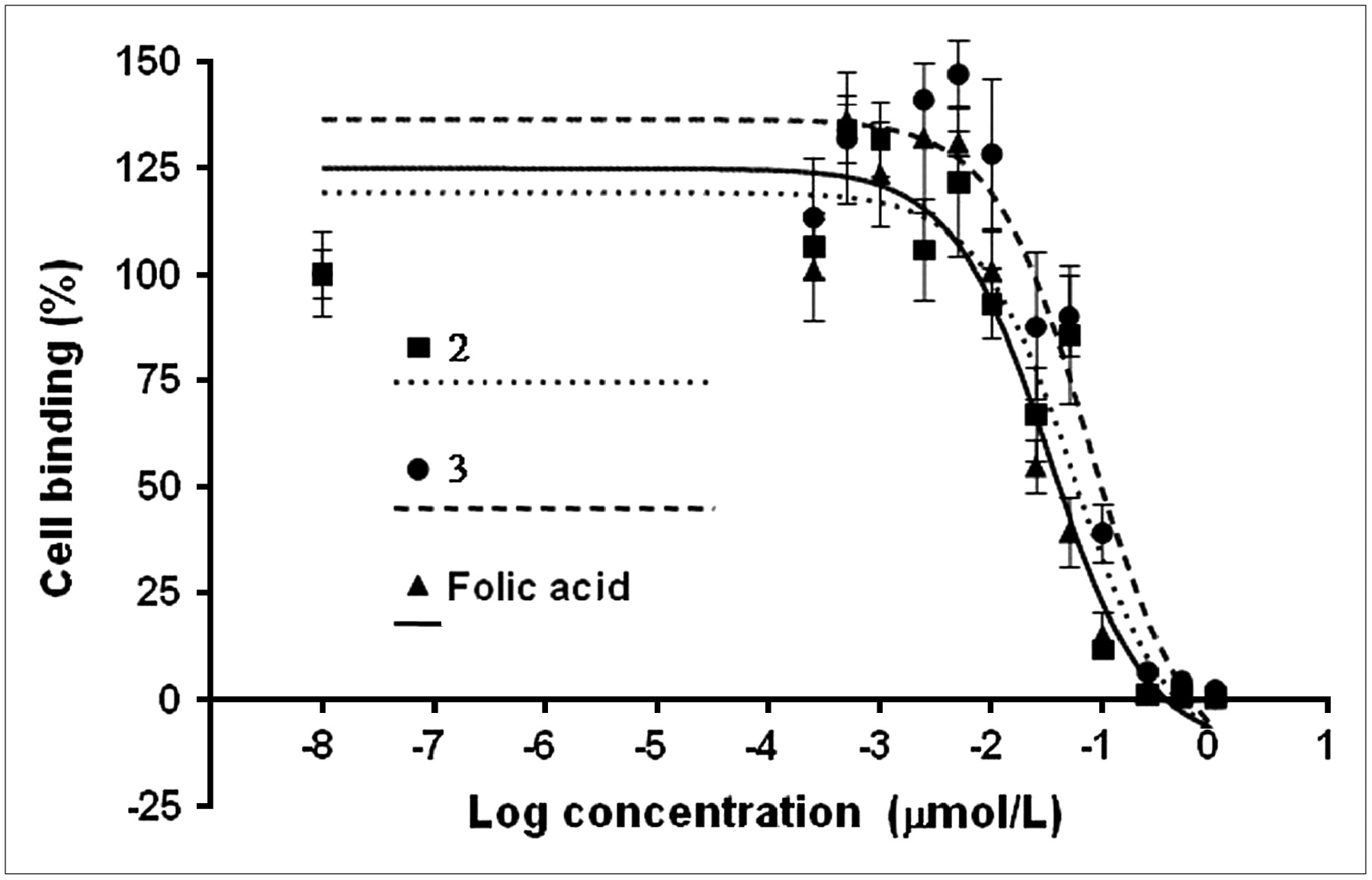
Determination of IC50 values using KB-31 cells (human nasopharyngeal epidermal carcinoma cell line overexpressing folate receptor): inhibition of 3H-folic acid with folic acid and folic acid derivatives 2 and 3. Curves do not correspond to values in Table 1 representing average of different experiments.
In Vivo Studies
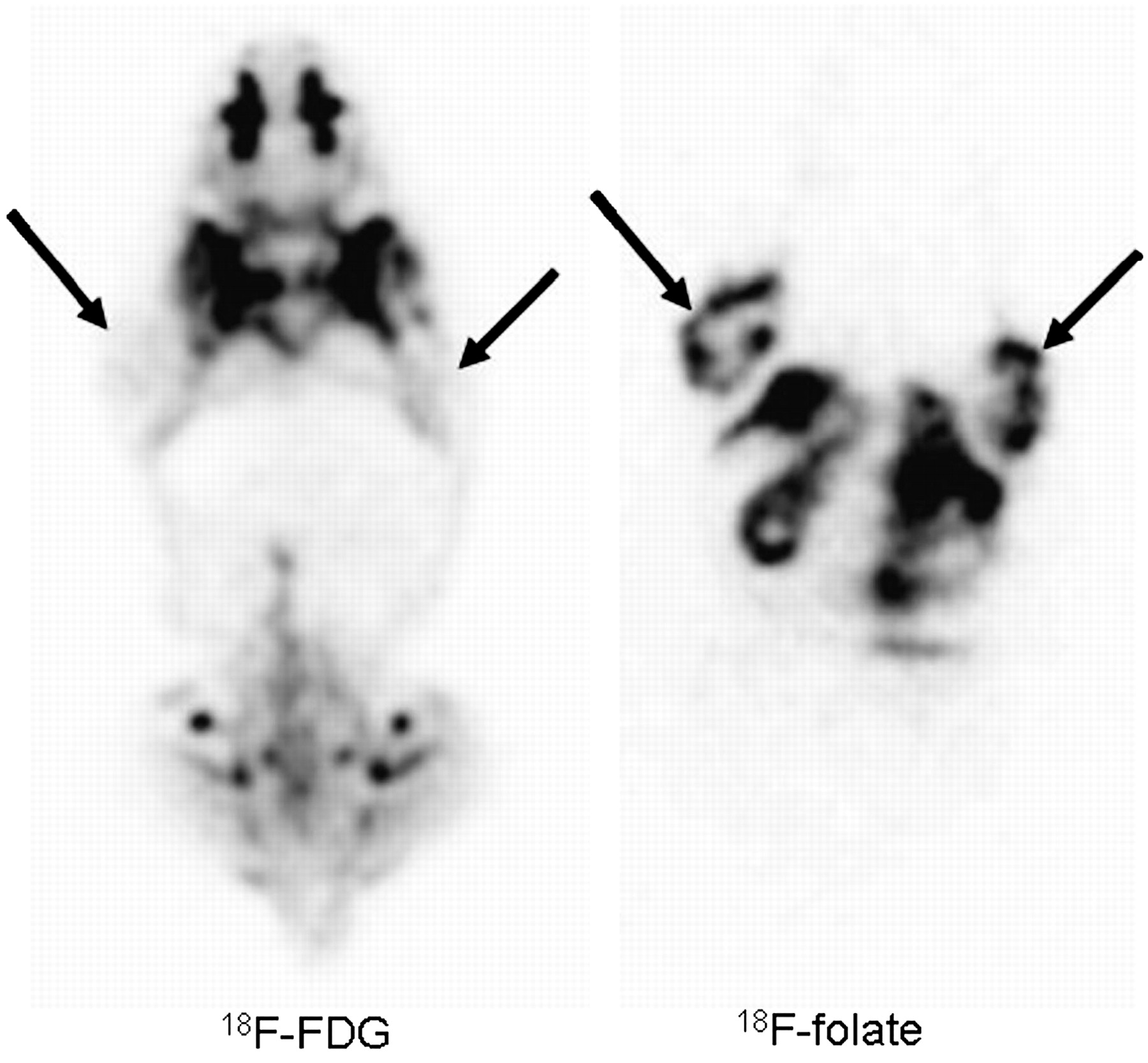
The high resolution of the quad-HIDAC camera allowed visualization of heterogeneous tracer uptake within the tumor (Fig. 5). Higher activity concentrations were observed in the tumor rim, suggesting increased perfusion or folate receptor expression in this region. For a direct comparison of 18F-FDG with 18F-α/γ-FBA-folate, a tumor-bearing mouse was scanned after injection of 18F-FDG and rescanned 2 d later after injection of the 18F-labeled folic acid derivative. The PET images in Figure 5 represent corresponding coronal whole-body sections through the tumor and clearly demonstrate the utility of the 18F-labeled folic acid derivative for visualizing KB-31 tumor xenografts. 18F-FDG accumulation in this KB-31 tumor model was only marginal (18).

PET imaging of 18F-FDG vs. 18F-α/γ-FBA-folate in same animal: Athymic nude mouse (24.5 g) on folate-free diet with 2 KB-31 tumors located dorsally on right and left sides of upper thorax (arrows) was scanned 30 min after injection of 16.9 MBq of 18F-FDG (scan duration, 30 min). Two days later, same animal was imaged again 75 min after injection of 13.3 MBq of 18F-labeled folate derivative (scan duration, 45 min). PET images represent corresponding coronal whole-body sections through tumor. Moderate 18F-FDG uptake was observed in various muscles, whereas for 18F-labeled folate derivative, highest accumulation of radioactivity was observed in liver and kidneys.
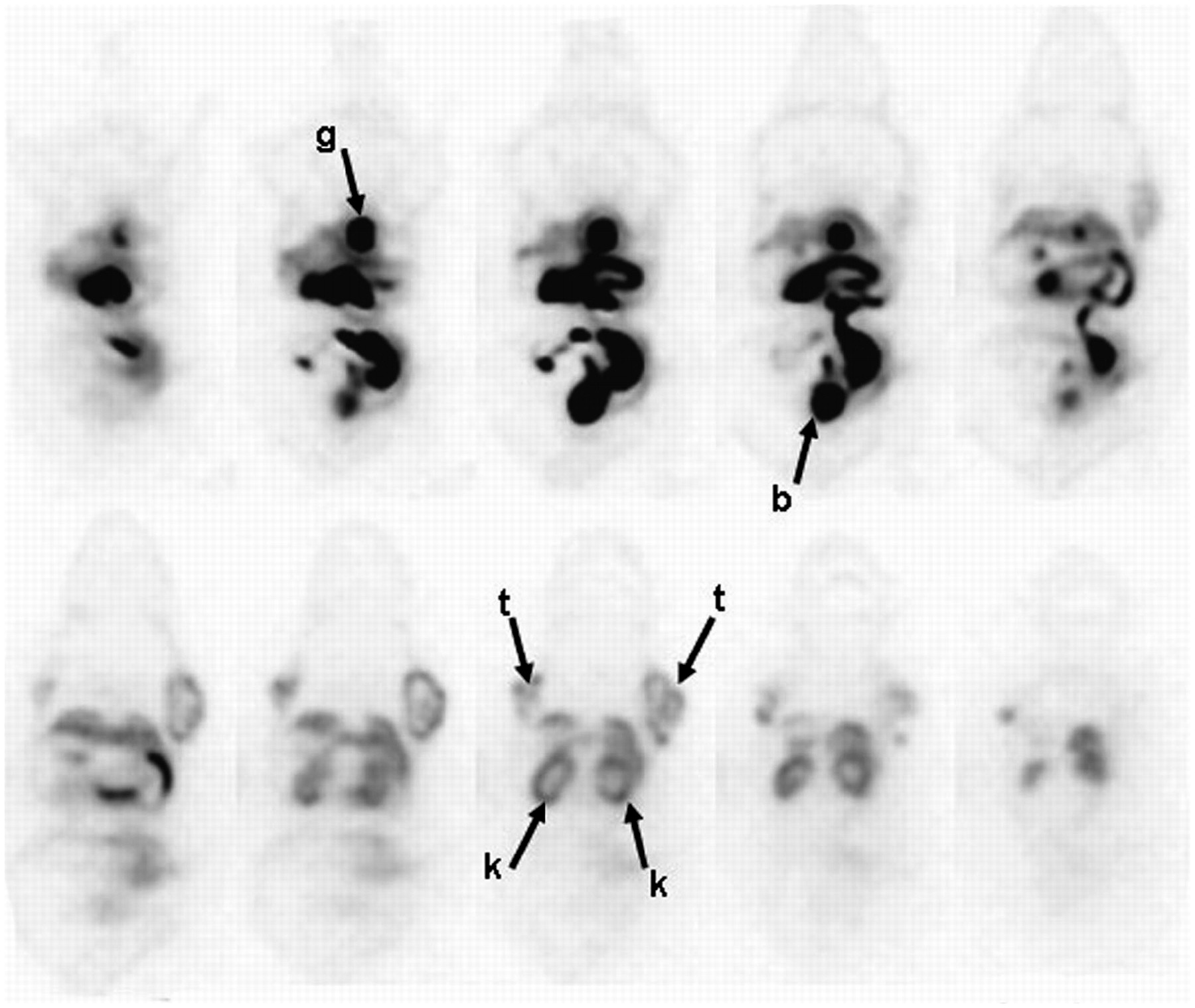
The whole-body distribution of 18F-α/γ-FBA-folate was then assessed by PET from 75 to 120 min after injection. Figure 6 shows a series of coronal slices from ventral to dorsal through the whole body of the mouse. Both the hepatobiliary and the renal elimination pathways of the radioligand dominated the whole-body distribution pattern, with the highest activity concentrations in the gallbladder, urinary bladder, and parts of the intestines. Moderate activity accumulation was evident in the kidneys, tumors, and liver.

Whole-body distribution of 18F-α/γ-FBA-folate visualized on PET: Athymic nude mouse (22.9 g) on folate-free diet was injected with 12.5 MBq of radiotracer and scanned for 45 min at 75 min after injection. PET images represent series of coronal slices of a single experiment from ventral to dorsal through whole body of mouse. b = bladder; g = gallbladder; k = kidneys; t = KB-31 tumors.
To demonstrate the specificity of radioligand binding to the folate receptor–positive KB-31 tumor xenografts, we performed classic postmortem biodistribution studies under baseline and blockage conditions. The blockage group received 200 μg of folic acid 10 min before the radioligand, and the control group was injected with a corresponding volume of phosphate buffer. Radioactivity uptake in tumors at 125 min after injection amounted to 6.56 %ID/g in control animals, whereas radioactivity accumulation in the tumors of blockage animals was significantly reduced by more than 80%—to 1.07 %ID/g (Table 1; P = 0.001). An even larger blockage effect (>97%) was observed in the kidneys, where radioactivity uptake was reduced from 40.65 to 1.16 %ID/g. The highest accumulation of activity in control animals was found in bile, urine, and feces, thus confirming the distribution pattern revealed by PET (Fig. 6).
Postmortem Biodistribution Study of 18F-α/γ-FBA-Folate at 125 Minutes After Injection in Athymic Nude Mice
The in vivo binding specificity of 18F-α/γ-FBA-folate to the folate receptor on KB-31 tumor cells was also demonstrated by PET. A nude mouse with 2 KB-31 tumors was scanned under control conditions and imaged 24 h later under blockage conditions. Both PET scans were initiated 75 min after injection of the radiotracer and lasted for 45 min. Figure 7 shows PET images of corresponding coronal whole-body sections through the KB-31 tumors and through the kidneys under both experimental conditions. Tumors and kidneys were visualized only in the absence of competing folic acid, supporting the specificity of 18F-α/γ-FBA-folate binding to the folate receptor in vivo.
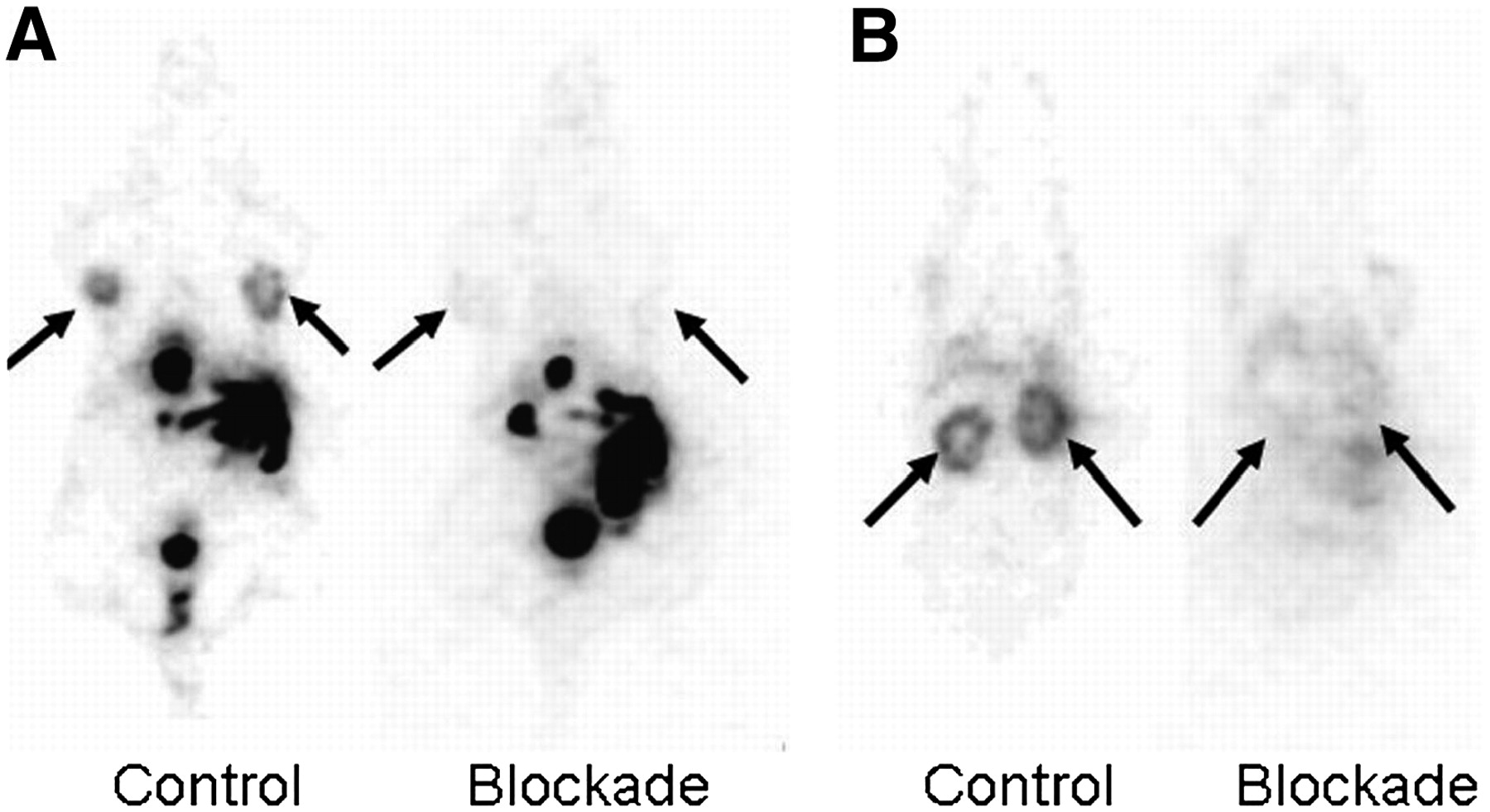
In vivo binding specificity of 18F-α/γ-FBA-folate: Athymic nude mouse (24.5 g) on folate-free diet with 2 KB-31 tumors was scanned after radiotracer injection under control conditions (100 μL of phosphate buffer injected 10 min before radiotracer) and 24 h later under blockage conditions (200 μg of folic acid injected 10 min before radiotracer). Both PET scans were initiated 75 min after injection of tracer and lasted for 45 min. PET images represent corresponding coronal whole-body sections through KB-31 tumors (A; arrows) and through kidneys (B; arrows) and are normalized to injected dose per body weight.
DISCUSSION
Tumor imaging with 18F-FDG, the most widely used PET tracer, is hampered by false-positive findings. A receptor-mediated uptake is an attractive approach for tumor imaging because this approach involves cell-selective radiopharmaceutical targeting. The folate receptor is known to be overexpressed in a variety of human epithelial carcinomas (2), making these tumors excellent targets for diagnostic imaging with radiolabeled folic acid conjugates. But for folate conjugates, it is still unclear whether a free α-carboxyl group is necessary for retaining binding to the folate receptor (12,22), and thus we found it necessary to prepare well-defined unlabeled γ- and α-folic acid derivatives to clarify this point. The binding experiments performed on the unlabeled γ- and α-FBA-folates using 3H-folic acid indicated that both regioisomers exhibited similar binding affinities to KB-31 cells, corroborating previous data obtained with 99mTc-folate derivatives (12) and suggesting further that linking a prosthetic group such as 4-fluorobenzylamine to folic acid via either the γ- or the α-carboxylate does not have a detrimental effect on the binding affinity of the corresponding folate conjugate. These data encouraged us to pursue the 18F labeling of folic acid. It is important to note that the synthetic procedure (Fig. 2) adopted for the 18F-labeled folic acid derivative is different from the method used in the synthesis of the unlabeled γ- and α-FBA-folates. The radiochemical yield obtained for 18F-4-flurobenzylamine was in the range of 8%−13%. The coupling step was accomplished using standard coupling reagents and gave, after HPLC purification, satisfactory radiochemical yields. However, the overall radiochemical yield was low because of material loss during purification of the prosthetic group. The use of a cartridge containing the same type of material but of reduced size may improve the yield of this step and, consequently, the overall radiochemical yield. The difference in the pKa value of the γ- and α-carboxyl-groups (23) allowed us to distinguish between the 2 regioisomers using analytic HPLC at pH 3.5 (Fig. 3), with the major peak being the γ-regioisomer. More detailed investigations are, however, required to determine whether the predominance of the γ-derivative is due to a rearrangement that progressively converts the α-isomer to the more thermodynamically stable γ-isomer. The analytic HPLC method used to distinguish the 2 regioisomers may potentially be used for the semipreparative purification; however, this use is not practicable because of the insolubility of the folic acid derivative at pH 3.5.
The 18F-FDG studies on a nude mouse were performed to compare 18F-FDG uptake with the new 18F-labeled folic acid derivative but found no significant accumulation of 18F-FDG in the KB-31 tumor–bearing mouse. With our new 18F-labeled folic acid derivative, KB-31 tumors were clearly visualized. The high-resolution quad-HIDAC tomograph permitted the delineation of heterogeneous tracer uptake within the tumor. The highest accumulation of activity was in the tumor rim, but even this peripheral zone of the tumor revealed a spotted distribution pattern (Fig. 5), suggesting heterogeneous folate receptor expression or perfusion in this region. Because of a prominent hepatobiliary elimination of the radiotracer, high activity concentrations were observed in the gallbladder and, consequently, in the intestines. These high concentrations may be problematic for clear visualization of abdominal tumors near the intestines, bladder, or kidneys. Hepatobiliary and renal elimination of the radioligand was also evident from postmortem dissection studies. As expected from the distribution pattern observed on PET, the highest activity concentrations were observed in bile, urine, feces, and kidneys (Table 1). Apart from these locations, the KB-31 tumors showed the highest radioactivity uptake (6.56 %ID/g at 125 min after injection). Tumor-to-blood and tumor-to-liver ratios amounted to 4.6 and 2.8, respectively. Other tumor-to-organ ratios gave even higher values. The in vivo specificity of the new ligand was confirmed by a repeated PET study on a single animal within 24 h, first under baseline conditions and then under blockage conditions. The comparison of normalized PET images proved the specificity of tracer binding in the tumors and the kidneys (Fig. 7). The specificity of binding to folate receptor was also clearly demonstrated by classic postmortem biodistribution studies. The dissection experiments revealed a highly significant difference in %ID/g values for tumors and kidneys, resulting in a blockage of 84% and 97%, respectively. The larger value for kidneys may be explained by a higher target protein density in kidneys or a higher nonspecific tracer accumulation in tumors. The high degree of specific binding observed in the folate receptor–positive tumors and the kidneys confirms previous data obtained with 99mTc-folate derivatives (9) and shows that this new PET ligand is also capable of efficiently imaging folate receptor–positive tumors in an animal model. Because folate receptors are significantly overexpressed in most human tumors, this new 18F-labeled folate derivative may be a promising diagnostic tool for investigating folate receptor–positive tumors in humans using PET. For some selected human tumors in which the use of 18F-FDG is unsatisfactory, folate receptor–targeted radiopharmaceuticals may show promise.
CONCLUSION
γ-FBA-folate and α-FBA-folate were evaluated in vitro. Both regioisomers exhibited similar binding affinities, which were comparable to that of parent folic acid. We also established a method for the 18F labeling of folic acid. The method uses commercially available folic acid as a precursor and circumvents time-consuming chemical modifications of the native compound such as protection/deprotection. In vivo studies indicated that the new radioligand binds selectively to folate receptor–positive KB-31 tumor xenografts. Therefore, this new 18F-labeled folic derivative is a promising tool for PET imaging of folate receptor–positive tumors.
Acknowledgments
We thank Andy Isenschmid and Yvonne Eichholzer for the 18F− production; Cecile Dumas, Alain Blanc, and Marie-Line Lehaire for the nuclear magnetic resonance and mass spectrometry measurements; Claudia Keller and Matthias Wyss for support with the in vivo studies; and Anass Johayem for technical support.
Footnotes
-
COPYRIGHT © 2006 by the Society of Nuclear Medicine, Inc.
References
- Received for publication October 7, 2005.
- Accepted for publication March 21, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Imaging the Folate Receptor on Cancer Cells with 99mTc-Etarfolatide: Properties, Clinical Use, and Future Potential of Folate Receptor Imaging
- Folic Acid Conjugates for Nuclear Imaging of Folate Receptor-Positive Cancer
- A New 18F-Labeled Folic Acid Derivative with Improved Properties for the PET Imaging of Folate Receptor-Positive Tumors
- Pemetrexed Improves Tumor Selectivity of 111In-DTPA-Folate in Mice with Folate Receptor-Positive Ovarian Cancer
- SPECT Study of Folate Receptor-Positive Malignant and Normal Tissues in Mice Using a Novel 99mTc-Radiofolate